
Formins in Human Disease


Centro de Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas, Universidad Autónoma de Madrid, 28049 Madrid, Spain
*
Author to whom correspondence should be addressed.
Cells 2021, 10(10), 2554; https://doi.org/10.3390/cells10102554
Submission received: 1 July 2021
/
Revised: 20 September 2021
/
Accepted: 22 September 2021
/
Published: 27 September 2021
(This article belongs to the Collection Feature Papers in ‘Cellular Pathology’)
Abstract
:Almost 25 years have passed since a mutation of a formin gene, DIAPH1, was identified as being responsible for a human inherited disorder: a form of sensorineural hearing loss. Since then, our knowledge of the links between formins and disease has deepened considerably. Mutations of DIAPH1 and six other formin genes (DAAM2, DIAPH2, DIAPH3, FMN2, INF2 and FHOD3) have been identified as the genetic cause of a variety of inherited human disorders, including intellectual disability, renal disease, peripheral neuropathy, thrombocytopenia, primary ovarian insufficiency, hearing loss and cardiomyopathy. In addition, alterations in formin genes have been associated with a variety of pathological conditions, including developmental defects affecting the heart, nervous system and kidney, aging-related diseases, and cancer. This review summarizes the most recent discoveries about the involvement of formin alterations in monogenic disorders and other human pathological conditions, especially cancer, with which they have been associated. In vitro results and experiments in modified animal models are discussed. Finally, we outline the directions for future research in this field.
1. Introduction
The human formin family consists of fifteen members (Figure 1A), divided into seven subfamilies [1], many of which are co-expressed in many tissues (Supplementary Tables S1 and S2). Formins are involved in the polymerization of monomeric actin into linear filaments [2,3]. All formins possess two characteristic domains: a formin homology (FH) 2 domain, which catalyzes actin polymerization, and an FH1 domain, which binds profilin to provide monomeric actin to the FH2 domain. The other regions and domains can differ between formin subfamilies and are involved in regulatory mechanisms or specific interactions with other proteins. In addition to regulating the actin cytoskeleton, formins bind to microtubules through the FH2 domain and regulate the acetylation and stability of microtubules, and their alignment with actin filaments [4,5].
Members of the human Diaphanous-related formin subfamily, which includes Diaphanous homolog (DIAPH) 1-3, are regulated through the interaction of the Diaphanous inhibitory domain (DID) at the N-terminal end and the Diaphanous autoregulatory domain (DAD) at the C-terminal region [1]. The transition between closed/inactive and open/active states is mediated by the interaction of the Rho-family GTPases with the DID, which releases its interaction with the DAD (Figure 1B). Other formins with similar regulation are Disheveled-associated activators of morphogenesis (DAAM) 1 and 2, formin-like (FMNL) 1-3, and FH1/FH2 domain-containing (FHOD) 1 and 3.
The mutation of some of the formin genes causes monogenic disorders, as is the case of DIAPH1, which was the first formin gene found to be linked to a human Mendelian disorder [6]. Alteration of seven formins genes (Figure 2) have been acknowledged to date by the Online Mendelian Inheritance in Man (OMIM®, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD, USA), https://omim.org; accessed on 20 September 2021) as meeting the criteria for consideration as a primary cause of human monogenic disorders [7,8]: DIAPH1-3 [6,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36] (Supplementary Tables S3–S5), DAMM2 [37] (Supplementary Table S6), FORMIN2 [38,39,40,41,42] (FMN2) (Supplementary Table S7), INVERTED FORMIN 2 (INF2) [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94] (Supplementary Table S8) and FHOD3 [95,96,97,98,99,100] (Supplementary Table S9). The mutations or dysregulation of the other formins have not been demonstrated to be the primary cause of the phenotype, although they probably contribute to it [101,102]. Mutant formins can alter specific organs by affecting the functioning of specific types of cell (Figure 3A,B). Here, we review the involvement of formin genes in human monogenic disorders, comment on the association of other formin genes with similar or other disorders, and discuss the experimental evidence (from in vitro experiments, modified animal models, etc.) of their roles in disease.
Figure 1.
The human formin family. (A) Tree of human formins. The FH2 domain sequence of the formins was aligned with BLAST and the alignment was used to construct the tree [103]. The UniProt accession numbers of the corresponding sequences were: DIAPH1 (O60610), DIAPH2 (O60879), DIAPH3 (Q9NSV4), DAAM1 (Q9Y4D1), DAAM2 (Q86T65), FMNL1 (O95466), FMNL2 (Q96PY5), FMNL3 (Q8IVF7), FHOD1 (Q9Y613), FHOD3 (Q2V2M9), FMN1 (Q68DA7), FMN2 (Q9NZ56), INF2 (Q27J81), FHDC1 (Q9C0D6) and Delphilin (A4D2P6). (B) Structure and regulation of Diaphanous-related formins. The interaction of the DID and the DAD maintains the formin in a closed, inactive conformation. The binding of a specific GTP-loaded Rho GTPase to the N-terminal region of the formin opens the molecule, rendering it in its active form. The FH1 domain recruits profilin, which feeds the FH2 domain with G-actin to form the actin filaments. The illustrated molecules are not drawn to scale.
Figure 1.
The human formin family. (A) Tree of human formins. The FH2 domain sequence of the formins was aligned with BLAST and the alignment was used to construct the tree [103]. The UniProt accession numbers of the corresponding sequences were: DIAPH1 (O60610), DIAPH2 (O60879), DIAPH3 (Q9NSV4), DAAM1 (Q9Y4D1), DAAM2 (Q86T65), FMNL1 (O95466), FMNL2 (Q96PY5), FMNL3 (Q8IVF7), FHOD1 (Q9Y613), FHOD3 (Q2V2M9), FMN1 (Q68DA7), FMN2 (Q9NZ56), INF2 (Q27J81), FHDC1 (Q9C0D6) and Delphilin (A4D2P6). (B) Structure and regulation of Diaphanous-related formins. The interaction of the DID and the DAD maintains the formin in a closed, inactive conformation. The binding of a specific GTP-loaded Rho GTPase to the N-terminal region of the formin opens the molecule, rendering it in its active form. The FH1 domain recruits profilin, which feeds the FH2 domain with G-actin to form the actin filaments. The illustrated molecules are not drawn to scale.

2. Monogenic Disorders Caused by Formin Mutation
2.1. Nephrotic Syndrome and Charcot-Marie-Tooth Disease
Blood filtration and the concentration of metabolic waste into urine take place in the renal glomeruli. Podocytes are terminally differentiated cells that wrap around endothelial cells of glomerular capillaries by means of elaborate projections known as foot processes (Figure 3B). The contact between two of these processes forms a slit diaphragm, which is the structure responsible for blood filtration [104]. Deficient blood filtration causes nephrotic syndrome, which is characterized by proteinuria, hypoalbulinemia, hyperlipidemia, and edema, and can end in renal failure.
Focal segmental glomerulosclerosis (FSGS) refers to a histological lesion of scarred appearance present in localized regions of some, but not all, glomeruli [105]. The INF2 gene is the formin for which the greatest number of pathogenic mutations has been described (Figure 2 and Supplementary Table S8) [106]. INF2 pathogenic mutations are autosomal dominant and produce FSGS (FSGS5, MIM: 613237) [43], which causes steroid-resistant nephrotic syndrome [107,108]. Depending on the specific mutation, FSGS co-occurs (or not) with Charcot–Marie–Tooth disease (CMTDIE, MIM: 614455) [48], which is a neuropathy affecting the functioning of the peripheral nerves that produces progressive distal muscle weakness [109]. All the INF2 disease-related mutations localize to the DID and the great majority are of the missense type. Genomic-wide screening (GWS) and whole-exome sequencing (WES) analyses in patients with renal disease identified a number of variants outside the INF2 DID [106] but, with the exception of a case of FSGS combined with CMT with a deletion in the DAD [94], it is not clear whether these variants are related to the pathogenic condition.
An in silico analysis of the effect of the pathogenic mutations indicates that they have a destabilizing structural effect in the DID [106]. This destabilization might affect the interaction of the DID with the DAD or with regulatory proteins [110,111], and results in gain-of-function of the actin polymerization activity of INF2. The case of a patient with combined FSGS and CMT has been described, in which a complete duplication of the INF2 gene occurred, which represents further evidence of a gain-of-function phenotype in INF2-linked disease [93]. It is of note that the mutations causing combined FSGS and CMT are generally more destabilizing than those producing only FSGS, and that these two types of mutation distribute in the DID in a different manner, with the former being concentrated in the N-terminal half of the DID, whereas those causing only FSGS are distributed throughout the DID [106].
FSGS patients suffer a progressive loss of podocytes, which decreases the filtration capacity of the kidney. INF2-linked FSGS starts to become clinically relevant in adolescence or adulthood, causing glomerular dysfunction [107,108]. It is still not clear how INF2 mutations affect podocytes but, consistent with the enhanced actin polymerization activity of the pathogenic INF2 mutants [110], aberrant actin bundles have been observed in a renal biopsy of an affected patient [43]. Knock-in mice expressing the most common mutation, p.Arg218Gln, exhibit no apparent alteration in podocyte structure unless they are exposed to acute kidney injury [112]. This finding is consistent with the degenerative nature of INF2-related disease and suggests that FSGS might be the result of repeated kidney insults in individuals in which INF2 mutation makes them more prone to developing the disease. In addition to FSGS, INF2 mutations have been found to contribute to, or be responsible for, other kidney conditions (Figure 2 and Supplementary Table S8). In patients with combined FSGS and CMT, CMT symptoms appear in childhood, and renal damage appears earlier in life than in patients with only FSGS. In the cases with CMT, pathogenic INF2 affects Schwann cell polarization (Figure 3B), leading to abnormal myelin formation and/or maintenance [113,114]. The manifestation of FSGS alone is common in individuals with pathogenic INF2, but only one case of CMT has been described to date that makes the absence of accompanying renal disease explicit [59].
Recessive mutations of DAAM2 (Figure 2 and Supplementary Table S6), have recently been involved in nephrotic syndrome, type 24 (NPHS24, MIM: 606627) [37]. All the affected individuals presented FSGS with no extra-renal manifestations. The mutations were found in homozygosity in three individuals from consanguineous families, and in one individual with two different missense mutations from an outbred family. The missense mutations map to the region encoding the DID, the FH1 or the DAD, whereas the nonsense mutation maps immediately downstream of the DID and generates a truncated DAAM2 protein. The mutations at the DID and DAD appear to cause increased autoinhibition and, consequently, loss-of-function of actin polymerization activity. DAAM2, which is expressed by podocytes, colocalizes and associates with INF2 [37], suggesting the existence of crosstalk between the two formins that, given the link between INF2 and renal disease, may explain the renal damage caused by pathogenic DAAM2. Other formins might be involved in other kidney disorders. For instance, Fmn1 has recently been identified as a candidate modifier gene in X-linked Alport syndrome in mice, which is a genetic disease characterized by hearing loss, hematuria and, eventually, renal failure [115].
2.2. Hearing Loss
Hearing depends on the correct mechanostransduction of sound vibrations into electrical signals. This takes place in the organ of Corti, which is located in the cochlea in the inner ear. The cells responsible for this process are sensory epithelial cells, known as hair cells (Figure 3B), which possess dozens of stereocilia on their apical surface, formed of bundles of actin filaments [116]. Outer hair cells amplify the signal, and the perturbation activates the opening of ion channels at the stereocilia tips of inner hair cells, depolarizing the plasma membrane. This perturbation is subsequently transmitted by neurotransmitters released at the synaptic ribbon between the basolateral surface of hair cells and the auditory nerve. This generates electrical impulses in the latter that are transmitted to the brain, where they are decoded and analyzed in the auditory cortex. Given the importance of actin in the architecture of stereocilia, mutations in actin, actin-binding proteins, and the machinery involved in actin filament formation and function, including formins, can all cause hearing loss [117].
Sensorineural hearing loss is caused by dysfunction of the inner ear or the auditory nerve. Mutations in DIAPH1 produce deafness, autosomal dominant 1 (DFNA1, MIM: 124900). In this disorder, auditory loss generally starts during the first decade of life, although there are cases with intrafamilial variability. Since the identification of a mutation in DIAPH1 as the cause of sensorineural hearing loss in a large Costa Rican family [6], more families with a dominant pedigree caused by DIAPH1 mutation have been described elsewhere in the world [11,12,13,18,19,21,22,23,24,25,26,27]. Affected individuals present frameshift or nonsense mutations or deletions near the DAD. These types of mutation create truncated forms of DIAPH1 that lack different segments of the carboxyl-terminal region of the molecule. In addition, more recently, missense mutations have been described at the DID and the coiled-coil downstream region and FH2 domain (Figure 2 and Supplementary Table S3). The pathogenic p.Arg1204X mutation [21] results in early termination immediately before a basic amino acid motif (RRKR1204–1207) present at the DAD C-terminus, which is important for the interaction with the DID [118]. This mutation partially relieves the autoinhibitory DID-DAD interaction, resulting in a mildly constitutive active molecule [21]. It is likely that this also occurs with other truncation mutations mapping around this site and with the missense mutations in the DID [119,120]. The DIAPH1 gene mouse homolog, mDia1, is expressed in the organ of Corti during and after cochlear maturation, and localizes at the apical junctional complexes between the supporting cells and the hair cells [121]. As further evidence that hearing loss caused by DIAPH1 mutations is due to gain- and not to loss-of-function, hearing progressively deteriorates in transgenic mice overexpressing wild-type mDia1 [121], whereas the hearing function of the mDia1 knock-out (KO) mice is not different from that of control mice [21]. The hearing defect in mice overexpressing mDia1 is associated with gradual loss of hair cells and the appearance of sparse and short or fused stereocilia cells [121]. A similar phenotype was observed in transgenic mice expressing the human Arg1204X mutant [21]. Increased gene dosage of DIAPH1 has been documented in several cases of sporadic sensorineural hearing loss in humans [28]. These findings are further evidence that deafness-associated mutations of DIAPH1 cause disease by increasing actin polymerization activity, which causes the disorganization and dysfunction of stereocilia.
Auditory neuropathy, autosomal dominant, 1 (AUNA1, MIM: 609129) is characterized by abnormal or absent auditory brainstem responses but preserved cochlear outer hair cell function. A mutation (c.-172G > A) in a highly conserved GC element at the exon encoding the 5′ untranslated region of DIAPH3, was the first to be described as being involved in AUNA1. This mutation, which is probably of the gain-of-function type, results in 2- to 3-fold overexpression of DIAPH3 mRNA and 1.5-fold overexpression of DIAPH3 protein levels. Consistent with increased levels of DIAPH3 as the cause of the auditory alterations, flies expressing a constitutively active form of Diaphanous, which encodes the sole Diaphanous-related formin in Drosophila, show an impaired response to sound [36]. Two reports found that mice overexpressing mouse mDia2, the murine homolog of human DIAPH3, present progressive impairment of inner hair cell stereocilia, whereas outer hair cells stereocilia and function were not generally affected in the specific mouse lines studied [122,123]. A reduction in the number of ribbon synapses was observed in one study [122], but not in the other [123]. Consistent with the role of formins in regulating microtubule dynamics, the microtubule meshwork undergoes aberrant targeting to the apical aspect of inner hair cells in transgenic mDia2 mice [123], probably contributing to stereocilia collapse. These mice also present early mortality due to cardiac defects, but no similar effect has yet been found in humans. In addition to the c.-172G > A mutation, other mutations causing AUNA1 have been described at the 5′ untranslated region of DIAPH3 mRNA [35] and the DID of DIAPH3 [15] (Figure 2, Supplementary Table S5). Missense variants mapping to the DIAPH3 FH2 domain have also been found in patients with auditory neuropathy spectrum disorders [15,124], but it is not clear whether there they are pathogenic or simply rare variants.
In the case of INF2 mutations associated with combined CMT plus FSGS, but not with FSGS alone, some of the patients also experience hearing loss [48,49,51,57]. Since INF2 mutations causing CMT affect peripheral nerve myelinization [113], auditory nerve damage is probably the cause of the hearing impairment, although hair cell stereocilia may also be affected, as in cases of DIAPH1 and DIAPH3 mutations.
2.3. Thrombocytopenia
The cell precursors of platelets, megakaryocytes, form extensions known as proplatelets, from which platelets are released into the circulatory system [125]. Macrothrombocytopenia is characterized by enlarged and reduced numbers of circulating platelets that can lead to inadequate clot formation and an increased risk of bleeding [125]. Platelet production begins with the extension of long membrane protrusions that are elongated by microtubule bundles to form proplatelet processes (Figure 3B). Amplification of the number of processes, which occurs by repeated bending and bifurcation, depends on actin filament formation [126,127,128]. It is controversial whether actin/microtubule crosstalk-induced proplatelet formation [126] or membrane budding without requiring proplatelet formation is the main mechanism of platelet formation in vivo [129].
Long after the discovery of the DIAPH1 mutation as the cause of DFNA1, affected individuals were found to present asymptomatic thrombocytopenia and, sometimes, asymptomatic mild neutropenia (Figure 2, Supplementary Table S3), which consists of abnormally low levels of neutrophils in the blood. The reduced platelet levels in these patients, the high content of polymerized actin, and the altered microtubule organization and stability observed in the platelets [27] are consistent with the requirement of DIAPH1 for proper proplatelet formation [130,131], and with previous works showing that DIAPH1 coordinates microtubules and the actin cytoskeleton [132,133]. It is of note that a moderate increase in the expression of DIAPH1 could be responsible for the thrombocytopenia associated with diseases caused by mutation in other genes, as may be the case for Roifman syndrome [134]. This is a rare, inherited disease (MIM: 616651) characterized by growth retardation, cognitive delays, skeletal malformations and immunodeficiency, and caused by mutation of the small non-coding RNA gene RNU4TAC [135].
Atypical hemolytic uremic syndrome (AHUS) is characterized by acute renal failure, thrombocytopenia, and microangiopathic hemolytic anemia (loss of blood cells through destruction). Two mutations in the DID of INF2 cause thrombocytopenia in the context of familial AHUS with (p.Val102Asp) or without (p.Arg177His) associated CMT [60]. In AHUS, the thrombocytopenia is due to platelet activation and consumption associated with blood cell destruction, rather than to an alteration in platelet production.
2.4. Microcephaly and Intellectual Disability
According to the Human Protein Atlas (http://www.proteinatlas.org; accessed on 30 August 2021), and consistent with the analyses of mouse brain [136], all the formins are expressed throughout the brain, generally with low regional specificity (Supplementary Table S2). Mutations of DIAPH1, FMN2, and INF2 are associated, to varying degrees, with intellectual disability and neurodevelopmental disorders [137].
DIAPH1 is expressed in neuronal progenitors during brain development [14]. Specific mutations of DIAPH1 cause seizures, cortical blindness (vision loss due to a damage or malfunction in the part of the brain cortex responsible for processing visual information), and microcephaly syndrome (SCBMS, MIM: 616632) (Figure 2, Supplementary Table S3). In contrast to DFNA1-related mutations, SCBMS-associated DIAPH1 mutations are generally of the nonsense type that affects the FH2 domain, are found in homozygosity, and are inherited with an autosomal recessive pattern, suggesting that they produce loss-of-function of DIAPH1 activity [10,14,16,17]. mDia1 KO mice are not microcephalic but, instead, some mice present unilateral dilatation of the ventricles, indicating that the effect of these mutations is species-specific [14]. Unlike mDia1 KO mice, mDia2 KO mice present microcephaly and also hydrocephalus (accumulation of cerebrospinal fluid within the brain) [138]. This phenotype seems to be due to incorrect spindle assembly checkpoint regulation in cortical progenitor cells, causing massive loss of cortical progenitor cells, with the subsequent depletion of neurons [138]. mDia1 and mDia3 double-KO mice present hydrocephalus, but not microcephaly, due to the formation of a periventricular dysplastic (abnormal) mass during brain development [139]. The alteration of the actin cytoskeleton affecting the adherens junctions and progenitors’ polarity seems to be the cause of the ectopic proliferation of neural stem cells in the double-KO mice. In addition to the characteristic SCBMS symptoms, some patients present pathologies related to immunodeficiency, such as recurrent infections, especially respiratory, bronchiectasis (enlargement of parts of the airways of the lung) and lymphoma [10,14,16]. Given that (i) mDia1 KO mice show defects in T cell migration and activation [140,141], (ii) DIAPH1 mutations are associated with mitochondrial dysfunction [10], and (iii) fibroblasts and some lymphocytes from SCBMS patients present mitochondrial alterations [10], it has been proposed that these additional symptoms are due to a defect in the mechanism of T cell activation [10].
A few cases of intellectual disability denominated mental retardation, autosomal recessive 47 (MRT47, MIM: 616193), are produced by mutations of FMN2 [38,39,41,42] (Figure 2, Supplementary Table S7). The genomic alterations consist of homozygous frameshift and nonsense mutations that are always found in consanguineous families, and large de novo heterozygous deletions. One case with sensory processing dysfunction was also associated with a de novo missense mutation [40]. This phenotype is consistent with the role of FMN2 in stabilizing filopodia tip adhesions and regulating the chemotaxis of neuronal grown cones [142,143]. Unlike the effect of FMN2 mutations in humans, Fmn2 KO mice do not present any alteration in the brain [144]. However, double-KO mice of FMN2 and filamin A show greater microcephaly severity and less neural progenitor proliferation compared with the phenotype of single filamin A KO mice. It has been suggested that this additive effect is a consequence of FMN2 and filamin A both forming part of the machinery of the endocytic route of the canonical Wnt pathway that regulates neural progenitor proliferation [145].
Mutations of other formin genes in addition to DIAPH1 and FMN2 have been associated with intellectual disability. In the case of INF2, some patients with FSGS and associated CMT, probably with severe mutations or an unfavorable genetic background, present intellectual disability and central nervous system anomalies [48,49,51]. An almost complete deletion of the FMNL2 gene has been associated with a case of mental retardation [146]. However, since the patient also presented haploinsufficiency in NR4A2, a gene involved in the cerebral dopaminergic system, it is difficult to ascertain whether the disorder is caused by one or both mutations.
2.5. Primary Ovarian Insufficiency
DIAPH2 has been implicated in premature ovarian failure (POF2A, MIM: 300511), also known as primary ovarian insufficiency, which manifests as premature menopause [29,30,31,32,33,34]. The patients generally present translocations of the X chromosome region that includes DIAPH2 (Figure 2, Supplementary Table S4). This gene might be involved in the development of gonads since it is expressed in the ovaries and testes of mouse embryos [29], and, indeed, some of the patients present ovarian dysgenesis (abnormal development) [33,34]. Underlining the importance of DIAPH2, Drosophila with mutations in Diaphanous are sterile due to cytokinesis failure that affects spermatogenesis in males and follicle cell division in females [147].
Consistent with the possibility that formin genes other than DIAPH2 are related to POF, FMN2 has been associated with POF and infertility in mice and in human patients [148,149]. Female Fmn2 KO mice exhibit defects in spindle positioning during meiosis I [144], which explains their low fertility. It has been proposed that upregulated levels of FMNL2 in humans also have a role in female infertility and gynecological health since they promote adenomyosis, which is characterized by the ectopic growth of the endometrium in the uterine walls, which are formed by the myometrium [150].
2.6. Cardiomyopathy
Thirteen out of the fifteen formins are expressed during postnatal development of the heart in mice within a specific timeframe that suggests a role for each formin in this process [151]. FHOD3, which is mainly expressed in the heart and regulates actin assembly in cardiomyocytes [152] (Figure 3B), has been linked to cardiac pathologies [153]. FHOD3 mutations have been associated with hypertrophic (CMH28, MIM: 619402) [95,96,97,98,99,100] and dilated cardiomyopathies [154] (Figure 2, Supplementary Table S9), which are conditions in which the walls of the heart becomes thicker and stiff, and where the heart is enlarged, respectively. As a consequence of these alterations, blood is pumped less effectively. Two intronic variants of FHOD3 have also been related to hypertrophic cardiomyopathy development [155] and a conservative substitution (p.Val1151Ile) with a reduced risk of dilated cardiomyopathy [156]. Fhod3 KO mice present embryonic lethality due to defects in cardiogenesis and in neural tube closure [157], whereas conditional KO mice show that the FHOD3 protein is needed not only for prenatal and postnatal heart development, but also for its maintenance, since adult mice present cardiomegaly and mild impairment of cardiac function [158]. Transgenic mice expressing FHOD3 defective in actin binding have a similar phenotype to that of dilated cardiomyopathy patients [157]. It is likely that the specific domain affected by the mutation, as well as the individual genetic background, could determine the appearance of one or other pathology, although both appear to be inherited in an autosomal-dominant manner. Angiotensin II is an important factor causing blood pressure overload-induced cardiac hypertrophy [159]. In cultured rat cardiomyocytes, angiotensin II signaling regulates FHOD3 activation through phosphorylation of its C-terminal region by ROCK kinase, raising the possibility that pathogenic FHOD3 causes heart hypertrophy by this mechanism [160].
3. Disorders Associated with Formin Mutation and Dysregulation
3.1. Developmental Cardiovascular Disorders
Myotonic dystrophies are multisystemic inherited disorders characterized by progressive muscle weakness, myotonia (prolonged muscle contraction), cardiac defects and early cataracts. Myotonic dystrophic types 1 (DM1; MIM: 160900) and 2 (DM2; MIM: 602668) are caused by the expansion of a trinucleotide (CTG)n or a tetranucleotide (CCTG)n in the 3′ untranslated region of, respectively, the dystrophia myotonica protein kinase (DMPK) and the nucleic acid binding protein (CNBP) genes, respectively [161]. The expression of the expanded transcripts causes aberrant splicing of many genes involved in muscle homeostasis and function [162]. FHOD1, the other member of the FHOD family, has not yet been associated directly with any human disease, but FHOD1 transcripts are known to undergo aberrant splicing in DM1 [163] and DM2 [164], raising the possibility that the alteration of FHOD1 is somehow involved in impaired cardiac function in these two diseases.
Deletions affecting DAAM1 have been implicated in congenital heart defects in humans [165,166]. Daam1 is expressed in the heart during murine embryonic development [167]. Daam1 KO mouse embryos present abnormalities that affect heart morphogenesis, and the cardiomyocytes exhibit aberrant organization of the actin cytoskeleton, fewer, underdeveloped sarcomeres (structures containing crosslinked bundles of actin filaments and interdigitating myosin filaments), and cell–cell junction defects [167].
3.2. Neurodevelopmental Disorders and Mental Diseases
Formin mutation has been linked not only to intellectual disability, but also to several neurodevelopmental disorders [137]. Autism spectrum disorder (ASD, MIM: 209850) is a neurodevelopmental condition characterized by young age of onset, impairment of communication and social abilities, and restricted interests and repetitive behaviors. The interaction between genetic and environmental conditions plays an important role in establishing this disorder [168]. DIAPH3 seems to be the formin gene most strongly linked to autism, as suggested by the identification of variants in autistic individuals. The first case to relate DIAPH3 with autism was that of a boy who inherited a variant (p.Pro614Thr) in the FH1 domain of one of the alleles and a large genomic deletion in the other, DIAPH3 being one of the genes considered in the study [169]. This double-hit case is very rare and indicates that, in the absence of a double mutation in DIAPH3, at least one extra mutation may be needed in one or more autism-related genes to produce the phenotype. This was found in a second case that presented a missense replacement in the region of DIAPH3 encoding the FH2 domain and a second mutation in SET2 [170], a gene associated with autism and other neurodevelopmental disorders. Another example was a case with infantile spasm that presented a missense mutation in DIAPH3 (p.Arg432Lys) and a nonsense mutation in the syntaxin-binding protein 1 gene (STXBP1) [171], whose mutation is known to cause developmental and epileptic encephalopathy 4 (MIM:602926) [172]. Another interesting case was that of a patient with a rare epileptic encephalopathy featuring a deletion at 13q21 that included part of DIAPH3 [173]. The specific KO of Diaph3 in the brain cortex of mice alters the spindle components, leading to fewer proliferative divisions and more neurogenic divisions in apical neural progenitor cells, and thereby to a thinner cortex (microcephaly). Externally, these mice show motor activity deficiency and autism-related behavior [174].
An FMN1 variant producing a missense substitution (p.Arg109Gly) was found in a search for genetic variants in high-risk ASD families [175]. A microdeletion affecting two exons of FMN1 was found in a whole-genome amplification study of copy number variation in patients with early-onset obsessive-compulsive disorder [176]. An FMN1 variant producing a missense mutation (p.Ser800Cys) was identified by WGS in a selected group of patients with schizophrenia. It displayed long homozygous runs throughout the genome due to recent inbreeding [177]. The possible involvement of FMN1 mutations in these mental disorders could be explained by the role of FMN1 in the glutamatergic synaptic terminals [178].
In a study of Wnt-pathway-related genes in genome regions previously linked to schizophrenia, six variants were found in DAAM2 [179]. DAAM2 mRNA levels were significantly higher in blood samples from patients suffering their first psychotic episode than in controls, and returned almost to normal when the patients entered remission [180]. Guillain–Barre Syndrome (GBS, MIM: 139393), acute inflammatory demyelinating polyneuropathy, is most commonly characterized by symmetric limb weakness and loss of tendon reflexes. DAAM2 protein levels are increased in the acute phase of GBS, and their reduction is associated with the improvement of symptoms [181]. Given the role of DAAM2 in Wnt signaling, it was proposed that its levels alter the nuclear transport of critical transcriptional factors involved in myelination. However, the role, if any, of DAAM2 in this group of disorders is currently unclear.
Two variants of FMNL3 that produce missense substitutions (p.Phe38Leu and p.Ile458Leu) were identified in a consanguineous family containing siblings with an infancy-onset, mixed central and peripheral neurodegenerative disorder. However, since they present a homozygous mutation in the STXBP5L gene, this alteration was considered the most likely cause of the phenotype [182], although a contribution from the FMNL3 variants cannot be ruled out.
3.3. Other Developmental Defects
Planar cell polarity (PCP) refers to the coordinated organization of cells or cell components across a tissue plane [183]. Defective PCP signaling is involved in the pathogenesis of developmental kidney disorders [184]. PCP is considered a noncanonical Wnt pathway (PCP/Wnt) due to the involvement of the secreted Wnt glycoproteins and the Frizzled family of Wnt receptors in the absence of the β-catenin-driven gene expression observed in the canonical Wnt cascade [185]. In the PCP/Wnt pathway, DAAM1 forms a complex with Disheveled that activates Rho, leading to cytoskeletal rearrangement [186]. In addition to the congenital heart defects mentioned above, DAAM1 gene variants encoding a missense replacement (p.Val233Met) and a deletion (p.Lys681_Val682del) were identified in an analysis of a set of genes selected by their possible involvement in developmental anomalies of the kidney and urinary tract [187]. A duplication of exons 1-4 of DAAM1 was identified in a study aimed at identifying copy number variants in cerebral palsy patients [188]. The developmental defects caused by the alterations in DAAM1 could be explained by the defective functioning of the PCP/Wnt pathway [189]. Consistent with the importance of DAAM1 in kidney development, DAAM1 knockdown (KD) impairs correct pronephric tubulogenesis in zebrafish and Xenopus laevis [190]. DAAM1 KD produces loss of primary cilia in kidney-derived cell lines [191], raising the possibility that the effect of reduced levels of DAAM1 on tubulogenesis is due to defective ciliogenesis. However, the effect on ciliogenesis is not observed in Xenopus nephric progenitor DAAM1 KD cells [190], making this explanation unlikely. Since DAAM1 is also expressed in the neural tubule during embryonic development [192], DAAM1 alterations could also affect developmental processes involving the PCP/Wnt pathway other than kidney tubulogenesis.
Similar to DAAM1, DAAM2 has been linked to pathologies that might involve dysregulation of the PCP/Wnt pathway. A variant (p.Arg987Leu) in the FH2 domain of DAAM2 has been found in a sporadic case of diffuse pulmonary ossification, which is a disorder characterized by unusual widespread bony metaplastic formations in the lung [193]. Since osteoclasts strongly express DAAM2, but not DAAM1, the mutation could alter bone resorption via Wnt/DAAM2 [194]. A duplication of DAAM2 was found in a family with a disorder of sexual development [195]. Since gene duplications and a mutation affecting other genes were also present, it is not possible to ascertain the involvement of DAAM2 in this disorder. One possibility is that DAAM2 is involved in gonadal development.
Limb development is a highly integrated process in which the cells involved must incorporate a variety of signals in a precise temporal manner. Defects in this process cause malformations in the extremities, as occurs Cenani–Lenz syndrome (MIM: 212780), which is a rare autosomal recessive disorder characterized by a complex syndactyly (two or more digits fused together) of the hands with malformations of the forearm bone and less severely affected lower limbs [196]. A de novo tandem duplication of the whole FMN1 gene was found in a patient with Cenani–Lenz-like non-syndromic oligosyndactyly. The same report described a homozygous deletion that affects the first twelve exons and the upstream region of FMN1 in a consanguineous family with similar malformations and with hearing loss [197]. However, it seems that those defects are not caused by the change in the FMN1 gene itself, since the altered genomic region also contains limb-specific regulatory sequences of GREM1 [198], which is a gene involved in kidney and limb development. Although Fmn1 KO mice in which the GREM1 gene was not affected had a phenotype very similar to that of the patient with a homozygous deletion of FMN1 [199], those mice had amplified GREM1 expression, suggesting that FMN1 regulates GREM1, and that the loss of FMN1 is not the direct cause of the disorder. It has also been reported that FMN1 in three patients with congenital anomalies of the kidney and urinary tract feature heterozygous missense (p.Arg752Trp or p.Pro529Arg) or frameshift (p.Ser1360Mfs*X19) variants [187]. However, the involvement of GREM1 expression in this phenotype cannot be discounted.
3.4. Aging-Related Diseases
Age-related macular degeneration, which is a major cause of blindness, involves the progressive degeneration of photoreceptors and retinal pigment epithelial cells. In two studies of X-chromosome-linked variants, several variants associated with age-related macular degeneration were found in DIAPH2 [200,201].
It was proposed that patients suffering neuropsychiatric disease, such as post-traumatic stress disorder (PTSD), at a young age have an increased risk of developing Alzheimer disease later in life [202,203]. Blood samples from PTSD patients and post mortem hippocampus samples from Alzheimer patients feature lower levels of FMN2 mRNA compared with controls [204], suggesting a possible association between these conditions and FMN2 downregulation. Consistent with this idea, it was observed that old Fmn2 KO mice were more likely to show early age-associative memory impairment compared with age-matched controls, suggesting that reduced FMN2 levels contribute to age-related memory impairment [205]. However, the significance of this association is not clear because FMN2 is expressed normally in old mice [205]. Higher levels of the DIAPH1 protein were found in myeloid cells from the brain of Alzheimer patients compared with age-matched controls, and it was suggested that this increase could potentiate neuroinflammation via advanced glycation end (AGE) receptor (RAGE)/DIAPH1 signaling, thereby contributing to the disease [206,207].
3.5. Other Disorders
POEMS syndrome is a rare, multisystem disorder that may include polyneuropathy, organomegaly, endocrinopathy, and other features, including aberrant plasma cells. A frameshift insertion and a nonframeshift deletion of FMNL2 were found by the WES of plasma cell DNA from a patient, although the role of these changes in the disease was not investigated [208]. Crohn’s disease is characterized by inflammation of parts of the digestive system and is associated with a failure of the innate immune system. Although the etiology of Crohn’s disease has not been fully determined, it clearly depends on genetic and environmental factors [209]. A heterozygous de novo mutation in the DID of FMNL2 (p.Leu136Pro) was found in a pediatric patient [210]. The mutation reduces the interaction between the DID and the DAD, with the mutant being more active than the wild-type protein. The stable expression of the mutant in THP-1 monocyte cells produces an increase in the number of podosome-like structures and a reduction in the percentage of THP-1 cells degrading the gelatin matrix in in vitro assays [210]. It is therefore plausible that FMNL2 regulates important processes dependent on actin dynamics in the cells of the innate immune system, since it is part of the genetic network that regulates the appearance of Crohn’s disease.
High levels of blood glucose stimulate the formation of AGE products, which are the result of non-enzymatic glycation and oxidation of proteins and lipids. AGEs accumulate in diabetic circulation and tissues and their interaction with RAGE, contributes to vascular alterations. The cytoplasmic domain of RAGE binds to DIAPH1, which is essential for RAGE ligand-mediated signal transduction [211]. It has been suggested that DIAPH1 could have a role in diabetes and obesity through its interaction with RAGE [212]. Indeed, studies in mDia1 KO diabetic mice suggest a role of DIAPH1 in the pathogenesis of diabetes-associated nephropathy [213]. FHOD3 mutations have been associated with type I diabetes [214], but further studies are needed to establish the relationship more convincingly. DAAM2 levels proved to be higher in diabetic nephropathy patients and in a mouse model of the disease [215], implying that a variety of formins may be involved in the diabetic condition.
DAAM2 is expressed throughout pregnancy, reaching its highest levels at term. Pregnancies with a fetal growth restriction complication have raised blood levels of DAAM2 mRNA and DAAM2 protein in preterm placental tissue [216]. This upregulation has been related to hypoxia, in which DAAM2 downregulates the expression of cytoprotective genes. The downregulation of DAAM2 may thereby have a beneficial effect during this type of abnormal pregnancy. DAAM1, but not DAAM2, KO mice are lethal during embryogenesis due to placental development defects [217]. Daam1 and Daam2 double-KO mice have a more severe phenotype, suggesting redundant functions of Daam formins in this process [217].
Three intronic variants found within a 27-base pair region and two missense variants of DAAM2 have been associated with osteoporosis [218,219]. As evidence for the role of DAAM2 in this process, it was found that hypomorphic Daam2 mice develop increased cortical porosity and reduced bone strength [218].
Ischemic stroke or brain ischemia is caused by a blockage in an artery supplying blood to the brain that reduces the flow of blood. Depending on whether circulation is restored, brain damage can be transient or permanent. Several formin genes are important in ischemic stroke. Thus, FMNL1 and FMNL2 appear to have a role in repairing damaged cardiomyofibrils [151]. This role might be evolutionary conserved since FMNLs genes are also expressed during heart development in zebrafish [220]. Consistent with their importance in this process, two FMNL2 variants were identified in a genome-wide association analysis aimed at identifying genetic loci predisposing to early-onset stroke. An intronic variant in DIAPH1 was associated with increased risk of ischemic stroke [221] and some DIAPH1 variants have also been associated with Moyamoya disease [222], which is a progressive vasculopathy that causes stroke in children. The possible role of altered DIAPH1 in ischemic stroke could be related to the involvement of RAGE/DIAPH1 in neuroinflammation and neurodegenerative diseases. Revascularization therapy is the primary modality for restoring the blood supply to infarcted brain tissues, but ischemia-reperfusion injury does run the risk of damaging the brain, worsening the stroke outcome. In cardiac ischemia-reperfusion injury, INF2 increases mitochondrial fission and represses mitochondrial fusion [223,224], contributing to cardiomyocyte death.
With regard to other variants of formin genes, several variants of FHOD3 have been associated with periodontal disease [225]. In addition, several FMNL2 variants have been associated with intraocular pressure and the subsequent development of glaucoma [226]. The latest finding could help explain some of the differences in glaucoma development observed between different ethnic populations [227].
4. Formins in Cancer
4.1. Expression of Formins in Cancer
According to the Human Protein Atlas (Supplementary Table S10), the mRNA levels of the different formins are generally of low specificity with regard to the type of cancer. However, DAAM2 and FMN1 expression is enhanced in melanoma, and that of FMN2 in glioma and testicular cancer. Based on ONCOMINETM data (https://www.oncomine.org; accessed on 30 August 2021; Supplementary Table S11), DIAPH1, DIAPH3, FMNL2, FHOD1, and INF2 transcripts are frequently upregulated in cancer, whereas those of DIAPH2, DAAM1, DAAM2, FHOD3, FMN2, and FHDC1 are generally downregulated. The other formins are upregulated or downregulated, depending on the type of cancer. Most reports are of cancers in which the expression of a given formin gene or protein is upregulated, although there are some exceptions, such as the DIAPH3 and FMNL2 proteins, in which the expression is downregulated in triple-negative breast cancer and hepatocellular carcinoma, respectively (Table 1).
In some cases, the expression levels of formin mRNA have been linked to the methylation status of their promoters. For instance, FHOD3 mRNA overexpression in thyroid cancer is associated with hypomethylation of the FHOD3 promoter and with gene copy number variation [315]. Another example is the downregulation of FMN2 expression by hypermethylation of its promoter during the early stages of colorectal cancer progression [269], possibly contributing to the initial steps of cancer development. It is also plausible that some formins can regulate the expression/activity of other formins. For instance, FMNL1 KD in glioblastoma cell lines downregulates DIAPH1 protein levels, whereas FMNL1 overexpression upregulates them [236], enabling malignant cells to migrate slower of faster, respectively, than control cells. Formins might also affect cancer progression through the expression of isoforms. For instance, in melanoma cell lines, the FMNL2 gene encodes different isoforms with distinct abilities to promote invasion [308].
4.2. The Role of Formins in Cancer
Formins have been associated with cancer by inducing epithelial-to-mesenchymal transition (EMT), migration, and invasion by their role in the actin and microtubule cytoskeletons (Figure 4, Table 1).
EMT is a cellular program by which epithelial cells progressively lose their typical polygonal appearance with segregated apical, lateral, and basal surfaces found in tightly packed cell monolayers, to adopt an elongated, mesenchymal morphology. EMT, which is normally reversible, has important roles at specific stages of embryogenesis and development [316]. At the molecular level, EMT-induced transcription factors orchestrate a profound change in gene expression. They repress the transcription of genes that maintain the epithelial state, such as those encoding E-cadherin and occludin, with the consequent disruption of cell–cell junctions. They also induce the expression of other genes that promote the mesenchymal state, such as those of N-cadherin and matrix metalloproteinases (MMPs). The malignant progression of carcinoma depends on the activation of EMT, which confers the capacity to migrate and invade neighboring tissues to neoplastic cells. Altered expression of formins affects cell morphology and can promote EMT. Examples are DIAPH3 in hepatocellular carcinoma cells [288]; FMNL1 in non-small cell lung cancer cells [293], clear cell renal cell carcinoma cells [281] and nasopharyngeal carcinoma cells [277]; FMNL2 in gastric cancer cells [312] and colorectal carcinoma cells [262]; FMNL3 in nasopharyngeal carcinoma cells and tumor xenografts [279]; and FHOD1 in lung carcinoma [294].
Migration and invasion are two cancerous processes that are closely connected with metastasis. Tumor cells can adopt two distinct modes of movement during migration in three-dimensional extracellular matrices [317,318]. One is the amoeboid mode, which allows round cells to squeeze forward through small gaps present in the extracellular matrix, which is driven by high Rho GTPase activity and actomyosin contractility in the absence of proteolytic activity. The alternative mode, called mesenchymal migration, is driven by the extension of Rac GTPase-dependent lamellipodia and requires proteolytic processing of the extracellular matrix to allow for the movement of elongated cells across wider gaps. The migration of cancer cells is generally studied in vitro by wound-healing experiments or assays in microperforated cell culture inserts in the absence of the extracellular matrix, whereas, for invasion, they are carried out in its presence (Table 1). Given that formins catalyze the formation of polymers of actin, it is not surprising that they play an important role in cancer cell migration. Examples of this function are DIAPH1-3 and DAAM1 in breast cancer [243,247], DIAPH1 in glioma [230], FMNL1 in nasopharyngeal carcinoma [277], FMNL2 in colorectal cancer [263], and FHOD1 in oral squamous carcinoma [280].
Invasion by the mesenchymal mode is facilitated by the formation of invadopodia, which are actin-based protrusions of the plasma membrane that contain metalloproteases, such as MMP2 and MMP9, that degrade the extracellular matrix [319]. DIAPH1-3 regulate invasion in cell cultures, and reduce metastasis and tumor volume in nude mice inoculated with breast cancer [243,244,245] and glioma [230]; FMNL1 does the same in glioblastoma and clear cell renal cell carcinoma cells [236,281]; FMNL2 [263] and FMNL3 [267] in colorectal cancer cells; FMNL3 in nasopharyngeal carcinoma cells [279]; and FHOD1 in oral squamous carcinoma cells [280]. As with the case of DIAPH1-3 [230,243,244], other formins could also control invasion by regulating the efficient formation of invadopodia, and the expression levels, localization and activity of metalloproteases [243].
During metastasis, tumor cells may switch between these two modes of invasion, depending on the activation status of specific Rho GTPases [317]. An interesting case is that of ovarian cancer and its relationship with DIAPH3. Exfoliated cells derived from the ovarian surface epithelium are detected as single cells, small aggregates, or highly ordered multicellular 3D spheroids. Spheroids promote ovarian cancer metastasis within the peritoneal cavity [320]. DIAPH3 expression is downregulated in early- to late-stage ovarian cancer, with this change being accompanied by disruption of the actin cytoskeleton and greater cell deformability [321,322]. While spheroids make use of both mesenchymal and amoeboid motility to invade, DIAPH3 KD disrupts spheroid architecture and enhances cell invasion by favoring ameboid migration [296]. Modulation of DIAPH3 expression also regulates the switch between the mesenchymal and ameboid migration modes in human MDA-MB-231 breast and DU145 prostate cancer cells [232,302,323]. DIAPH1 and FMNL2 have also been involved in regulating the migratory plasticity of breast tumor cell lines [241,242].
Other mechanisms by which formins can contribute to cancer progression include alteration of mitochondrial dynamics and of the microtubule cytoskeleton. An example of the former is found in INF2, which can regulate cancer progression through its role in mitochondrial fission [324,325]. Speckle-type POZ protein (SPOP), which is one of the most frequently mutated genes in prostate cancer [326], is an adapter that brings INF2 [305] and other substrates [327] to the proximity of specific E3 ubiquitin ligase complexes for ubiquitination. INF2 ubiquitination does not lead to degradation, as occurs with other proteins, but instead reduces its localization at the ER, abrogating its ability to facilitate mitochondrial fission [305], which is one of the best-characterized functions of INF2 [324,325]. A dominant negative effect probably causes SPOP mutant forms associated with prostate cancer to be defective in promoting INF2 ubiquitination, resulting in increased localization of INF2 at the endoplasmic reticulum. In turn, this facilitates augmented mitochondrial fission, which is known to promote cell migration [328], resulting in enhanced migration and invasion [305]. However, INF2 activation appears to promote apoptosis of human papillary thyroid cancer MDA-T32 cells [329]. Examples of formin levels altering cancer cells is the regulation of microtubule dynamics are the cases of DIAPH1 and DIAPH2, whose silencing impairs chromosome alignment during mitosis in colorectal cancer cell lines, giving rise to aneuploidy, and of DIAPH3, which regulates the activation of the spindle assembly checkpoint and microtubule attachment to kinetochores [174,256,258,330]. DIAPH3 downregulation increases the sensitivity of breast and prostate carcinoma cells to taxanes [138,331], which are inhibitors of microtubule dynamics.
The tumor microenvironment is an important element in cancer development. It includes cancer stem cells and the network formed by the cells, signals and extracellular matrix around the tumor [332]. Formins can promote cancer development through this network. One in vitro example is DIAPH3 in breast cancer cells, in which cancer-associated fibroblasts (CAFs) regulate tumor cell migration and invasion by secreting protein factors that reduce DIAPH3 protein levels in the tumor cells [333]. In turn, cancer-associated fibroblasts upregulate their own DIAPH3 levels to remodel the extracellular matrix and promote invasion [334]. Cancer cell inoculation models using nude mice, which are immunocompromised and often metastasize early on, do not reproduce the tumor microenvironment, which is necessary to study cancer progression under normal conditions. In contrast, genetically modified mouse models (GEMMs) develop de novo tumors in a natural immune-proficient microenvironment and are able to spontaneously progress toward metastatic disease, mimicking the process in humans. Therefore, GEMMs are of great help for basic cancer research in general, and specifically for investigating the influence of the tumor microenvironment [335,336]. Using different types of GEMMs, it was observed that enhanced levels of activin A, which is a member of the transforming growth factor β family, in the skin promote skin tumor formation and their malignant progression through the induction of a pro-tumorigenic microenvironment [337] through transcriptional activation of mDia2 in CAFs [338]. In turn, mDia2 interacts with the transcription factor p53, which is a tumor-suppressor protein [339], and prevents the accumulation of p53 in the nucleus and, consequently, p53-dependent transcriptional control of CAF-derived secreted factors that promote tumor formation [338]. These works are an example of how the use of GEMMs can help to study the role of the tumor microenvironment and the contribution of formins to this microenvironment in cancer.
Formins can also affect the tumor microenvironment through the mechanism of extracellular vesicle secretion. Thus, DIAPH3 silencing in prostate cancer cells promotes the formation of large membrane blebs that can be shed as extracellular vesicles, and the release of exosome-sized particles that enhance the proliferation of recipient tumor cells and inhibit proliferation of immune cells, thereby helping to remodel the tumor cell microenvironment [302,340]. Another mechanism is exemplified by FMNLs, whose expression is positively correlated with the levels of tumor-infiltrated immune cells in gastric cancer patients [311].
4.3. Formins as Prognostic Biomarkers and Therapeutical Targets in Cancer
The expression levels of some formins are associated with a favorable or unfavorable prognosis, depending on the type of cancer (Supplementary Table S10). For instance, in renal cancer, a high level of transcripts of DIAPH1, DAAM1, FHOD3 and FHDC1 has a favorable prognosis, whereas in DIAPH2, DAAM2, FMNL1, FMNL3, and FHOD1, this corresponds to an unfavorable prognosis. Some studies investigated the mRNA or protein levels of a given formin in cancer cell lines and the metastatic capacity of those cells in nude mice (Table 1). These and other studies also examined the relative levels of formin, determined by immunohistochemical analysis of biobank tumor samples relative to paired normal tissue, or levels of formin mRNA, to identify associations with the clinicopathological characteristics of the corresponding patient, such as tumor size, presence of distant metastasis, and outcome. Some examples of strong correlations are DAAM1 in breast cancer [248]; FMNL1 in nasopharyngeal cancer [277], non-small cell lung cancer [293], and clear cell renal cell cancer [281]; and of FMNL3 in colorectal cancer [268].
Not only have formin levels been associated with cancer prognosis, but gene variants and deletions have also been associated with the risk of cancer progression. For example, specific FMN1 variants have been linked to a higher risk of developing pancreatic [300] and prostate cancer [304]. In lung cancer, a variant in the FH2 domain of DAAM2 (p.Asp762Gly) has been associated with a protective effect, and, although not a statistically significant result, a variant in the DID (p.Arg172His) was found to be enriched in healthy controls [292]. The frequency of FMN2 mutations markedly differs between the stages of colorectal cancer. This finding qualifies FMN2 for inclusion in the list of useful biomarkers for diagnosis, pathological classification, staging and prognosis of this type of cancer [270]. DIAPH3 deletions are common to several carcinomas and accumulate during cancer progression, as occurs in prostate, hepatocellular, and breast carcinoma. The deletions promote motility and invasion, and are associated with aggressive tumors and with metastatic prostate cancer [232].
Given their involvement in cancer, formins might have a use as therapeutic targets. Combined treatment of the formin inhibitor SIMFH2 [341] with the anti-cancer drugs taxol and cisplatin, which, respectively, stabilize microtubules and damage DNA, is useful for reducing human ovarian cancer spheroid viability in vitro [342]. However, since SIMFH2 has some off-target effects, such as reduction in the expression and activity of p53 [343] and inhibition of myosin ATPase activity [344], these should also be considered for the doses and the duration of the treatment in order to avoid harmful effects. Phenanthriplatin is a platinum (II) complex that binds to only one DNA strand (unlike cisplatin, which binds to both strands) and causes cancer-cell death by blocking gene transcription. Treatment of non-small cell lung cancer cells with phenanthriplatin, but not with cisplatin, downregulates DIAPH2 expression, with this effect being potentially useful for treating this type of cancer [345]. Silencing DIAPH3 increases microtubule dynamics and enhances the responsiveness to taxol in prostate DU145 and LNCaP cancer cells, and low levels of DIAPH3 mRNA improve the sensitivity to taxol and other taxanes in prostate and breast cancer cell lines. Consistent with these results, a low level of DIAPH3 expression was correlated with improved clinical outcomes after taxane-containing chemotherapy in a clinical trial of breast cancer patients [331]. Methotrexate is a chemotherapy agent used to treat acute lymphoblastic leukemia. The ability of cells to polyglutamylate methotrexate is a major factor determining whether the drug is retained for a longer period and, consequently, its in vivo efficacy and toxicity [346]. Variants of FHOD3 and six other genes influence the formation of methotrexate polyglutamates in leukemia treatment, making them important for methotrexate responsiveness [286].
The overexpression of FMNL1 in blood-derived tumor cells has led to the development of novel approaches to cancer treatment, such as the use of cytotoxic T cell clones with T cell receptors specific to an FMNL1 peptide to treat lymphomas and other malignancies of cells of the immune system [347]. Mitochondrial fission is a novel target for modulating cancer development and progression because increased fission produces apoptosis [328]. Thus, treatment with interleukin-2 and the anti-tumor drug tanshinone IIA of SW480 colorectal cancer cells upregulates INF2, which increases mitochondrial fission and apoptosis [348]. This work provides a further example of how the manipulation of formin expression can be exploited to treat cancer. A better understanding of the role of the various formins in cancer could clear the way for the development of better and more specific cancer treatments in the near future.
5. Future Directions
Formins control not only actin filament remodeling and microtubule dynamics, but also the crosstalk between these two types of cytoskeletal structures [4,5,132,133,349]. The correct functioning of formins is essential to many cellular processes, including the formation of actin-based structures such as lamellipodia, filopodia and microvilli, and of specialized microtubule arrays, membrane trafficking, mitochondrial fission, and cell division, migration, and invasion [2,3,138,325,349]. In addition, formins can regulate gene expression, for instance, as in the case of DAAMs, by forming part of important signaling pathways [189], or, in general, by modulating the nuclear levels of the myocardin-related transcription factor coactivator (MRTF), which associates with the serum response factor (SRF), and forms an MRTF–SRF complex that controls the transcription of a myriad of genes, many of which encode proteins related to the cytoskeleton [349,350,351]. The list of MRTF-SRF target genes overlaps with gene signatures associated with cancer cell invasiveness and metastasis, response to extracellular matrix, stiffness, or response to FAK or TGFβ signaling [352,353]. Therefore, altering the activity of formins, by modulating their expression levels, or as a result of mutations in regulatory or catalytic domains, may have important consequences for the function of differentiated cells and for embryonic development and cancer. The growing list of human disorders caused by, or associated with, formin mutation illustrates the importance of formins in ensuring the normal function of cells.
An extensive analysis of multiple independent pedigrees with affected individuals is available for some cases of monogenic disorders caused by formin mutations. However, in other cases, the information is scarce and further work is needed to find more examples and mutations. The increasing use of GWS and WES approaches in Human Medical Genetics Units will certainly help identify novel variants responsible for, or associated with, pathological conditions, and novel disorders involving formin alterations. This work will facilitate diagnosis and, it is to be hoped, contribute to the development of pharmacological agents to treat these diseases.
Despite the advances in our knowledge of formins, further work is needed to fully understand the molecular mechanism leading to disease. The use of in vitro systems and animal models has undoubtedly helped us gain insights into the function of formins in health and disease. However, in most cases, the molecular mechanism by which a formin alteration leads to the pathological condition is not clear. In some cases, the primary consequence of the alteration is an increase in the actin polymerization of the affected formin, for instance, in the case of the DIAPH1 mutants with truncated carboxyl-terminal regulatory sequences. However, in cases in which the mutation maps to other formin domains, for example, the FH2 domain, the effect on the activity is less evident and requires in vitro analysis. This is also true for the pathogenic mutations in the DID of INF2, which are known to increase the actin polymerization activity of the formin. Nevertheless, despite the considerable advances, the exact mechanism underlying its activation is yet to be described. In some cases, the alteration in a formin produces disease in humans but not in mice and vice versa. The use of GEMMs will provide important insights into how formins function in vivo and how formin alterations lead to or contribute to disease, especially cancer, and hopefully to validate formin-specific drugs and develop preclinical trials to evaluate their therapeutic response.
Changes in formin levels and the emergence of formin mutations play an important role in cancer progression, although, despite the established role of formins in building invadopodia and regulating cell migration and invasion, the effect of formin alterations in cancer has yet to be fully established. Given their importance, the inclusion of formins in cancer biomarker panels might be useful for prognostic purposes. The development of new formin inhibitors with higher specificity and tolerance than SIMFH2 could be exploited to prevent chromosome segregation errors causing aneuploidy as a result of deficient mitotic spindle assembly [354]. The activation of the MRTF-SRF transcriptional complex by formins, in addition to control the expression of a large number of protein coding mRNAs, regulates the expression of non-coding RNAs. Examples of such regulation are the expression of miR-21, which promotes fibrosis and EMT transition and has been implicated in cancer, and the lncRNA Malat1/Neat2, which has been implicated in lung cancer invasion and metastasis [354]. The involvement of non-coding RNA, as well the role of extracellular vesicles preparing the niche for metastasis, are new and developing fields in cancer research [355,356]. We hope that all these combined efforts will help us fully understand the role of formins in vivo and the mechanism by which their alteration causes, or contributes to, cell dysfunction, and that they will stimulate the development of novel strategies for treating formin-related diseases.
Supplementary Materials
The following are available online at www.mdpi.com/article/10.3390/cells10102554/s1. Table S1: Expression of formin genes in different human tissues. Table S2: RNA expression of formins in brain and blood cells. Supplementary Tables S3–S9: Pathogenic mutations of DIAPH1/DIAPH2/DIAPH3/DAAM2/FMN2/INF2/FHOD3. Table S10: Prognostic value of formin mRNA levels in cancer. Table S11: Analysis of mRNA expression of the formin genes in cancerous versus normal tissue.
Author Contributions
Writing—original draft preparation, figure design and preparation, L.L.-d.-H.; writing—review, editing, supervision, and funding acquisition, M.A.A. Both authors have read and agreed to the published version of the manuscript.
Funding
Research in the laboratory of M.A.A. was supported by a grant (PGC2018-095643-B-I00) from the Spanish Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación, and the Fondo Europeo de Desarrollo Regional, European Union (MICINN/AEI/FEDER, EU). L.L.-d.-H. was supported by a contract (FPU16/00935) from the Spanish Ministerio de Educación y Formación Profesional (MEFP).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Phil Mason for revising the English language of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schönichen, A.; Geyer, M. Fifteen Formins for an Actin Filament: A Molecular View on the Regulation of Human Formins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing Formins to Remodel the Actin and Microtubule Cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Bartolini, F.; Gundersen, G.G. Formins and Microtubules. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Barrera, J.; Alonso, M.A. Coordination of Microtubule Acetylation and the Actin Cytoskeleton by Formins. Cell. Mol. Life Sci. 2018, 75, 3181–3191. [Google Scholar] [CrossRef]
- Lynch, E.D.; Lee, M.K.; Morrow, J.E.; Welcsh, P.L.; León, P.E.; King, M.-C. Nonsyndromic Deafness DFNA1 Associated with Mutation of a Human Homolog of the Drosophila Gene Diaphanous. Science 1997, 278, 1315–1318. [Google Scholar] [CrossRef] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Hamosh, A.; Amberger, J.S.; Bocchini, C.; Scott, A.F.; Rasmussen, S.A. Online Mendelian Inheritance in Man (OMIM®): Victor McKusick’s Magnum Opus. Am. J. Med. Genet. Part A 2021. [Google Scholar] [CrossRef]
- Iwasa, Y.; Nishio, S.; Usami, S. Comprehensive Genetic Analysis of Japanese Autosomal Dominant Sensorineural Hearing Loss Patients. PLoS ONE 2016, 11, e0166781. [Google Scholar] [CrossRef]
- Kaustio, M.; Nayebzadeh, N.; Hinttala, R.; Tapiainen, T.; Åström, P.; Mamia, K.; Pernaa, N.; Lehtonen, J.; Glumoff, V.; Rahikkala, E.; et al. Loss of DIAPH1 Causes SCBMS, Combined Immunodeficiency, and Mitochondrial Dysfunction. J. Allergy Clin. Immunol. 2021, 148, 599–611. [Google Scholar] [CrossRef]
- Brozkova, D.S.; Marková, S.P.; Mészárosová, A.U.; Jenčík, J.; Čejnová, V.; Čada, Z.; Laštůvková, J.; Rašková, D.; Seeman, P. Spectrum and Frequencies of Non GJB2 Gene Mutations in Czech Patients with Early Non-Syndromic Hearing Loss Detected by Gene Panel NGS and Whole-Exome Sequencing. Clin. Genet. 2020, 98, 548–554. [Google Scholar] [CrossRef]
- Kim, B.J.; Ueyama, T.; Miyoshi, T.; Lee, S.; Han, J.H.; Park, H.-R.; Kim, A.R.; Oh, J.; Kim, M.Y.; Kang, Y.S.; et al. Differential Disruption of Autoinhibition and Defect in Assembly of Cytoskeleton during Cell Division Decide the Fate of Human DIAPH1-Related Cytoskeletopathy. J. Med. Genet. 2019, 56, 818–827. [Google Scholar] [CrossRef]
- Kang, T.-H.; Baek, J.-I.; Sagong, B.; Park, H.-J.; Park, C.I.; Lee, K.-Y.; Kim, U.-K. A Novel Missense Variant in the DIAPH1 Gene in a Korean Family with Autosomal Dominant Nonsyndromic Hearing Loss. Genes Genet. Syst. 2016, 91, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Ercan-Sencicek, A.G.; Jambi, S.; Franjic, D.; Nishimura, S.; Li, M.; El-Fishawy, P.; Morgan, T.M.; Sanders, S.J.; Bilguvar, K.; Suri, M.; et al. Homozygous Loss of DIAPH1 Is a Novel Cause of Microcephaly in Humans. Eur. J. Hum. Genet. 2015, 23, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; van den Ende, J.; Boudewyns, A.; De Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef]
- Al-Maawali, A.; Barry, B.J.; Rajab, A.; El-Quessny, M.; Seman, A.; Coury, S.N.; Barkovich, A.J.; Yang, E.; Walsh, C.A.; Mochida, G.H.; et al. Novel Loss-of-Function Variants in DIAPH1 Associated with Syndromic Microcephaly, Blindness, and Early Onset Seizures. Am. J. Med. Genet. Part A 2016, 170, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Yavarna, T.; Al-Dewik, N.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; Lakhani, S.; AlMulla, M.; Nawaz, Z.; et al. High Diagnostic Yield of Clinical Exome Sequencing in Middle Eastern Patients with Mendelian Disorders. Hum. Genet. 2015, 134, 967–980. [Google Scholar] [CrossRef]
- Wu, K.; Wang, H.; Guan, J.; Lan, L.; Zhao, C.; Zhang, M.; Wang, D.; Wang, Q. A Novel Variant in Diaphanous Homolog 1 (DIAPH1) as the Cause of Auditory Neuropathy in a Chinese Family. Int. J. Pediatr. Otorhinolaryngol. 2020, 133, 109947. [Google Scholar] [CrossRef]
- Westbury, S.K.; Downes, K.; Burney, C.; Lozano, M.L.; Obaji, S.G.; Toh, C.H.; Sevivas, T.; Morgan, N.V.; Erber, W.N.; Kempster, C.; et al. Phenotype Description and Response to Thrombopoietin Receptor Agonist in DIAPH1-Related Disorder. Blood Adv. 2018, 2, 2341–2346. [Google Scholar] [CrossRef]
- Shearer, A.E.; Black-Ziegelbein, E.A.; Hildebrand, M.S.; Eppsteiner, R.W.; Ravi, H.; Joshi, S.; Guiffre, A.C.; Sloan, C.M.; Happe, S.; Howard, S.D.; et al. Advancing Genetic Testing for Deafness with Genomic Technology. J. Med. Genet. 2013, 50, 627–634. [Google Scholar] [CrossRef]
- Ueyama, T.; Ninoyu, Y.; Nishio, S.; Miyoshi, T.; Torii, H.; Nishimura, K.; Sugahara, K.; Sakata, H.; Thumkeo, D.; Sakaguchi, H.; et al. Constitutive Activation of DIA1 (DIAPH1) via C-terminal Truncation Causes Human Sensorineural Hearing Loss. EMBO Mol. Med. 2016, 8, 1310–1324. [Google Scholar] [CrossRef]
- Neuhaus, C.; Lang-Roth, R.; Zimmermann, U.; Heller, R.; Eisenberger, T.; Weikert, M.; Markus, S.; Knipper, M.; Bolz, H.J. Extension of the Clinical and Molecular Phenotype of DIAPH1-Associated Autosomal Dominant Hearing Loss (DFNA1). Clin. Genet. 2017, 91, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing High-Throughput Sequencing into Mainstream Genetic Diagnosis Practice in Inherited Platelet Disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Ganaha, A.; Kaname, T.; Shinjou, A.; Chinen, Y.; Yanagi, K.; Higa, T.; Kondo, S.; Suzuki, M. Progressive Macrothrombocytopenia and Hearing Loss in a Large Family with DIAPH1 Related Disease. Am. J. Med. Genet. 2017, 173, 2826–2830. [Google Scholar] [CrossRef] [PubMed]
- Karki, N.R.; Ajebo, G.; Savage, N.; Kutlar, A. DIAPH1 Mutation as a Novel Cause of Autosomal Dominant Macrothrombocytopenia and Hearing Loss. Acta Haematol. 2021, 144, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Rabbolini, D.; Connor, D.; Morel-Kopp, M.-C.; Donikian, D.; Kondo, M.; Chen, W.; Alessi, M.-C.; Stevenson, W.; Chen, V.; Joseph, J.; et al. An Integrated Approach to Inherited Platelet Disorders: Results from a Research Collaborative, the Sydney Platelet Group. Pathology 2020, 52, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Stritt, S.; Nurden, P.; Turro, E.; Greene, D.; Jansen, S.B.; Westbury, S.K.; Petersen, R.; Astle, W.J.; Marlin, S.; Bariana, T.K.; et al. A Gain-of-Function Variant in DIAPH1 Causes Dominant Macrothrombocytopenia and Hearing Loss. Blood 2016, 127, 2903–2914. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Lu, J.; Wang, J.; Li, H.; Lin, X. Combined Examination of Sequence and Copy Number Variations in Human Deafness Genes Improves Diagnosis for Cases of Genetic Deafness. BMC Ear Nose Throat Disord. 2014, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Bione, S.; Sala, C.; Manzini, C.; Arrigo, G.; Zuffardi, O.; Banfi, S.; Borsani, G.; Jonveaux, P.; Philippe, C.; Zuccotti, M.; et al. A Human Homologue of the Drosophila Melanogaster Diaphanous Gene Is Disrupted in a Patient with Premature Ovarian Failure: Evidence for Conserved Function in Oogenesis and Implications for Human Sterility. Am. J. Hum. Genet. 1998, 62, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Sala, C.; Arrigo, G.; Torri, G.; Martinazzi, F.; Riva, P.; Larizza, L.; Philippe, C.; Jonveaux, P.; Sloan, F.; Labella, T.; et al. Eleven X Chromosome Breakpoints Associated with Premature Ovarian Failure (POF) Map to a 15-Mb YAC Contig Spanning Xq21. Genomics 1997, 40, 123–131. [Google Scholar] [CrossRef]
- Marozzi, A.; Manfredini, E.; Tibiletti, M.; Furlan, D.; Villa, N.; Vegetti, W.; Crosignani, P.; Ginelli, E.; Meneveri, R.; Dalprà, L. Molecular Definition of Xq Common-Deleted Region in Patients Affected by Premature Ovarian Failure. Hum. Genet. 2000, 107, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Rødningen, O.K.; Barøy, T.; Sorte, H.; Mellembakken, J.R.; Strømme, P.; Fannemel, M.; Frengen, E. A Translocation between Xq21.33 and 22q13.33 Causes an Intragenic SHANK3 Deletion in a Woman with Phelan-McDermid Syndrome and Hypergonadotropic Hypogonadism. Am. J. Med. Genet. 2011, 155, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Genesio, R.; Mormile, A.; Licenziati, M.R.; De Brasi, D.; Leone, G.; Balzano, S.; Izzo, A.; Bonfiglio, F.; Conti, A.; Fioretti, G.; et al. Short Stature and Primary Ovarian Insufficiency Possibly Due to Chromosomal Position Effect in a Balanced X;1 Translocation. Mol. Cytogenet. 2015, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Bestetti, I.; Castronovo, C.; Sironi, A.; Caslini, C.; Sala, C.; Rossetti, R.; Crippa, M.; Ferrari, I.; Pistocchi, A.; Toniolo, D.; et al. High-Resolution Array-CGH Analysis on 46, XX Patients Affected by Early Onset Primary Ovarian Insufficiency Discloses New Genes Involved in Ovarian Function. Hum. Reprod. 2019, 34, 574–583. [Google Scholar] [CrossRef]
- Sánchez-Martínez, A.; Benito-Orejas, J.I.; Tellería-Orriols, J.J.; Alonso-Ramos, M.J. Autosomal Dominant Auditory Neuropathy and Variant DIAPH3 (c.-173C>T). Acta Otorrinolaringol. Esp. 2017, 68, 183–185. [Google Scholar] [CrossRef]
- Schoen, C.J.; Emery, S.B.; Thorne, M.C.; Ammana, H.R.; Śliwerska, E.; Arnett, J.; Hortsch, M.; Hannan, F.; Burmeister, M.; Lesperance, M.M. Increased Activity of Diaphanous Homolog 3 (DIAPH3)/Diaphanous Causes Hearing Defects in Humans with Auditory Neuropathy and in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 13396–13401. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Deutsch, K.; Hoeprich, G.J.; Marquez, J.; Hermle, T.; Braun, D.A.; Seltzsam, S.; Kitzler, T.M.; Mao, Y.; Buerger, F.; et al. DAAM2 Variants Cause Nephrotic Syndrome via Actin Dysregulation. Am. J. Hum. Genet. 2020, 107, 1113–1128. [Google Scholar] [CrossRef]
- Law, R.; Dixon-Salazar, T.; Jerber, J.; Cai, N.; Abbasi, A.A.; Zaki, M.S.; Mittal, K.; Gabriel, S.B.; Rafiq, M.A.; Khan, V.; et al. Biallelic Truncating Mutations in FMN2, Encoding the Actin-Regulatory Protein Formin 2, Cause Nonsyndromic Autosomal-Recessive Intellectual Disability. Am. J. Hum. Genet. 2014, 95, 721–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorukmez, O.; Gorukmez, O.; Ekici, A. A Novel Nonsense FMN2 Mutation in Nonsyndromic Autosomal Recessive Intellectual Disability Syndrome. Fetal Pediatr. Pathol. 2020, 39, 1–5. [Google Scholar] [CrossRef]
- Marco, E.J.; Aitken, A.B.; Nair, V.P.; da Gente, G.; Gerdes, M.R.; Bologlu, L.; Thomas, S.; Sherr, E.H. Burden of de Novo Mutations and Inherited Rare Single Nucleotide Variants in Children with Sensory Processing Dysfunction. BMC Med. Genom. 2018, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.D.; Rocca, M.S.; Bruno, I.; Faletra, F.; Pecile, V.; Gasparini, P. De Novo 911 kb Interstitial Deletion on Chromosome 1q43 in a Boy with Mental Retardation and Short Stature. Eur. J. Med. Genet. 2012, 55, 117–119. [Google Scholar] [CrossRef]
- Almuqbil, M.; Hamdan, F.F.; Mathonnet, G.; Rosenblatt, B.; Srour, M. De Novo Deletion of FMN2 in a Girl with Mild Non-Syndromic Intellectual Disability. Eur. J. Med. Genet. 2013, 56, 686–688. [Google Scholar] [CrossRef]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the Formin Protein INF2 Cause Focal Segmental Glomerulosclerosis. Nat. Genet. 2010, 42, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Nagano, C.; Yamamura, T.; Horinouchi, T.; Aoto, Y.; Ishiko, S.; Sakakibara, N.; Shima, Y.; Nakanishi, K.; Nagase, H.; Iijima, K.; et al. Comprehensive Genetic Diagnosis of Japanese Patients with Severe Proteinuria. Sci. Rep. 2020, 10, 270. [Google Scholar] [CrossRef] [Green Version]
- Varner, J.D.; Chryst-Stangl, M.; Esezobor, C.I.; Solarin, A.; Wu, G.; Lane, B.; Hall, G.; Abeyagunawardena, A.; Matory, A.; Hunley, T.E.; et al. Genetic Testing for Steroid-Resistant-Nephrotic Syndrome in an Outbred Population. Front. Pediatr. 2018, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Pinto, E.; Vairo, F.; Hogan, M.C.; Erickson, S.B.; El Ters, M.; Bentall, A.J.; Kukla, A.; Greene, E.L.; Hernandez, L.H.; et al. Identification of Genetic Causes of Focal Segmental Glomerulosclerosis Increases with Proper Patient Selection. Mayo Clin. Proc. 2021, 96, 2342–2353. [Google Scholar] [CrossRef]
- Laššuthová, P.; Šafka Brožková, D.; Krůtová, M.; Neupauerová, J.; Haberlová, J.; Mazanec, R.; Dřímal, P.; Seeman, P. Improving Diagnosis of Inherited Peripheral Neuropathies through Gene Panel Analysis. Orphanet J. Rare Dis. 2016, 11, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Huynh Cong, E.; Arrondel, C.; Tête, M.-J.; Montjean, R.; et al. INF2 Mutations in Charcot-Marie-Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.; Weis, J.; Korinthenberg, R.; Fehrenbach, H.; Häusler, M.; Züchner, S.; Mache, C.; Hubmann, H.; Auer-Grumbach, M.; Senderek, J. Inverted Formin 2-Related Charcot-Marie-Tooth Disease: Extension of the Mutational Spectrum and Pathological Findings in Schwann Cells and Axons. J. Peripher. Nerv. Syst. 2015, 20, 52–59. [Google Scholar] [CrossRef]
- Toyota, K.; Ogino, D.; Hayashi, M.; Taki, M.; Saito, K.; Abe, A.; Hashimoto, T.; Umetsu, K.; Tsukaguchi, H.; Hayasaka, K. INF2 Mutations in Charcot-Marie-Tooth Disease Complicated with Focal Segmental Glomerulosclerosis. J. Peripher. Nerv. Syst. 2013, 18, 97–98. [Google Scholar] [CrossRef]
- Mademan, I.; Deconinck, T.; Dinopoulos, A.; Voit, T.; Schara, U.; Devriendt, K.; Meijers, B.; Lerut, E.; De Jonghe, P.; Baets, J. De Novo INF2 Mutations Expand the Genetic Spectrum of Hereditary Neuropathy with Glomerulopathy. Neurology 2013, 81, 1953–1958. [Google Scholar] [CrossRef]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 Gene Account for a Significant Proportion of Familial but Not Sporadic Focal and Segmental Glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Snoek, R.; Nguyen, T.Q.; van der Zwaag, B.; van Zuilen, A.D.; Kruis, H.M.E.; van Gils-Verrij, L.A.; Goldschmeding, R.; Knoers, N.V.A.M.; Rookmaaker, M.B.; van Eerde, A.M. Importance of Genetic Diagnostics in Adult-Onset Focal Segmental Glomerulosclerosis. Nephron 2019, 142, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome Sequencing and Identification of Phenocopies in Patients with Clinically Presumed Hereditary Nephropathies. Am. J. Kidney Dis. 2020, 76, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Benoit, G.; Gribouval, O.; Nevo, F.; Tête, M.-J.; Dantal, J.; Gilbert-Dussardier, B.; Touchard, G.; Karras, A.; Presne, C.; et al. Mutations in INF2 Are a Major Cause of Autosomal Dominant Focal Segmental Glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Lee, C.; Kim, N.K.D.; Ahn, Y.H.; Park, Y.S.; Lee, J.H.; Kim, S.H.; Cho, M.H.; Cho, H.; Yoo, K.H.; et al. Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis. J. Clin. Med. 2020, 9, 2013. [Google Scholar] [CrossRef]
- Rodriguez, P.Q.; Lohkamp, B.; Celsi, G.; Mache, C.J.; Auer-Grumbach, M.; Wernerson, A.; Hamajima, N.; Tryggvason, K.; Patrakka, J. Novel INF2 Mutation p. L77P in a Family with Glomerulopathy and Charcot-Marie-Tooth Neuropathy. Pediatr. Nephrol. 2013, 28, 339–343. [Google Scholar] [CrossRef]
- Xie, J.; Hao, X.; Azeloglu, E.U.; Ren, H.; Wang, Z.; Ma, J.; Liu, J.; Ma, X.; Wang, W.; Pan, X.; et al. Novel Mutations in the Inverted Formin 2 Gene of Chinese Families Contribute to Focal Segmental Glomerulosclerosis. Kidney Int. 2015, 88, 593–604. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Latour, P. A Cryptic Splicing Mutation in the INF2 Gene Causing Charcot-Marie-Tooth Disease with Minimal Glomerular Dysfunction. J. Peripher. Nerv. Syst. 2019, 24, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Challis, R.C.; Ring, T.; Xu, Y.; Wong, E.K.S.; Flossmann, O.; Roberts, I.S.D.; Ahmed, S.; Wetherall, M.; Salkus, G.; Brocklebank, V.; et al. Thrombotic Microangiopathy in Inverted Formin 2-Mediated Renal Disease. J. Am. Soc. Nephrol. 2017, 28, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Bacquet, J.; Stojkovic, T.; Boyer, A.; Martini, N.; Audic, F.; Chabrol, B.; Salort-Campana, E.; Delmont, E.; Desvignes, J.-P.; Verschueren, A.; et al. Molecular Diagnosis of Inherited Peripheral Neuropathies by Targeted Next-Generation Sequencing: Molecular Spectrum Delineation. BMJ Open 2018, 8, e021632. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, G.; Neemeh, J.A. Charcot-Marie-Tooth Disease and Nephritis. Can. Med. Assoc. J. 1967, 97, 1193–1198. [Google Scholar]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caridi, G.; Lugani, F.; Dagnino, M.; Gigante, M.; Iolascon, A.; Falco, M.; Graziano, C.; Benetti, E.; Dugo, M.; Del Prete, D.; et al. Novel INF2 Mutations in an Italian Cohort of Patients with Focal Segmental Glomerulosclerosis, Renal Failure and Charcot-Marie-Tooth Neuropathy. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 4), iv80–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Ma, M.; Pang, M.; Yang, L.; Li, G.; Song, J.; Zhang, J. Analysis of a pedigree with autosomal dominant intermediate Charcot-Marie-Tooth disease type E and nephropathy. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2019, 36, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Wang, W.; Wang, R.; Lv, H.; Zhang, W.; Wang, Z.; Jiao, J.; Yuan, Y. INF2 Mutations Associated with Dominant Inherited Intermediate Charcot-Marie-Tooth Neuropathy with Focal Segmental Glomerulosclerosis in Two Chinese Patients. Clin. Neuropathol. 2015, 34, 275–281. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, H.J.; Hong, Y.B.; Nam, S.H.; Chung, K.W.; Choi, B.-O. A Novel INF2 Mutation in a Korean Family with Autosomal Dominant Intermediate Charcot-Marie-Tooth Disease and Focal Segmental Glomerulosclerosis. J. Peripher. Nerv. Syst. 2014, 19, 175–179. [Google Scholar] [CrossRef]
- Laurin, L.-P.; Lu, M.; Mottl, A.K.; Blyth, E.R.; Poulton, C.J.; Weck, K.E. Podocyte-Associated Gene Mutation Screening in a Heterogeneous Cohort of Patients with Sporadic Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2014, 29, 2062–2069. [Google Scholar] [CrossRef]
- Wang, M.; Chun, J.; Genovese, G.; Knob, A.U.; Benjamin, A.; Wilkins, M.S.; Friedman, D.J.; Appel, G.B.; Lifton, R.P.; Mane, S.; et al. Contributions of Rare Gene Variants to Familial and Sporadic FSGS. J. Am. Soc. Nephrol. 2019, 30, 1625–1640. [Google Scholar] [CrossRef]
- Münch, J.; Grohmann, M.; Lindner, T.H.; Bergmann, C.; Halbritter, J. Diagnosing FSGS without Kidney Biopsy—A Novel INF2-Mutation in a Family with ESRD of Unknown Origin. BMC Med. Genet. 2016, 17, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büscher, A.K.; Celebi, N.; Hoyer, P.F.; Klein, H.-G.; Weber, S.; Hoefele, J. Mutations in INF2 May Be Associated with Renal Histology other than Focal Segmental Glomerulosclerosis. Pediatr. Nephrol. 2018, 33, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Braunisch, M.C.; Riedhammer, K.M.; Herr, P.-M.; Draut, S.; Günthner, R.; Wagner, M.; Weidenbusch, M.; Lungu, A.; Alhaddad, B.; Renders, L.; et al. Identification of Disease-Causing Variants by Comprehensive Genetic Testing with Exome Sequencing in Adults with Suspicion of Hereditary FSGS. Eur. J. Hum. Genet. 2021, 29, 262–270. [Google Scholar] [CrossRef]
- Rood, I.M.; Bongers, E.M.H.F.; Lugtenberg, D.; Klein, I.H.H.T.; Steenbergen, E.J.; Wetzels, J.F.M.; Deegens, J.K.J. Familial Focal Segmental Glomerulosclerosis: Mutation in Inverted Formin 2 Mimicking Alport Syndrome. Neth. J. Med. 2016, 74, 82–85. [Google Scholar] [PubMed]
- Gbadegesin, R.A.; Lavin, P.J.; Hall, G.; Bartkowiak, B.; Homstad, A.; Jiang, R.; Wu, G.; Byrd, A.; Lynn, K.; Wolfish, N.; et al. Inverted Formin 2 Mutations with Variable Expression in Patients with Sporadic and Hereditary Focal and Segmental Glomerulosclerosis. Kidney Int. 2012, 81, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef]
- Tan, W.; Lovric, S.; Ashraf, S.; Rao, J.; Schapiro, D.; Airik, M.; Shril, S.; Gee, H.Y.; Baum, M.; Daouk, G.; et al. Analysis of 24 Genes Reveals a Monogenic Cause in 11.1% of Cases with Steroid-Resistant Nephrotic Syndrome at a Single Center. Pediatr. Nephrol. 2018, 33, 305–314. [Google Scholar] [CrossRef]
- Ogino, D.; Hashimoto, T.; Hattori, M.; Sugawara, N.; Akioka, Y.; Tamiya, G.; Makino, S.; Toyota, K.; Mitsui, T.; Hayasaka, K. Analysis of the Genes Responsible for Steroid-Resistant Nephrotic Syndrome and/or Focal Segmental Glomerulosclerosis in Japanese Patients by Whole-Exome Sequencing Analysis. J. Hum. Genet. 2016, 61, 137–141. [Google Scholar] [CrossRef]
- Shang, S.; Peng, F.; Wang, T.; Wu, X.; Li, P.; Li, Q.; Chen, X.M. Genotype-Phenotype Correlation and Prognostic Impact in Chinese Patients with Alport Syndrome. Mol. Genet. Genom. Med. 2019, 7, e00741. [Google Scholar] [CrossRef] [Green Version]
- Gribouval, O.; Boyer, O.; Hummel, A.; Dantal, J.; Martinez, F.; Sberro-Soussan, R.; Etienne, I.; Chauveau, D.; Delahousse, M.; Lionet, A.; et al. Identification of Genetic Causes for Sporadic Steroid-Resistant Nephrotic Syndrome in Adults. Kidney Int. 2018, 94, 1013–1022. [Google Scholar] [CrossRef]
- Singh, A.; Singh, A.; Mishra, O.P.; Prasad, R.; Narayan, G.; Batra, V.V.; Tabatabaeifar, M.; Schaefer, F. Molecular Study of Childhood Steroid-Resistant Nephrotic Syndrome: A Hospital-Based Study. J. Pediatr. Genet. 2021. [Google Scholar] [CrossRef]
- Santín, S.; Bullich, G.; Tazón-Vega, B.; García-Maset, R.; Giménez, I.; Silva, I.; Ruíz, P.; Ballarín, J.; Torra, R.; Ars, E. Clinical Utility of Genetic Testing in Children and Adults with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2011, 6, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.P.; Durfee, T.; Wilson, J.D.; Beggs, M.L. A Custom Targeted Next-Generation Sequencing Gene Panel for the Diagnosis of Genetic Nephropathies. Am. J. Kidney Dis. 2016, 67, 992–993. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Ma, X.; Zhang, X.; Luo, D.; Zhang, J.; Li, M.; Ye, Z.; Peng, H. INF2 p.Arg214Cys Mutation in a Chinese Family with Rapidly Progressive Renal Failure and Follow-up of Renal Transplantation: Case Report and Literature Review. BMC Nephrol. 2021, 22, 51. [Google Scholar] [CrossRef]
- Safarikova, M.; Stekrova, J.; Honsova, E.; Horinova, V.; Tesar, V.; Reiterova, J. Mutational Screening of Inverted Formin 2 in Adult-Onset Focal Segmental Glomerulosclerosis or Minimal Change Patients from the Czech Republic. BMC Med. Genet. 2018, 19, 147. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) Mutations Are the Most Frequent Mutations Underlying Adult Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipska, B.S.; Iatropoulos, P.; Maranta, R.; Caridi, G.; Ozaltin, F.; Anarat, A.; Balat, A.; Gellermann, J.; Trautmann, A.; Erdogan, O.; et al. Genetic Screening in Adolescents with Steroid-Resistant Nephrotic Syndrome. Kidney Int. 2013, 84, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullich, G.; Trujillano, D.; Santín, S.; Ossowski, S.; Mendizábal, S.; Fraga, G.; Madrid, Á.; Ariceta, G.; Ballarín, J.; Torra, R.; et al. Targeted Next-Generation Sequencing in Steroid-Resistant Nephrotic Syndrome: Mutations in Multiple Glomerular Genes May Influence Disease Severity. Eur. J. Hum. Genet. 2015, 23, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Han, K.H.; Jung, Y.H.; Kang, H.G.; Moon, K.C.; Ha, I.S.; Choi, Y.; Cheong, H.I. Variable Renal Phenotype in a Family with an INF2 Mutation. Pediatr. Nephrol. 2011, 26, 73–76. [Google Scholar] [CrossRef]
- Sanchez-Ares, M.; Garcia-Vidal, M.; Antucho, E.-E.; Julio, P.; Eduardo, V.-M.; Lens, X.M.; Garcia-Gonzalez, M.A. A Novel Mutation, Outside of the Candidate Region for Diagnosis, in the Inverted Formin 2 Gene Can Cause Focal Segmental Glomerulosclerosis. Kidney Int. 2013, 83, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büscher, A.K.; Beck, B.B.; Melk, A.; Hoefele, J.; Kranz, B.; Bamborschke, D.; Baig, S.; Lange-Sperandio, B.; Jungraithmayr, T.; Weber, L.T.; et al. Rapid Response to Cyclosporin A and Favorable Renal Outcome in Nongenetic Versus Genetic Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Weber, S.; Büscher, A.K.; Hagmann, H.; Liebau, M.C.; Heberle, C.; Ludwig, M.; Rath, S.; Alberer, M.; Beissert, A.; Zenker, M.; et al. Dealing with the Incidental Finding of Secondary Variants by the Example of SRNS Patients Undergoing Targeted Next-Generation Sequencing. Pediatr. Nephrol. 2016, 31, 73–81. [Google Scholar] [CrossRef]
- Dohrn, M.F.; Glöckle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hörtnagel, K.; et al. Frequent Genes in Rare Diseases: Panel-Based next Generation Sequencing to Disclose Causal Mutations in Hereditary Neuropathies. J. Neurochem. 2017, 143, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.; Maiti, R.; Das, D.; Mandal, K. Steroid Resistant Nephrotic Syndrome with Clumsy Gait Associated with INF2 Mutation. Indian Pediatr. 2020, 57, 764. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ruan, J.; Liu, J.; Zhang, C.; Kang, L.; Wang, J.; Zou, Y.; Song, L. Variant Spectrum of Formin Homology 2 Domain-Containing 3 Gene in Chinese Patients with Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e018236. [Google Scholar] [CrossRef]
- Huang, S.; Pu, T.; Wei, W.; Xu, R.; Wu, Y. Exome Sequencing Identifies a FHOD3 p.S527del Mutation in a Chinese Family with Hypertrophic Cardiomyopathy. J. Gene Med. 2020, 22, e3146. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Bagnall, R.D. Revisiting Genome Sequencing Data in Light of Novel Disease Gene Associations. J. Am. Coll. Cardiol. 2019, 73, 1365–1366. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Lopes, L.R.; Perez-Barbeito, M.; Cazón-Varela, L.; de la Torre-Carpente, M.M.; Sonicheva-Paterson, N.; Uña-Iglesias, D.D.; Quinn, E.; Kuzmina-Krutetskaya, S.; Garrote, J.A.; et al. Deletions of Specific Exons of FHOD3 Detected by Next-Generation Sequencing Are Associated with Hypertrophic Cardiomyopathy. Clin. Genet. 2020, 98, 86–90. [Google Scholar] [CrossRef]
- Hayashi, T.; Tanimoto, K.; Hirayama-Yamada, K.; Tsuda, E.; Ayusawa, M.; Nunoda, S.; Hosaki, A.; Kimura, A. Genetic Background of Japanese Patients with Pediatric Hypertrophic and Restrictive Cardiomyopathy. J. Hum. Genet. 2018, 63, 989–996. [Google Scholar] [CrossRef]
- DeWard, A.D.; Eisenmann, K.M.; Matheson, S.F.; Alberts, A.S. The Role of Formins in Human Disease. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, T.S.; Ehler, E. A Formin-g Role during Development and Disease. Eur. J. Cell Biol. 2014, 93, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Scott, R.P.; Quaggin, S.E. The Cell Biology of Renal Filtration. J. Cell Biol. 2015, 209, 199–210. [Google Scholar] [CrossRef]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef] [Green Version]
- Labat-de-Hoz, L.; Alonso, M.A. The Formin INF2 in Disease: Progress from 10 Years of Research. Cell. Mol. Life Sci. 2020, 77, 4581–4600. [Google Scholar] [CrossRef]
- Bose, B.; Cattran, D. Glomerular Diseases: FSGS. Clin. J. Am. Soc. Nephrol. 2014, 9, 626–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogo, A.B. Causes and Pathogenesis of Focal Segmental Glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical Implications of Genetic Advances in Charcot-Marie-Tooth Disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef]
- Bayraktar, S.; Nehrig, J.; Menis, E.; Karli, K.; Janning, A.; Struk, T.; Halbritter, J.; Michgehl, U.; Krahn, M.P.; Schuberth, C.E.; et al. A Deregulated Stress Response Underlies Distinct INF2-Associated Disease Profiles. J. Am. Soc. Nephrol. 2020, 31, 1296–1313. [Google Scholar] [CrossRef]
- Mu, A.; Fung, T.S.; Kettenbach, A.N.; Chakrabarti, R.; Higgs, H.N. A Complex Containing Lysine-Acetylated Actin Inhibits the Formin INF2. Nat. Cell Biol. 2019, 21, 592–602. [Google Scholar] [CrossRef]
- Subramanian, B.; Sun, H.; Yan, P.; Charoonratana, V.T.; Higgs, H.N.; Wang, F.; Lai, K.-M.V.; Valenzuela, D.M.; Brown, E.J.; Schlöndorff, J.S.; et al. Mice with Mutant Inf2 Show Impaired Podocyte and Slit Diaphragm Integrity in Response to Protamine-Induced Kidney Injury. Kidney Int. 2016, 90, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, S.; Funalot, B.; Boyer, O.; Lacroix, C.; Marcorelles, P.; Magy, L.; Richard, L.; Antignac, C.; Vallat, J.-M. Neuropathologic Characterization of INF2-Related Charcot-Marie-Tooth Disease: Evidence for a Schwann Cell Actinopathy. J. Neuropathol. Exp. Neurol. 2014, 73, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Tricaud, N. Myelinating Schwann Cell Polarity and Mechanically-Driven Myelin Sheath Elongation. Front. Cell. Neurosci. 2018, 11, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemon, Y.; Wright, V.; Davenport, B.; Gatti, D.M.; Sheehan, S.M.; Letson, K.; Savage, H.S.; Lennon, R.; Korstanje, R. Uncovering Modifier Genes of X-Linked Alport Syndrome Using a Novel Multiparent Mouse Model. J. Am. Soc. Nephrol. 2021, 32, 1961–1973. [Google Scholar] [CrossRef]
- Schwander, M.; Kachar, B.; Müller, U. Review Series: The Cell Biology of Hearing. J. Cell Biol. 2010, 190, 9–20. [Google Scholar] [CrossRef]
- Drummond, M.C.; Belyantseva, I.A.; Friderici, K.H.; Friedman, T.B. Actin in Hair Cells and Hearing Loss. Hear Res. 2012, 288, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallar, B.J.; Stropich, B.N.; Schoenherr, J.A.; Holman, H.A.; Kitchen, S.M.; Alberts, A.S. The Basic Region of the Diaphanous-Autoregulatory Domain (DAD) Is Required for Autoregulatory Interactions with the Diaphanous-Related Formin Inhibitory Domain. J. Biol. Chem. 2006, 281, 4300–4307. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, T.; Belyantseva, I.A.; Kitajiri, S.-I.; Miyajima, H.; Nishio, S.-Y.; Usami, S.-I.; Kim, B.J.; Choi, B.Y.; Omori, K.; Shroff, H.; et al. Human Deafness-Associated Variants Alter the Dynamics of Key Molecules in Hair Cell Stereocilia F-Actin Cores. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Lakha, R.; Montero, A.M.; Jabeen, T.; Costeas, C.C.; Ma, J.; Vizcarra, C.L. Variable Autoinhibition among Deafness-Associated Variants of Diaphanous 1 (DIAPH1). Biochemistry 2021, 60, 2320–2329. [Google Scholar] [CrossRef]
- Ninoyu, Y.; Sakaguchi, H.; Lin, C.; Suzuki, T.; Hirano, S.; Hisa, Y.; Saito, N.; Ueyama, T. The Integrity of Cochlear Hair Cells Is Established and Maintained through the Localization of Dia1 at Apical Junctional Complexes and Stereocilia. Cell Death Dis. 2020, 11, 536. [Google Scholar] [CrossRef] [PubMed]
- Schoen, C.J.; Burmeister, M.; Lesperance, M.M. Diaphanous Homolog 3 (Diap3) Overexpression Causes Progressive Hearing Loss and Inner Hair Cell Defects in a Transgenic Mouse Model of Human Deafness. PLoS ONE 2013, 8, e56520. [Google Scholar] [CrossRef] [Green Version]
- Surel, C.; Guillet, M.; Lenoir, M.; Bourien, J.; Sendin, G.; Joly, W.; Delprat, B.; Lesperance, M.M.; Puel, J.-L.; Nouvian, R. Remodeling of the Inner Hair Cell Microtubule Meshwork in a Mouse Model of Auditory Neuropathy AUNA1. eNeuro 2016, 3, 0295-16.2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.-H.; Baek, J.-I.; Lee, J.D.; Song, M.H.; Kwon, T.-J.; Oh, S.-K.; Jeong, J.Y.; Choi, J.Y.; Lee, K.-Y.; Kim, U.-K. Genetic Analysis of Auditory Neuropathy Spectrum Disorder in the Korean Population. Gene 2013, 522, 65–69. [Google Scholar] [CrossRef]
- Almazni, I.; Stapley, R.; Morgan, N.V. Inherited Thrombocytopenia: Update on Genes and Genetic Variants which May Be Associated with Bleeding. Front. Cardiovasc. Med. 2019, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, I.C.; Scheller, I.; Wackerbarth, L.M.; Beck, S.; Heib, T.; Aurbach, K.; Manukjan, G.; Gross, C.; Spindler, M.; Nagy, Z.; et al. Actin/Microtubule Crosstalk during Platelet Biogenesis in Mice Is Critically Regulated by Twinfilin1 and Cofilin1. Blood Adv. 2020, 4, 2124–2134. [Google Scholar] [CrossRef]
- Italiano, J.E.; Lecine, P.; Shivdasani, R.A.; Hartwig, J.H. Blood Platelets Are Assembled Principally at the Ends of Proplatelet Processes Produced by Differentiated Megakaryocytes. J. Cell Biol. 1999, 147, 1299–1312. [Google Scholar] [CrossRef]
- Patel, S.R. The Biogenesis of Platelets from Megakaryocyte Proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef] [Green Version]
- Potts, K.S.; Farley, A.; Dawson, C.A.; Rimes, J.; Biben, C.; de Graaf, C.; Potts, M.A.; Stonehouse, O.J.; Carmagnac, A.; Gangatirkar, P.; et al. Membrane Budding Is a Major Mechanism of in Vivo Platelet Biogenesis. J. Exp. Med. 2020, 217, e20191206. [Google Scholar] [CrossRef]
- Pan, J.; Lordier, L.; Meyran, D.; Rameau, P.; Lecluse, Y.; Kitchen-Goosen, S.; Badirou, I.; Mokrani, H.; Narumiya, S.; Alberts, A.S.; et al. The Formin DIAPH1 (MDia1) Regulates Megakaryocyte Proplatelet Formation by Remodeling the Actin and Microtubule Cytoskeletons. Blood 2014, 124, 3967–3977. [Google Scholar] [CrossRef] [Green Version]
- Zuidscherwoude, M.; Green, H.L.H.; Thomas, S.G. Formin Proteins in Megakaryocytes and Platelets: Regulation of Actin and Microtubule Dynamics. Platelets 2019, 30, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizaki, T.; Morishima, Y.; Okamoto, M.; Furuyashiki, T.; Kato, T.; Narumiya, S. Coordination of Microtubules and the Actin Cytoskeleton by the Rho Effector MDia1. Nat. Cell Biol. 2001, 3, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Cook, T.A.; Alberts, A.S.; Gundersen, G.G. mDia Mediates Rho-Regulated Formation and Orientation of Stable Microtubules. Nat. Cell Biol. 2001, 3, 723–729. [Google Scholar] [CrossRef]
- Heremans, J.; Garcia-Perez, J.E.; Turro, E.; Schlenner, S.M.; Casteels, I.; Collin, R.; de Zegher, F.; Greene, D.; Humblet-Baron, S.; Lesage, S.; et al. Abnormal Differentiation of B Cells and Megakaryocytes in Patients with Roifman Syndrome. J. Allergy Clin. Immunol. 2018, 142, 630–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merico, D.; Roifman, M.; Braunschweig, U.; Yuen, R.K.C.; Alexandrova, R.; Bates, A.; Reid, B.; Nalpathamkalam, T.; Wang, Z.; Thiruvahindrapuram, B.; et al. Compound Heterozygous Mutations in the Noncoding RNU4ATAC Cause Roifman Syndrome by Disrupting Minor Intron Splicing. Nat. Commun. 2015, 6, 8718. [Google Scholar] [CrossRef]
- Dutta, P.; Maiti, S. Expression of Multiple Formins in Adult Tissues and during Developmental Stages of Mouse Brain. Gene Expr. Patterns 2015, 19, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kawabata Galbraith, K.; Kengaku, M. Multiple Roles of the Actin and Microtubule-Regulating Formins in the Developing Brain. Neurosci. Res. 2019, 138, 59–69. [Google Scholar] [CrossRef]
- Damiani, D.; Goffinet, A.M.; Alberts, A.; Tissir, F. Lack of Diaph3 Relaxes the Spindle Checkpoint Causing the Loss of Neural Progenitors. Nat. Commun. 2016, 7, 13509. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Shinohara, R.; Watanabe, K.; Takebayashi, H.; Toyoda, Y.; Tohyama, K.; Ishizaki, T.; Furuyashiki, T.; Narumiya, S. Deficiency of mDia, an Actin Nucleator, Disrupts Integrity of Neuroepithelium and Causes Periventricular Dysplasia. PLoS ONE 2011, 6, e25465. [Google Scholar] [CrossRef] [Green Version]
- Eisenmann, K.M.; West, R.A.; Hildebrand, D.; Kitchen, S.M.; Peng, J.; Sigler, R.; Zhang, J.; Siminovitch, K.A.; Alberts, A.S. T Cell Responses in Mammalian Diaphanous-Related Formin MDia1 Knock-out Mice. J. Biol. Chem. 2007, 282, 25152–25158. [Google Scholar] [CrossRef] [Green Version]
- Sakata, D.; Taniguchi, H.; Yasuda, S.; Adachi-Morishima, A.; Hamazaki, Y.; Nakayama, R.; Miki, T.; Minato, N.; Narumiya, S. Impaired T Lymphocyte Trafficking in Mice Deficient in an Actin-Nucleating Protein, MDia1. J. Exp. Med. 2007, 204, 2031–2038. [Google Scholar] [CrossRef] [Green Version]
- Kundu, T.; Dutta, P.; Nagar, D.; Maiti, S.; Ghose, A. Coupling of Dynamic Microtubules to F-Actin by Fmn2 Regulates Chemotaxis of Neuronal Growth Cones. J. Cell Sci. 2021, 134, jcs252916. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, A.; Ghate, K.; Mutalik, S.; Jacob, A.; Ghose, A. Formin 2 Regulates the Stabilization of Filopodial Tip Adhesions in Growth Cones and Affects Neuronal Outgrowth and Pathfinding in Vivo. Development 2016, 143, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leader, B.; Lim, H.; Carabatsos, M.J.; Harrington, A.; Ecsedy, J.; Pellman, D.; Maas, R.; Leder, P. Formin-2, Polyploidy, Hypofertility and Positioning of the Meiotic Spindle in Mouse Oocytes. Nat. Cell Biol. 2002, 4, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Dettenhofer, M.; Lu, J.; Downing, M.; Chenn, A.; Wong, T.; Sheen, V. Filamin A- and Formin 2-Dependent Endocytosis Regulates Proliferation via the Canonical Wnt Pathway. Development 2016, 143, 4509–4520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lybaek, H.; Ørstavik, K.H.; Prescott, T.; Hovland, R.; Breilid, H.; Stansberg, C.; Steen, V.M.; Houge, G. An 8.9 Mb 19p13 Duplication Associated with Precocious Puberty and a Sporadic 3.9 Mb 2q23.3q24.1 Deletion Containing NR4A2 in Mentally Retarded Members of a Family with an Intrachromosomal 19p-into-19q between-Arm Insertion. Eur. J. Hum. Genet. 2009, 17, 904–910. [Google Scholar] [CrossRef]
- Castrillon, D.H.; Wasserman, S.A. Diaphanous Is Required for Cytokinesis in Drosophila and Shares Domains of Similarity with the Products of the Limb Deformity Gene. Development 1994, 120, 3367–3377. [Google Scholar] [CrossRef]
- Ryley, D.A.; Wu, H.-H.; Leader, B.; Zimon, A.; Reindollar, R.H.; Gray, M.R. Characterization and Mutation Analysis of the Human Formin-2 (FMN2) Gene in Women with Unexplained Infertility. Fertil. Steril. 2005, 83, 1363–1371. [Google Scholar] [CrossRef]
- Tšuiko, O.; Nõukas, M.; Žilina, O.; Hensen, K.; Tapanainen, J.S.; Mägi, R.; Kals, M.; Kivistik, P.A.; Haller-Kikkatalo, K.; Salumets, A.; et al. Copy Number Variation Analysis Detects Novel Candidate Genes Involved in Follicular Growth and Oocyte Maturation in a Cohort of Premature Ovarian Failure Cases. Hum. Reprod. 2016, 31, 1913–1925. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Q.; Liu, F.; Zhang, Z.; Zou, Y.; Yang, B.; Luo, Y.; Wang, L.; Huang, O. Inhibition of Formin like 2 Promotes the Transition of Ectopic Endometrial Stromal Cells to Epithelial Cells in Adenomyosis through a MET-like Process. Gene 2019, 710, 186–192. [Google Scholar] [CrossRef]
- Rosado, M.; Barber, C.F.; Berciu, C.; Feldman, S.; Birren, S.J.; Nicastro, D.; Goode, B.L. Critical Roles for Multiple Formins during Cardiac Myofibril Development and Repair. Mol. Biol. Cell 2014, 25, 811–827. [Google Scholar] [CrossRef]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-o, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian Formin Fhod3 Regulates Actin Assembly and Sarcomere Organization in Striated Muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef] [Green Version]
- Ehler, E. Actin-Associated Proteins and Cardiomyopathy-the “unknown” beyond Troponin and Tropomyosin. Biophys. Rev. 2018, 10, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated Cardiomyopathy-Associated FHOD3 Variant Impairs the Ability to Induce Activation of Transcription Factor Serum Response Factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, U.; Garnier, S.; Korniat, A.; Proust, C.; Kararigas, G.; Müller-Nurasyid, M.; Empana, J.-P.; Morley, M.P.; Perret, C.; Stark, K.; et al. Exome-Wide Association Study Reveals Novel Susceptibility Genes to Sporadic Dilated Cardiomyopathy. PLoS ONE 2017, 12, e0172995. [Google Scholar] [CrossRef]
- Kan, O.M.; Takeya, R.; Abe, T.; Kitajima, N.; Nishida, M.; Tominaga, R.; Kurose, H.; Sumimoto, H. Mammalian Formin Fhod3 Plays an Essential Role in Cardiogenesis by Organizing Myofibrillogenesis. Biol. Open 2012, 1, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan, O.M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The Actin-Organizing Formin Protein Fhod3 Is Required for Postnatal Development and Functional Maintenance of the Adult Heart in Mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heineke, J.; Molkentin, J.D. Regulation of Cardiac Hypertrophy by Intracellular Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Zhou, Q.; Wei, S.-S.; Wang, H.; Wang, Q.; Li, W.; Li, G.; Hou, J.-W.; Chen, X.-M.; Chen, J.; Xu, W.-P.; et al. Crucial Role of ROCK2-Mediated Phosphorylation and Upregulation of FHOD3 in the Pathogenesis of Angiotensin II-Induced Cardiac Hypertrophy. Hypertension 2017, 69, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Hilton-Jones, D. Myotonic Dystrophy: Diagnosis, Management and New Therapies. Curr. Opin. Neurol. 2014, 27, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Oliveira, R.; Sznajder, Ł.J.; Swanson, M.S. Myotonic Dystrophy and Developmental Regulation of RNA Processing. In Comprehensive Physiology; Terjung, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 509–553. ISBN 978-0-470-65071-4. [Google Scholar]
- Dixon, D.M.; Choi, J.; El-Ghazali, A.; Park, S.Y.; Roos, K.P.; Jordan, M.C.; Fishbein, M.C.; Comai, L.; Reddy, S. Loss of Muscleblind-like 1 Results in Cardiac Pathology and Persistence of Embryonic Splice Isoforms. Sci. Rep. 2015, 5, 9932. [Google Scholar] [CrossRef]
- Perfetti, A.; Greco, S.; Fasanaro, P.; Bugiardini, E.; Cardani, R.; Garcia-Manteiga, J.M.; Manteiga, J.M.G.; Riba, M.; Cittaro, D.; Stupka, E.; et al. Genome Wide Identification of Aberrant Alternative Splicing Events in Myotonic Dystrophy Type 2. PLoS ONE 2014, 9, e93983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, B.; Zhang, L.; Hu, H.; Yin, S.; Liang, Z. Deletion of a Single-Copy DAAM1 Gene in Congenital Heart Defect: A Case Report. BMC Med. Genet. 2012, 13, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehalle, D.; Sanlaville, D.; Guimier, A.; Plouvier, E.; Leblanc, T.; Galmiche, L.; Radford, I.; Romana, S.; Colleaux, L.; de Pontual, L.; et al. Multiple Congenital Anomalies-Intellectual Disability (MCA-ID) and Neuroblastoma in a Patient Harboring a de Novo 14q23.1q23.3 Deletion. Am. J. Med. Genet. 2014, 164, 1310–1317. [Google Scholar] [CrossRef]
- Li, D.; Hallett, M.A.; Zhu, W.; Rubart, M.; Liu, Y.; Yang, Z.; Chen, H.; Haneline, L.S.; Chan, R.J.; Schwartz, R.J.; et al. Dishevelled-Associated Activator of Morphogenesis 1 (Daam1) Is Required for Heart Morphogenesis. Development 2011, 138, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheroni, C.; Caporale, N.; Testa, G. Autism Spectrum Disorder at the Crossroad between Genes and Environment: Contributions, Convergences, and Interactions in ASD Developmental Pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef]
- Vorstman, J.; van Daalen, E.; Jalali, G.; Schmidt, E.; Pasterkamp, R.; de Jonge, M.; Hennekam, E.; Janson, E.; Staal, W.; van der Zwaag, B.; et al. A Double Hit Implicates DIAPH3 as an Autism Risk Gene. Mol. Psychiatry 2011, 16, 442–451. [Google Scholar] [CrossRef]
- Xie, J.; Li, H.; Zhu, H.; Huang, L.; Li, H.; Zhang, X.; Zhou, Y.; Zhou, Q.; Xu, W. Analysis of DIAPH3 gene mutation in a boy with autism spectrum disorder. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 481–484. [Google Scholar] [CrossRef]
- Liu, L.; Liu, F.; Wang, Q.; Xie, H.; Li, Z.; Lu, Q.; Wang, Y.; Zhang, M.; Zhang, Y.; Picker, J.; et al. Confirming the Contribution and Genetic Spectrum of de Novo Mutation in Infantile Spasms: Evidence from a Chinese Cohort. Mol. Genet. Genom. Med. 2021, 9, e1689. [Google Scholar] [CrossRef]
- Abramov, D.; Guiberson, N.G.L.; Burré, J. STXBP1 Encephalopathies: Clinical Spectrum, Disease Mechanisms, and Therapeutic Strategies. J. Neurochem. 2020, 157, 165–178. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Labalme, A.; Hirsch, E.; Arzimanoglou, A.; Genton, P.; Motte, J.; de Saint Martin, A.; Valenti, M.-P.; Boulay, C.; et al. Epileptic Encephalopathies of the Landau-Kleffner and Continuous Spike and Waves during Slow-Wave Sleep Types: Genomic Dissection Makes the Link with Autism. Epilepsia 2012, 53, 1526–1538. [Google Scholar] [CrossRef]
- Lau, E.O.-C.; Damiani, D.; Chehade, G.; Ruiz-Reig, N.; Saade, R.; Jossin, Y.; Aittaleb, M.; Schakman, O.; Tajeddine, N.; Gailly, P.; et al. DIAPH3 Deficiency Links Microtubules to Mitotic Errors, Defective Neurogenesis, and Brain Dysfunction. eLife 2021, 10, e61974. [Google Scholar] [CrossRef]
- Matsunami, N.; Hensel, C.H.; Baird, L.; Stevens, J.; Otterud, B.; Leppert, T.; Varvil, T.; Hadley, D.; Glessner, J.T.; Pellegrino, R.; et al. Identification of Rare DNA Sequence Variants in High-Risk Autism Families and Their Prevalence in a Large Case/Control Population. Mol. Autism 2014, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Cappi, C.; Hounie, A.G.; Mariani, D.B.; Diniz, J.B.; Silva, A.R.T.; Reis, V.N.S.; Busso, A.F.; Silva, A.G.; Fidalgo, F.; Rogatto, S.R.; et al. An Inherited Small Microdeletion at 15q13.3 in a Patient with Early- Onset Obsessive-Compulsive Disorder. PLoS ONE 2014, 9, e110198. [Google Scholar] [CrossRef] [Green Version]
- Giacopuzzi, E.; Gennarelli, M.; Minelli, A.; Gardella, R.; Valsecchi, P.; Traversa, M.; Bonvicini, C.; Vita, A.; Sacchetti, E.; Magri, C. Exome Sequencing in Schizophrenic Patients with High Levels of Homozygosity Identifies Novel and Extremely Rare Mutations in the GABA/Glutamatergic Pathways. PLoS ONE 2017, 12, e0182778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Areces, J.; Dopazo, A.; Dettenhofer, M.; Rodriguez-Tebar, A.; Garcia-Segura, L.M.; Arevalo, M.-A. Formin1 Mediates the Induction of Dendritogenesis and Synaptogenesis by Neurogenin3 in Mouse Hippocampal Neurons. PLoS ONE 2011, 6, e21825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proitsi, P.; Li, T.; Hamilton, G.; Di Forti, M.; Collier, D.; Killick, R.; Chen, R.; Sham, P.; Murray, R.; Powell, J.; et al. Positional Pathway Screen of Wnt Signaling Genes in Schizophrenia: Association with DKK4. Biol. Psychiatry 2008, 63, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Kuzman, M.R.; Medved, V.; Terzic, J.; Krainc, D. Genome-Wide Expression Analysis of Peripheral Blood Identifies Candidate Biomarkers for Schizophrenia. J. Psychiatr. Res. 2009, 43, 1073–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.; Xie, P. Correlation Between Daam2 Expression Changes and Demyelination in Guillain–Barre Syndrome. Cell. Mol. Neurobiol. 2016, 36, 683–688. [Google Scholar] [CrossRef]
- Kumar, R.; Corbett, M.A.; Smith, N.J.C.; Jolly, L.A.; Tan, C.; Keating, D.J.; Duffield, M.D.; Utsumi, T.; Moriya, K.; Smith, K.R.; et al. Homozygous Mutation of STXBP5L Explains an Autosomal Recessive Infantile-Onset Neurodegenerative Disorder. Hum. Mol. Genet. 2015, 24, 2000–2010. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.T.; Wallingford, J.B. Planar Cell Polarity in Development and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Torban, E.; Sokol, S.Y. Planar Cell Polarity Pathway in Kidney Development, Function and Disease. Nat. Rev. Nephrol. 2021, 17, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Koni, M.; Pinnarò, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef]
- Liu, W.; Sato, A.; Khadka, D.; Bharti, R.; Diaz, H.; Runnels, L.W.; Habas, R. Mechanism of Activation of the Formin Protein Daam1. Proc. Natl. Acad. Sci. USA 2008, 105, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, N.; Pulit, S.L.; Nijman, I.J.; Monroe, G.R.; Feitz, W.F.J.; Schreuder, M.F.; van Eerde, A.M.; de Jong, T.P.V.M.; Giltay, J.C.; van der Zwaag, B.; et al. Prioritization and Burden Analysis of Rare Variants in 208 Candidate Genes Suggest They Do Not Play a Major Role in CAKUT. Kidney Int. 2016, 89, 476–486. [Google Scholar] [CrossRef] [Green Version]
- McMichael, G.; Girirajan, S.; Moreno-De-Luca, A.; Gecz, J.; Shard, C.; Nguyen, L.S.; Nicholl, J.; Gibson, C.; Haan, E.; Eichler, E.; et al. Rare Copy Number Variation in Cerebral Palsy. Eur. J. Hum. Genet. 2014, 22, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Habas, R.; Kato, Y.; He, X. Wnt/Frizzled Activation of Rho Regulates Vertebrate Gastrulation and Requires a Novel Formin Homology Protein Daam1. Cell 2001, 107, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.K.; de la Torre Canny, S.G.; Jang, C.-W.; Cho, K.; Ji, H.; Wagner, D.S.; Jones, E.A.; Habas, R.; McCrea, P.D. Pronephric Tubulogenesis Requires Daam1-Mediated Planar Cell Polarity Signaling. J. Am. Soc. Nephrol. 2011, 22, 1654–1664. [Google Scholar] [CrossRef] [Green Version]
- Corkins, M.E.; Krneta-Stankic, V.; Kloc, M.; McCrea, P.D.; Gladden, A.B.; Miller, R.K. Divergent Roles of the Wnt/PCP Formin Daam1 in Renal Ciliogenesis. PLoS ONE 2019, 14, e0221698. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.; Shiraishi, T.; Ogura, T. Identification of Chick and Mouse Daam1 and Daam2 Genes and Their Expression Patterns in the Central Nervous System. Dev. Brain Res. 2004, 153, 143–150. [Google Scholar] [CrossRef]
- Sun, S.-W.; Zhou, M.; Chen, L.; Wu, J.-H.; Meng, Z.-J.; Miao, S.-Y.; Han, H.-L.; Zhu, C.-C.; Xiong, X.-Z. Whole Exome Sequencing Identifies a Rare Variant in DAAM2 as a Potential Candidate in Idiopathic Pulmonary Ossification. Ann. Transl. Med. 2019, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Udagawa, N.; Mukai, H.; Ishihara, A.; Maeda, K.; Yamashita, T.; Murakami, K.; Nishita, M.; Nakamura, T.; Kato, S.; et al. Protein Kinase N3 Promotes Bone Resorption by Osteoclasts in Response to Wnt5a-Ror2 Signaling. Sci. Signal. 2017, 10, eaan0023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvel, P.; Kusz-Zamelczyk, K.; Makrythanasis, P.; Janecki, D.; Borel, C.; Conne, B.; Vannier, A.; Béna, F.; Gimelli, S.; Fichna, P.; et al. A Case of Wiedemann-Steiner Syndrome Associated with a 46, XY Disorder of Sexual Development and Gonadal Dysgenesis. Sex Dev. 2015, 9, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Cenani, A.; Lenz, W. Total syndactylia and total radioulnar synostosis in 2 brothers. A contribution on the genetics of syndactylia. Z. Kinder-Heilk. 1967, 101, 181–190. [Google Scholar] [CrossRef]
- Dimitrov, B.I.; Voet, T.; De Smet, L.; Vermeesch, J.R.; Devriendt, K.; Fryns, J.-P.; Debeer, P. Genomic Rearrangements of the GREM1-FMN1 Locus Cause Oligosyndactyly, Radio-Ulnar Synostosis, Hearing Loss, Renal Defects Syndrome and Cenani-Lenz-like Non-Syndromic Oligosyndactyly. J. Med. Genet. 2010, 47, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Pavel, E.; Zhao, W.; Powell, K.A.; Weinstein, M.; Kirschner, L.S. Analysis of a New Allele of Limb Deformity (Ld) Reveals Tissue- and Age-Specific Transcriptional Effects of the Ld Global Control Region. Int. J. Dev. Biol. 2007, 51, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Leder, P.; Zuniga, A.; Dettenhofer, M. Formin1 Disruption Confers Oligodactylism and Alters Bmp Signaling. Hum. Mol. Genet. 2009, 18, 2472–2482. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Dong, J.; Joo, J.; Geller, N.L.; Zheng, G. Simple Strategies for Haplotype Analysis of the X Chromosome with Application to Age-Related Macular Degeneration. Eur. J. Hum. Genet. 2011, 19, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Joo, J.; Zhang, C.; Geller, N.L. Testing association for markers on the X chromosome. Genet. Epidemiol. 2007, 31, 834–843. [Google Scholar] [CrossRef]
- Burri, A.; Maercker, A.; Krammer, S.; Simmen-Janevska, K. Childhood Trauma and PTSD Symptoms Increase the Risk of Cognitive Impairment in a Sample of Former Indentured Child Laborers in Old Age. PLoS ONE 2013, 8, e57826. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, K.; Vittinghoff, E.; Lindquist, K.; Barnes, D.; Covinsky, K.E.; Neylan, T.; Kluse, M.; Marmar, C. Posttraumatic Stress Disorder and Risk of Dementia Among US Veterans. Arch. Gen. Psychiatry 2010, 67, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agís-Balboa, R.C.; Pinheiro, P.S.; Rebola, N.; Kerimoglu, C.; Benito, E.; Gertig, M.; Bahari-Javan, S.; Jain, G.; Burkhardt, S.; Delalle, I.; et al. Formin 2 Links Neuropsychiatric Phenotypes at Young Age to an Increased Risk for Dementia. EMBO J. 2017, 36, 2815–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered Histone Acetylation Is Associated with Age-Dependent Memory Impairment in Mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derk, J.; Hernandez, K.B.; Rodriguez, M.; He, M.; Koh, H.; Abedini, A.; Li, H.; Fenyö, D.; Schmidt, A.M. Diaphanous 1 (DIAPH1) Is Highly Expressed in the Aged Human Medial Temporal Cortex and Upregulated in Myeloid Cells During Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 995. [Google Scholar] [CrossRef]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) and DIAPH1: Implications for Vascular and Neuroinflammatory Dysfunction in Disorders of the Central Nervous System. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Chen, J.; Gao, X.-M.; Zhao, H.; Cai, H.; Zhang, L.; Cao, X.-X.; Zhou, D.-B.; Li, J. A Highly Heterogeneous Mutational Pattern in POEMS Syndrome. Leukemia 2021, 35, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.J.; Harbord, M.W.; MacAllister, R.; Rahman, F.Z.; Young, J.; Al-Lazikani, B.; Lees, W.; Novelli, M.; Bloom, S.; Segal, A.W. Defective Acute Inflammation in Crohn’s Disease: A Clinical Investigation. Lancet 2006, 367, 668–678. [Google Scholar] [CrossRef]
- Trefzer, R.; Elpeleg, O.; Gabrusskaya, T.; Stepensky, P.; Mor-Shaked, H.; Grosse, R.; Brandt, D.T. Characterization of a L136P Mutation in Formin-like 2 (FMNL2) from a Patient with Chronic Inflammatory Bowel Disease. PLoS ONE 2021, 16, e0252428. [Google Scholar] [CrossRef]
- Rai, V.; Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Yan, S.F.; Schmidt, A.M.; Shekhtman, A. Signal Transduction in Receptor for Advanced Glycation End Products (RAGE): Solution Structure of C-Terminal Rage (CtRAGE) and Its Binding to mDia1. J. Biol. Chem. 2012, 287, 5133–5144. [Google Scholar] [CrossRef] [Green Version]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights from Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Friedman, R.A.; Ramasamy, R.; D’Agati, V.; Schmidt, A.M. Deletion of the Formin Diaph1 Protects from Structural and Functional Abnormalities in the Murine Diabetic Kidney. Am. J. Physiol. Ren. Physiol. 2018, 315, F1601–F1612. [Google Scholar] [CrossRef]
- Cooper, J.D.; Walker, N.M.; Smyth, D.J.; Downes, K.; Healy, B.C.; Todd, J.A. Type I Diabetes Genetics Consortium Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium Genome-Wide Association Study in Type I Diabetes Families. Genes Immun. 2009, 10 (Suppl. 1), S85–S94. [Google Scholar] [CrossRef] [Green Version]
- Qi, C.; Al Somali, F.; Zhong, J.; Harris, R.C.; Kon, V.; Yang, H.; Fogo, A.B. Increased DAAM2, a New Podocyte-Associated Protein, in Diabetic Nephropathy. Nephrol. Dial. Transplant. 2021, 36, 1006–1016. [Google Scholar] [CrossRef]
- De Alwis, N.; Beard, S.; Binder, N.K.; Pritchard, N.; Kaitu’u-Lino, T.J.; Walker, S.P.; Stock, O.; Groom, K.; Petersen, S.; Henry, A.; et al. DAAM2 Is Elevated in the Circulation and Placenta in Pregnancies Complicated by Fetal Growth Restriction and Is Regulated by Hypoxia. Sci. Rep. 2021, 11, 5540. [Google Scholar] [CrossRef]
- Nakaya, M.-A.; Gudmundsson, K.O.; Komiya, Y.; Keller, J.R.; Habas, R.; Yamaguchi, T.P.; Ajima, R. Placental Defects Lead to Embryonic Lethality in Mice Lacking the Formin and PCP Proteins Daam1 and Daam2. PLoS ONE 2020, 15, e0232025. [Google Scholar] [CrossRef]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An Atlas of Genetic Influences on Osteoporosis in Humans and Mice. Nat. Genet. 2019, 51, 258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Deng, H.-W.; Shen, H.; Ehrlich, M. Prioritization of Osteoporosis-Associated Genome-wide Association Study (GWAS) Single-Nucleotide Polymorphisms (SNPs) Using Epigenomics and Transcriptomics. JBMR Plus 2021, 5, e10481. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ledo, A.; Jenny, A.; Marlow, F.L. Comparative Gene Expression Analysis of the Fmnl Family of Formins during Zebrafish Development and Implications for Tissue Specific Functions. Gene Expr. Patterns 2013, 13, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Chen, X.; Tang, W.; Li, J.; Yang, S.; Chen, Y.; Zhao, X.; Zong, H.; Liu, C.; Shen, C. Association of DIAPH1 Gene Polymorphisms with Ischemic Stroke. Aging 2020, 12, 416–435. [Google Scholar] [CrossRef]
- Kundishora, A.J.; Peters, S.T.; Pinard, A.; Duran, D.; Panchagnula, S.; Barak, T.; Miyagishima, D.F.; Dong, W.; Smith, H.; Ocken, J.; et al. DIAPH1 Variants in Non-East Asian Patients With Sporadic Moyamoya Disease. JAMA Neurol. 2021, 78, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J. Nurr1 Exacerbates Cerebral Ischemia-Reperfusion Injury via Modulating YAP-INF2-Mitochondrial Fission Pathways. Int. J. Biochem. Cell Biol. 2018, 104, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, W.; Chen, L.; Luo, Y.; Xu, R. Nur77 Promotes Cerebral Ischemia-Reperfusion Injury via Activating INF2-Mediated Mitochondrial Fragmentation. J. Mol. Histol. 2018, 49, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.R.; Polk, D.E.; Wang, X.; Feingold, E.; Weeks, D.E.; Lee, M.-K.; Cuenco, K.T.; Weyant, R.J.; Crout, R.J.; McNeil, D.W.; et al. Genome-Wide Association Study of Periodontal Health Measured by Probing Depth in Adults Ages 18–49 Years. G3 Genes Genomes Genet. 2014, 4, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Choquet, H.; Thai, K.K.; Yin, J.; Hoffmann, T.J.; Kvale, M.N.; Banda, Y.; Schaefer, C.; Risch, N.; Nair, K.S.; Melles, R.; et al. A Large Multi-Ethnic Genome-Wide Association Study Identifies Novel Genetic Loci for Intraocular Pressure. Nat. Commun. 2017, 8, 2108. [Google Scholar] [CrossRef]
- Shin, H.-T.; Yoon, B.W.; Seo, J.H. Analysis of Risk Allele Frequencies of Single Nucleotide Polymorphisms Related to Open-Angle Glaucoma in Different Ethnic Groups. BMC Med. Genom. 2021, 14, 80. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Y.; Zhang, C.; Liu, X.; Jiang, L.; Chen, F. Mammalian Diaphanous-Related Formin 1 Is Required for Motility and Invadopodia Formation in Human U87 Glioblastoma Cells. Int. J. Mol. Med. 2014, 33, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Qiu, Y.; Ren, W.; Gong, J.; Chen, F. An Identification of Stem Cell-Resembling Gene Expression Profiles in High-Grade Astrocytomas. Mol. Carcinog. 2008, 47, 893–903. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, L.; Chen, J.; Liang, J.; Xu, Y.; Li, Z.; Chen, F.; Du, D. Knockdown of Diaph1 Expression Inhibits Migration and Decreases the Expression of MMP2 and MMP9 in Human Glioma Cells. Biomed. Pharmacother. 2017, 96, 596–602. [Google Scholar] [CrossRef]
- Yamana, N.; Arakawa, Y.; Nishino, T.; Kurokawa, K.; Tanji, M.; Itoh, R.E.; Monypenny, J.; Ishizaki, T.; Bito, H.; Nozaki, K.; et al. The Rho-MDia1 Pathway Regulates Cell Polarity and Focal Adhesion Turnover in Migrating Cells through Mobilizing Apc and c-Src. Mol. Cell. Biol. 2006, 26, 6844–6858. [Google Scholar] [CrossRef] [Green Version]
- Hager, M.H.; Morley, S.; Bielenberg, D.R.; Gao, S.; Morello, M.; Holcomb, I.N.; Liu, W.; Mouneimne, G.; Demichelis, F.; Kim, J.; et al. DIAPH3 Governs the Cellular Transition to the Amoeboid Tumour Phenotype. EMBO Mol. Med. 2012, 4, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yan, T.; Li, X.; Sun, J.; Zhang, B.; Wang, H.; Zhu, Y. Daam1 Activates RhoA to Regulate Wnt5a-induced Glioblastoma Cell Invasion. Oncol. Rep. 2018, 39, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, V.; Arnotskaya, N.; Zaitsev, S.; Sharma, A.; Sharma, H.S.; Bryukhovetskiy, A.; Pak, O.; Khotimchenko, Y.; Bryukhovetskiy, I. Proteins of Wnt Signaling Pathway in Cancer Stem Cells of Human Glioblastoma. Int. Rev. Neurobiol. 2020, 151, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Krishna, S.; Garcia, C.; Lin, C.-C.J.; Mitchell, B.D.; Scott, K.L.; Mohila, C.A.; Creighton, C.J.; Yoo, S.-H.; Lee, H.K.; et al. Daam2 Driven Degradation of VHL Promotes Gliomagenesis. eLife 2017, 6, e31926. [Google Scholar] [CrossRef] [PubMed]
- Higa, N.; Shinsato, Y.; Kamil, M.; Hirano, T.; Takajo, T.; Shimokawa, M.; Minami, K.; Yamamoto, M.; Kawahara, K.; Yonezawa, H.; et al. Formin-like 1 (FMNL1) Is Associated with Glioblastoma Multiforme Mesenchymal Subtype and Independently Predicts Poor Prognosis. Int. J. Mol. Sci. 2019, 20, 6355. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Monzo, P.; Chong, Y.K.; Guetta-Terrier, C.; Krishnasamy, A.; Sathe, S.R.; Yim, E.K.F.; Ng, W.H.; Ang, B.T.; Tang, C.; Ladoux, B.; et al. Mechanical Confinement Triggers Glioma Linear Migration Dependent on Formin FHOD3. Mol. Biol. Cell 2016, 27, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Shi, W.; Zhao, R.; Shen, W.; Li, H. FHOD3 Promotes Carcinogenesis by Regulating RhoA/ROCK1/LIMK1 Signaling Pathway in Medulloblastoma. Clin. Transl. Oncol. 2020, 22, 2312–2323. [Google Scholar] [CrossRef]
- Heuser, V.D.; Kiviniemi, A.; Lehtinen, L.; Munthe, S.; Kristensen, B.W.; Posti, J.P.; Sipilä, J.O.T.; Vuorinen, V.; Carpén, O.; Gardberg, M. Multiple Formin Proteins Participate in Glioblastoma Migration. BMC Cancer 2020, 20, 710. [Google Scholar] [CrossRef]
- Kitzing, T.M.; Sahadevan, A.S.; Brandt, D.T.; Knieling, H.; Hannemann, S.; Fackler, O.T.; Großhans, J.; Grosse, R. Positive Feedback between Dia1, LARG, and RhoA Regulates Cell Morphology and Invasion. Genes Dev. 2007, 21, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Kitzing, T.M.; Wang, Y.; Pertz, O.; Copeland, J.W.; Grosse, R. Formin-like 2 Drives Amoeboid Invasive Cell Motility Downstream of RhoC. Oncogene 2010, 29, 2441–2448. [Google Scholar] [CrossRef] [Green Version]
- Lizárraga, F.; Poincloux, R.; Romao, M.; Montagnac, G.; Le Dez, G.; Bonne, I.; Rigaill, G.; Raposo, G.; Chavrier, P. Diaphanous-Related Formins Are Required for Invadopodia Formation and Invasion of Breast Tumor Cells. Cancer Res. 2009, 69, 2792–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Jung, J.; You, E.; Ko, P.; Oh, S.; Rhee, S. MDia1 Regulates Breast Cancer Invasion by Controlling Membrane Type 1-Matrix Metalloproteinase Localization. Oncotarget 2016, 7, 17829–17843. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J. Diaphanous-Related Formin-3 Overexpression Inhibits the Migration and Invasion of Triple-Negative Breast Cancer by Inhibiting RhoA-GTP Expression. Biomed. Pharmacother. 2017, 94, 439–445. [Google Scholar] [CrossRef]
- Reis-Sobreiro, M.; Chen, J.-F.; Novitskaya, T.; You, S.; Morley, S.; Steadman, K.; Gill, N.K.; Eskaros, A.; Rotinen, M.; Chu, C.-Y.; et al. Emerin Deregulation Links Nuclear Shape Instability to Metastatic Potential. Cancer Res. 2018, 78, 6086–6097. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Liu, Y.; Yu, X.; Zhu, Y.; Zhu, Y. Formin Homology Domains of Daam1 Bind to Fascin and Collaboratively Promote Pseudopodia Formation and Cell Migration in Breast Cancer. Cell Prolif. 2021, 54, e12994. [Google Scholar] [CrossRef]
- Mei, J.; Xu, B.; Hao, L.; Xiao, Z.; Liu, Y.; Yan, T.; Zhu, Y. Overexpressed DAAM1 Correlates with Metastasis and Predicts Poor Prognosis in Breast Cancer. Pathol. Res. Pract. 2020, 216, 152736. [Google Scholar] [CrossRef]
- Yan, T.; Zhang, A.; Shi, F.; Chang, F.; Mei, J.; Liu, Y.; Zhu, Y. Integrin Avβ3-Associated DAAM1 Is Essential for Collagen-Induced Invadopodia Extension and Cell Haptotaxis in Breast Cancer Cells. J. Biol. Chem. 2018, 293, 10172–10185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Tian, Y.; Du, J.; Hu, Z.; Yang, L.; Liu, J.; Gu, L. Dvl2-Dependent Activation of Daam1 and RhoA Regulates Wnt5a-Induced Breast Cancer Cell Migration. PLoS ONE 2012, 7, e37823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, V.D.; Mansuri, N.; Mogg, J.; Kurki, S.; Repo, H.; Kronqvist, P.; Carpén, O.; Gardberg, M. Formin Proteins FHOD1 and INF2 in Triple-Negative Breast Cancer: Association With Basal Markers and Functional Activities. Breast Cancer 2018, 12, 1178223418792247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Shen, Y.; Yan, T.; Zhang, A. Wnt5a/ROR1 Activates DAAM1 and Promotes the Migration in Osteosarcoma Cells. Oncol. Rep. 2020, 43, 601–608. [Google Scholar] [CrossRef]
- Wang, Q. Identification of Biomarkers for Metastatic Osteosarcoma Based on DNA Microarray Data. Neoplasma 2015, 62, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Xing, Y.; Lv, J.; Gu, D.; Pan, J.; Zhang, Y.; Liu, J. Construction of an Immune-Related Gene Signature for Prediction of Prognosis in Patients with Cervical Cancer. Int. Immunopharmacol. 2020, 88, 106882. [Google Scholar] [CrossRef]
- Lin, Y.-N.; Izbicki, J.R.; König, A.; Habermann, J.K.; Blechner, C.; Lange, T.; Schumacher, U.; Windhorst, S. Expression of DIAPH1 Is Up-Regulated in Colorectal Cancer and Its down-Regulation Strongly Reduces the Metastatic Capacity of Colon Carcinoma Cells. Int. J. Cancer 2014, 134, 1571–1582. [Google Scholar] [CrossRef]
- Miao, S.; Schäfer, P.; Nojszewski, J.; Meyer, F.; Windhorst, S. DIAPH1 Regulates Chromosomal Instability of Cancer Cells by Controlling Microtubule Dynamics. Eur. J. Cell Biol. 2021, 100, 151156. [Google Scholar] [CrossRef] [PubMed]
- Sho, S.; Court, C.M.; Winograd, P.; Russell, M.M.; Tomlinson, J.S. A Prognostic Mutation Panel for Predicting Cancer Recurrence in Stages II and III Colorectal Cancer. J. Surg. Oncol. 2017, 116, 996–1004. [Google Scholar] [CrossRef]
- Grueb, S.S.; Muhs, S.; Popp, Y.; Schmitt, S.; Geyer, M.; Lin, Y.-N.; Windhorst, S. The Formin Drosophila Homologue of Diaphanous2 (Diaph2) Controls Microtubule Dynamics in Colorectal Cancer Cells Independent of Its FH2-Domain. Sci. Rep. 2019, 9, 5352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, J.; Liu, Z.; Liu, X.; Ma, Y.; Zhang, B.; Chen, Y.; Li, X.; Feng, Z.; Yang, N.; et al. Identification of Hub Genes in Colorectal Cancer Based on Weighted Gene Co-Expression Network Analysis and Clinical Data from The Cancer Genome Atlas. Biosci. Rep. 2021, 41, BSR20211280. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Albers, C.G.; Holcombe, R.F. Differentiation of Tubular and Villous Adenomas Based on Wnt Pathway-Related Gene Expression Profiles. Int. J. Mol. Med. 2010, 26, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Liu, Z.; Wu, Q.; Li, H. Disheveled-Associated Activator of Morphogenesis 2 Promotes Invasion of Colorectal Cancer by Activating PAK1 and Promoting MMP7 Expression. Genes Genom. 2021, 43, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, X.; Zeng, Y.; Wang, J.; Zhang, X.; Ding, Y.; Liang, L. FMNL2 Enhances Invasion of Colorectal Carcinoma by Inducing Epithelial-Mesenchymal Transition. Mol. Cancer Res. 2010, 8, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.L.; Qiao, Y.D.; Li, J.Y.; Li, X.M.; Zhang, D.; Zhang, X.J.; Zhu, X.H.; Zhou, W.J.; Shi, J.; Wang, W.; et al. Cortactin Recruits FMNL2 to Promote Actin Polymerization and Endosome Motility in Invadopodia Formation. Cancer Lett. 2018, 419, 245–256. [Google Scholar] [CrossRef]
- Yang, S.S.; Li, X.M.; Yang, M.; Ren, X.L.; Hu, J.L.; Zhu, X.H.; Wang, F.F.; Zeng, Z.C.; Li, J.Y.; Cheng, Z.Q.; et al. FMNL2 Destabilises COMMD10 to Activate NF-ΚB Pathway in Invasion and Metastasis of Colorectal Cancer. Br. J. Cancer 2017, 117, 1164–1175. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Zeng, Y.-F.; Guan, J.; Li, Y.-F.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q.; Liang, L. FMNL2 Is a Positive Regulator of Cell Motility and Metastasis in Colorectal Carcinoma. J. Pathol. 2011, 224, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-L.; Liang, L.; Ding, Y.-Q. Overexpression of FMNL2 Is Closely Related to Metastasis of Colorectal Cancer. Int. J. Colorectal Dis. 2008, 23, 1041–1047. [Google Scholar] [CrossRef]
- Zeng, Y.-F.; Xiao, Y.-S.; Liu, Y.; Luo, X.-J.; Wen, L.-D.; Liu, Q.; Chen, M. Formin-like 3 Regulates RhoC/FAK Pathway and Actin Assembly to Promote Cell Invasion in Colorectal Carcinoma. World J. Gastroenterol. 2018, 24, 3884–3897. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.-F.; Xiao, Y.-S.; Lu, M.-Z.; Luo, X.-J.; Hu, G.-Z.; Deng, K.-Y.; Wu, X.-M.; Xin, H.-B. Increased Expression of Formin-like 3 Contributes to Metastasis and Poor Prognosis in Colorectal Carcinoma. Exp. Mol. Pathol. 2015, 98, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-J.; Feng, Z.-C.; Li, X.-R.; Hu, G. Involvement of Methylation-Associated Silencing of Formin 2 in Colorectal Carcinogenesis. World J. Gastroenterol. 2018, 24, 5013–5024. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, H.; Jiang, D.; Li, Y.; Feng, L.; Tian, C.; Pu, M.; Wang, X.; Zhang, J.; Hu, Y.; et al. Multi Gene Mutation Signatures in Colorectal Cancer Patients: Predict for the Diagnosis, Pathological Classification, Staging and Prognosis. BMC Cancer 2021, 21, 380. [Google Scholar] [CrossRef]
- Kawamata, H.; Furihata, T.; Omotehara, F.; Sakai, T.; Horiuchi, H.; Shinagawa, Y.; Imura, J.; Ohkura, Y.; Tachibana, M.; Kubota, K.; et al. Identification of Genes Differentially Expressed in a Newly Isolated Human Metastasizing Esophageal Cancer Cell Line, T.Tn-AT1, by CDNA Microarray. Cancer Sci. 2003, 94, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Yan, T.; Hao, L.; Zhu, Y. Wnt5a Induces ROR1 and ROR2 to Activate RhoA in Esophageal Squamous Cell Carcinoma Cells. Cancer Manag. Res. 2019, 11, 2803–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Zhang, Y.; Mao, X.; Cheng, Z.; Wu, S.; Wu, S.; Zhou, L. Expression of Formin-like 2 and Cortactin in Gallbladder Adenocarcinoma and Their Clinical Significance. Int. J. Clin. Exp. Pathol. 2020, 13, 1655–1661. [Google Scholar] [PubMed]
- Yang, J.; Zhou, L.; Zhang, Y.; Zheng, J.; Zhou, J.; Wei, Z.; Zou, J. DIAPH1 Is Upregulated and Inhibits Cell Apoptosis through ATR/P53/Caspase-3 Signaling Pathway in Laryngeal Squamous Cell Carcinoma. Dis. Markers 2019, 2019, 6716472. Available online: https://www.hindawi.com/journals/dm/2019/6716472 (accessed on 30 August 2021). [CrossRef] [PubMed] [Green Version]
- Kostrzewska-Poczekaj, M.; Byzia, E.; Soloch, N.; Jarmuz-Szymczak, M.; Janiszewska, J.; Kowal, E.; Paczkowska, J.; Kiwerska, K.; Wierzbicka, M.; Bartochowska, A.; et al. DIAPH2 Alterations Increase Cellular Motility and May Contribute to the Metastatic Potential of Laryngeal Squamous Cell Carcinoma. Carcinogenesis 2019, 40, 1251–1259. [Google Scholar] [CrossRef]
- Śnit, M.; Misiołek, M.; Ścierski, W.; Koniewska, A.; Stryjewska-Makuch, G.; Okła, S.; Grzeszczak, W. DIAPH2, PTPRD and HIC1 Gene Polymorphisms and Laryngeal Cancer Risk. Int. J. Environ. Res. Public Health 2021, 18, 7486. [Google Scholar] [CrossRef]
- Chen, W.-H.; Cai, M.-Y.; Zhang, J.-X.; Wang, F.-W.; Tang, L.-Q.; Liao, Y.-J.; Jin, X.-H.; Wang, C.-Y.; Guo, L.; Jiang, Y.-G.; et al. FMNL1 Mediates Nasopharyngeal Carcinoma Cell Aggressiveness by Epigenetically Upregulating MTA1. Oncogene 2018, 37, 6243–6258. [Google Scholar] [CrossRef]
- Liu, J.; Chen, S.; Chen, Y.; Geng, N.; Feng, C. High Expression of FMNL3 Associates with Cancer Cell Migration, Invasion, and Unfavorable Prognosis in Tongue Squamous Cell Carcinoma. J. Oral Pathol. Med. 2019, 48, 459–467. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, Z.; Wang, K.; Ha, Y.; Lei, H.; Jia, Y.; Ding, R.; Wu, D.; Gan, S.; Li, R.; et al. High FMNL3 Expression Promotes Nasopharyngeal Carcinoma Cell Metastasis: Role in TGF-Β1-Induced Epithelia-to-Mesenchymal Transition. Sci. Rep. 2017, 7, 42507. [Google Scholar] [CrossRef] [Green Version]
- Gardberg, M.; Kaipio, K.; Lehtinen, L.; Mikkonen, P.; Heuser, V.D.; Talvinen, K.; Iljin, K.; Kampf, C.; Uhlen, M.; Grénman, R.; et al. FHOD1, a Formin Upregulated in Epithelial-Mesenchymal Transition, Participates in Cancer Cell Migration and Invasion. PLoS ONE 2013, 8, e74923. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.-F.; Li, Q.-L.; Yang, Y.-F.; Cao, Y.; Zhang, C.Z. FMNL1 Exhibits Pro-Metastatic Activity via CXCR2 in Clear Cell Renal Cell Carcinoma. Front. Oncol. 2020, 10, 564614. [Google Scholar] [CrossRef]
- Hirata, H.; Hinoda, Y.; Nakajima, K.; Kikuno, N.; Yamamura, S.; Kawakami, K.; Suehiro, Y.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Wnt Antagonist Gene Polymorphisms and Renal Cancer. Cancer 2009, 115, 4488–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.B.; Wigton, E.J.; Krovi, S.H.; Chung, J.W.; Long, R.A.; Jacobelli, J. The Formin mDia1 Regulates Acute Lymphoblastic Leukemia Engraftment, Migration, and Progression in Vivo. Front. Oncol. 2018, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Favaro, P.; Traina, F.; Machado-Neto, J.A.; Lazarini, M.; Lopes, M.R.; Pereira, J.K.N.; Costa, F.F.; Infante, E.; Ridley, A.J.; Saad, S.T.O. FMNL1 Promotes Proliferation and Migration of Leukemia Cells. J. Leukoc. Biol. 2013, 94, 503–512. [Google Scholar] [CrossRef]
- Bergamo Favaro, P.; de Souza Medina, S.; Traina, F.; Sanchez Bassères, D.; Ferreira Costa, F.; Olalla Saad, S.T. Human Leukocyte Formin: A Novel Protein Expressed in Lymphoid Malignancies and Associated with Akt. Available online: https://pubmed.ncbi.nlm.nih.gov/14592423/ (accessed on 26 January 2021).
- French, D.; Yang, W.; Cheng, C.; Raimondi, S.C.; Mullighan, C.G.; Downing, J.R.; Evans, W.E.; Pui, C.-H.; Relling, M.V. Acquired Variation Outweighs Inherited Variation in whole Genome Analysis of Methotrexate Polyglutamate Accumulation in Leukemia. Blood 2009, 113, 4512–4520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, P.M.B.; Traina, F.; Vassallo, J.; Brousset, P.; Delsol, G.; Costa, F.F.; Saad, S.T.O. High Expression of FMNL1 Protein in T Non-Hodgkin’s Lymphomas. Leuk. Res. 2006, 30, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Z.; Xue, L.; Li, G.; Zhang, C.; Cai, Z.; Li, H.; Guo, R. DIAPH3 Promoted the Growth, Migration and Metastasis of Hepatocellular Carcinoma Cells by Activating Beta-Catenin/TCF Signaling. Mol. Cell. Biochem. 2018, 438, 183–190. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, D.; Zhao, W.; Gao, L.; Wang, L. Dishevelled Associated Activator Of Morphogenesis (DAAM) Facilitates Invasion of Hepatocellular Carcinoma by Upregulating Hypoxia-Inducible Factor 1α (HIF-1α) Expression. Med. Sci. Monit. 2020, 26, e924670. [Google Scholar] [CrossRef]
- Liang, L.; Guan, J.; Zeng, Y.; Wang, J.; Li, X.; Zhang, X.; Ding, Y. Down-Regulation of Formin-like 2 Predicts Poor Prognosis in Hepatocellular Carcinoma. Hum. Pathol. 2011, 42, 1603–1612. [Google Scholar] [CrossRef]
- Xiang, G.; Weiwei, H.; Erji, G.; Haitao, M. DIAPH3 Promotes the Tumorigenesis of Lung Adenocarcinoma. Exp. Cell Res. 2019, 385, 111662. [Google Scholar] [CrossRef]
- Liu, Y.; Lusk, C.M.; Cho, M.H.; Silverman, E.K.; Qiao, D.; Zhang, R.; Scheurer, M.E.; Kheradmand, F.; Wheeler, D.A.; Tsavachidis, S.; et al. Rare Variants in Known Susceptibility Loci and Their Contribution to Risk of Lung Cancer. J. Thorac. Oncol. 2018, 13, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-Y.; Liao, J.-J.; Xue, W.-R. FMNL1 Down-Regulation Suppresses Bone Metastasis through Reducing TGF-Β1 Expression in Non-Small Cell Lung Cancer (NSCLC). Biomed. Pharmacother. 2019, 117, 109126. [Google Scholar] [CrossRef]
- Hałas-Wiśniewska, M.; Izdebska, M.; Zielińska, W.; Grzanka, A. Downregulation of FHOD1 Inhibits Metastatic Potential in A549 Cells. Cancer Manag. Res. 2021, 13, 91–106. [Google Scholar] [CrossRef]
- Schiewek, J.; Schumacher, U.; Lange, T.; Joosse, S.A.; Wikman, H.; Pantel, K.; Mikhaylova, M.; Kneussel, M.; Linder, S.; Schmalfeldt, B.; et al. Clinical Relevance of Cytoskeleton Associated Proteins for Ovarian Cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 2195–2205. [Google Scholar] [CrossRef]
- Pettee, K.M.; Dvorak, K.M.; Nestor-Kalinoski, A.L.; Eisenmann, K.M. An MDia2/ROCK Signaling Axis Regulates Invasive Egress from Epithelial Ovarian Cancer Spheroids. PLoS ONE 2014, 9, e90371. [Google Scholar] [CrossRef] [Green Version]
- Paul, N.R.; Allen, J.L.; Chapman, A.; Morlan-Mairal, M.; Zindy, E.; Jacquemet, G.; Fernandez del Ama, L.; Ferizovic, N.; Green, D.M.; Howe, J.D.; et al. A5β1 Integrin Recycling Promotes Arp2/3-Independent Cancer Cell Invasion via the Formin FHOD3. J. Cell Biol. 2015, 210, 1013–1031. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Gao, J.; Kuang, T.; Chen, J.; Li, J.-A.; Huang, Y.; Xin, H.; Fang, Y.; Han, X.; Sun, L.-Q.; et al. DIAPH3 Promotes Pancreatic Cancer Progression by Activating Selenoprotein TrxR1-Mediated Antioxidant Effects. J. Cell. Mol. Med. 2020, 25, 2163–2175. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Liu, H.; Luo, S.; Moorman, P.G.; Walsh, K.M.; Li, W.; Wei, Q. Associations between Genetic Variants of KIF5B, FMN1, and MGAT3 in the Cadherin Pathway and Pancreatic Cancer Risk. Cancer Med. 2020, 9, 9620–9631. [Google Scholar] [CrossRef]
- Raja, A.; Malik, M.F.A.; Haq, F. Genomic Relevance of FGF14 and Associated Genes on the Prognosis of Pancreatic Cancer. PLoS ONE 2021, 16, e0252344. [Google Scholar] [CrossRef]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome Formation in Prostate Cancer: Association with a Region of Frequent Chromosomal Deletion in Metastatic Disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef] [Green Version]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. RhoA and RhoC Have Distinct Roles in Migration and Invasion by Acting through Different Targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Lisitskaia, K.V.; Krakhmaleva, I.N.; Shishkin, S.S. A study of the single nucleotide polymorphism in seven genes (GHR, IGFBP3, IGFR1, IRS1, FMN1, ANXA2, TaGLN) in ethnic Russians and in patients with prostate cancer. Mol. Gen. Mikrobiol. Virusol. 2010, 2, 34–37. [Google Scholar] [CrossRef]
- Jin, X.; Wang, J.; Gao, K.; Zhang, P.; Yao, L.; Tang, Y.; Tang, L.; Ma, J.; Xiao, J.; Zhang, E.; et al. Dysregulation of INF2-Mediated Mitochondrial Fission in SPOP-Mutated Prostate Cancer. PLoS Genet. 2017, 13, e1006748. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Hernandez, I.; Maiques, O.; Kohlhammer, L.; Cantelli, G.; Perdrix-Rosell, A.; Monger, J.; Fanshawe, B.; Bridgeman, V.L.; Karagiannis, S.N.; Penin, R.M.; et al. WNT11-FZD7-DAAM1 Signalling Supports Tumour Initiating Abilities and Melanoma Amoeboid Invasion. Nat. Commun. 2020, 11, 5315. [Google Scholar] [CrossRef]
- Gardberg, M.; Heuser, V.D.; Koskivuo, I.; Koivisto, M.; Carpén, O. FMNL2/FMNL3 Formins Are Linked with Oncogenic Pathways and Predict Melanoma Outcome. J. Pathol. Clin. Res. 2016, 2, 41–52. [Google Scholar] [CrossRef]
- Péladeau, C.; Heibein, A.; Maltez, M.T.; Copeland, S.J.; Copeland, J.W. A Specific FMNL2 Isoform Is Up-Regulated in Invasive Cells. BMC Cell Biol. 2016, 17, 32. [Google Scholar] [CrossRef] [Green Version]
- Koka, S.; Neudauer, C.L.; Li, X.; Lewis, R.E.; McCarthy, J.B.; Westendorf, J.J. The Formin-Homology-Domain-Containing Protein FHOD1 Enhances Cell Migration. J. Cell Sci. 2003, 116, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Peippo, M.; Gardberg, M.; Lamminen, T.; Kaipio, K.; Carpén, O.; Heuser, V.D. FHOD1 Formin Is Upregulated in Melanomas and Modifies Proliferation and Tumor Growth. Exp. Cell Res. 2017, 350, 267–278. [Google Scholar] [CrossRef]
- Nie, H.; Mei, J.; Zhang, Q.; An, F.; Zhan, Q. Systematic Characterization of the Expression and Prognostic Values of Formin-Like Gene Family in Gastric Cancer. DNA Cell Biol. 2020, 39, 1664–1677. [Google Scholar] [CrossRef]
- Zhong, B.; Wang, K.; Xu, H.; Kong, F. Silencing Formin-like 2 Inhibits Growth and Metastasis of Gastric Cancer Cells through Suppressing Internalization of Integrins. Cancer Cell Int. 2018, 18, 79. [Google Scholar] [CrossRef]
- Mansuri, N.; Heuser, V.D.; Birkman, E.-M.; Lintunen, M.; Ålgars, A.; Sundström, J.; Ristamäki, R.; Carpén, O.; Lehtinen, L. FHOD1 and FMNL1 Formin Proteins in Intestinal Gastric Cancer: Correlation with Tumor-Infiltrating T Lymphocytes and Molecular Subtypes. Gastric Cancer 2021, 1–10. [Google Scholar] [CrossRef]
- Liang, L.; Ye, L.; Cui, D.; Yang, D. Gene Expression Signature Associated with Metastasis of Stomach Adenocarcinoma. Int. J. Clin. Exp. Med. 2017, 10, 3016–3026. [Google Scholar]
- Chai, L.; Li, J.; Lv, Z. An Integrated Analysis of Cancer Genes in Thyroid Cancer. Oncol. Rep. 2016, 35, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Marshall, C.J. The Plasticity of Cytoskeletal Dynamics Underlying Neoplastic Cell Migration. Curr. Opin. Cell Biol. 2010, 22, 690–696. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions That Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. Compact Spheroid Formation by Ovarian Cancer Cells Is Associated with Contractile Behavior and an Invasive Phenotype. Int. J. Cancer 2009, 124, 2060–2070. [Google Scholar] [CrossRef]
- Creekmore, A.L.; Silkworth, W.T.; Cimini, D.; Jensen, R.V.; Roberts, P.C.; Schmelz, E.M. Changes in Gene Expression and Cellular Architecture in an Ovarian Cancer Progression Model. PLoS ONE 2011, 6, e17676. [Google Scholar] [CrossRef]
- Ketene, A.N.; Schmelz, E.M.; Roberts, P.C.; Agah, M. The Effects of Cancer Progression on the Viscoelasticity of Ovarian Cell Cytoskeleton Structures. Nanomedicine 2012, 8, 391. [Google Scholar] [CrossRef]
- Wyse, M.M.; Lei, J.; Nestor-Kalinoski, A.L.; Eisenmann, K.M. Dia-Interacting Protein (DIP) Imposes Migratory Plasticity in MDia2-Dependent Tumor Cells in Three-Dimensional Matrices. PLoS ONE 2012, 7, e45085. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Ji, W.-K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-Mediated Actin Polymerization at the ER Stimulates Mitochondrial Calcium Uptake, Inner Membrane Constriction, and Division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.; Burleson, M. SPOP and Cancer: A Systematic Review. Am. J. Cancer Res. 2020, 10, 704–726. [Google Scholar]
- Da Silva, A.F.; Mariotti, F.R.; Máximo, V.; Campello, S. Mitochondria Dynamism: Of Shape, Transport and Cell Migration. Cell. Mol. Life Sci. 2014, 71, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, M.; An, D.; Yuan, H.; Li, Z.; Song, Y.; Liu, Z. Suppression of Tafazzin Promotes Thyroid Cancer Apoptosis via Activating the JNK Signaling Pathway and Enhancing INF2-Mediated Mitochondrial Fission. J. Cell. Physiol. 2019, 234, 16238–16251. [Google Scholar] [CrossRef]
- Yasuda, S.; Oceguera-Yanez, F.; Kato, T.; Okamoto, M.; Yonemura, S.; Terada, Y.; Ishizaki, T.; Narumiya, S. Cdc42 and mDia3 Regulate Microtubule Attachment to Kinetochores. Nature 2004, 428, 767–771. [Google Scholar] [CrossRef]
- Morley, S.; You, S.; Pollan, S.; Choi, J.; Zhou, B.; Hager, M.H.; Steadman, K.; Spinelli, C.; Rajendran, K.; Gertych, A.; et al. Regulation of Microtubule Dynamics by DIAPH3 Influences Amoeboid Tumor Cell Mechanics and Sensitivity to Taxanes. Sci. Rep. 2015, 5, 12136. [Google Scholar] [CrossRef] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, K.M.; Pettee, K.M.; Rubinic-Minotti, K.; Su, R.; Nestor-Kalinoski, A.; Eisenmann, K.M. Carcinoma Associated Fibroblasts (CAFs) Promote Breast Cancer Motility by Suppressing Mammalian Diaphanous-Related Formin-2 (mDia2). PLoS ONE 2018, 13, e0195278. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-Dependent Matrix Remodelling Is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically Engineered Mouse Models in Oncology Research and Cancer Medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Antsiferova, M.; Huber, M.; Meyer, M.; Piwko-Czuchra, A.; Ramadan, T.; MacLeod, A.S.; Havran, W.L.; Dummer, R.; Hohl, D.; Werner, S. Activin Enhances Skin Tumourigenesis and Malignant Progression by Inducing a Pro-Tumourigenic Immune Cell Response. Nat. Commun. 2011, 2, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cangkrama, M.; Wietecha, M.; Mathis, N.; Okumura, R.; Ferrarese, L.; Al-Nuaimi, D.; Antsiferova, M.; Dummer, R.; Innocenti, M.; Werner, S. A Paracrine Activin A-mDia2 Axis Promotes Squamous Carcinogenesis via Fibroblast Reprogramming. EMBO Mol. Med. 2020, 12, e11466. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a Guardian of the Cancer Cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Kim, J.; Morley, S.; Le, M.; Bedoret, D.; Umetsu, D.T.; Di Vizio, D.; Freeman, M.R. Enhanced Shedding of Extracellular Vesicles from Amoeboid Prostate Cancer Cells: Potential Effects on the Tumor Microenvironment. Cancer Biol. Ther. 2014, 15, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.A.; Neidt, E.M.; Cui, J.; Feiger, Z.; Skau, C.T.; Gardel, M.L.; Kozmin, S.A.; Kovar, D.R. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem. Biol. 2009, 16, 1158–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziske, M.A.; Pettee, K.M.; Khaing, M.; Rubinic, K.; Eisenmann, K.M. SMIFH2-Mediated mDia Formin Functional Inhibition Potentiates Chemotherapeutic Targeting of Human Ovarian Cancer Spheroids. Biochem. Biophys. Res. Commun. 2016, 472, 33–39. [Google Scholar] [CrossRef]
- Isogai, T.; van der Kammen, R.; Innocenti, M. SMIFH2 has Effects on Formins and p53 that Perturb the Cell Cytoskeleton. Sci. Rep. 2015, 5, 9802. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Shi, S.; Zhang, F.; Liu, R.; Takagi, Y.; Bershadsky, A.D.; Viasnoff, V.; Sellers, J.R. The Formin Inhibitor SMIFH2 Inhibits Members of the Myosin Superfamily. J. Cell Sci. 2021, 134, jcs253708. [Google Scholar] [CrossRef]
- Monroe, J.D.; Moolani, S.A.; Irihamye, E.N.; Speed, J.S.; Gibert, Y.; Smith, M.E. RNA-Seq Analysis of Cisplatin and the Monofunctional Platinum(II) Complex, Phenanthriplatin, in A549 Non-Small Cell Lung Cancer and IMR90 Lung Fibroblast Cell Lines. Cells 2020, 9, 2637. [Google Scholar] [CrossRef] [PubMed]
- Fabre, I.; Fabre, G.; Goldman, I.D. Polyglutamylation, an Important Element in Methotrexate Cytotoxicity and Selectivity in Tumor versus Murine Granulocytic Progenitor Cells in Vitro. Cancer Res. 1984, 44, 3190–3195. [Google Scholar] [PubMed]
- Schuster, I.G.; Busch, D.H.; Eppinger, E.; Kremmer, E.; Milosevic, S.; Hennard, C.; Kuttler, C.; Ellwart, J.W.; Frankenberger, B.; Nössner, E.; et al. Allorestricted T Cells with Specificity for the FMNL1-Derived Peptide PP2 Have Potent Antitumor Activity against Hematologic and Other Malignancies. Blood 2007, 110, 2931–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Fang, D.; Lu, H.; Cao, Y.; Zhang, J.; Ding, R.; Li, L.; Huo, J. Tanshinone IIA Promotes IL2-Mediated SW480 Colorectal Cancer Cell Apoptosis by Triggering INF2-Related Mitochondrial Fission and Activating the Mst1-Hippo Pathway. Biomed. Pharmacother. 2018, 108, 1658–1669. [Google Scholar] [CrossRef]
- Fernandez-Barrera, J.; Bernabe-Rubio, M.; Casares-Arias, J.; Rangel, L.; Fernandez-Martin, L.; Correas, I.; Alonso, M.A. The Actin-MRTF-SRF Transcriptional Circuit Controls Tubulin Acetylation via α-TAT1 Gene Expression. J. Cell Biol. 2018, 217, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N.; Nordheim, A. Linking Actin Dynamics and Gene Transcription to Drive Cellular Motile Functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, S.F.; Kulacz, W.A.; Shaikh, S.; Lee, J.M.; Copeland, J.W. The Ability to Induce Microtubule Acetylation Is a General Feature of Formin Proteins. PLoS ONE 2012, 7, e48041. [Google Scholar] [CrossRef]
- Gualdrini, F.; Esnault, C.; Horswell, S.; Stewart, A.; Matthews, N.; Treisman, R. SRF Co-Factors Control the Balance between Cell Proliferation and Contractility. Mol. Cell 2016, 64, 1048–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-Actin Signaling to the MRTF Coactivators Dominates the Immediate Transcriptional Response to Serum in Fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-C.; Jo, Y.-J.; Kim, N.-H.; Namgoong, S. Small Molecule Inhibitor of Formin Homology 2 Domains (SMIFH2) Reveals the Roles of the Formin Family of Proteins in Spindle Assembly and Asymmetric Division in Mouse Oocytes. PLoS ONE 2015, 10, e0123438. [Google Scholar] [CrossRef] [Green Version]
- Ghafouri-Fard, S.; Shoorei, H.; Anamag, F.T.; Taheri, M. The Role of Non-Coding RNAs in Controlling Cell Cycle Related Proteins in Cancer Cells. Front. Oncol. 2020, 10, 608975. [Google Scholar] [CrossRef]
- Lin, C.-P.; He, L. Noncoding RNAs in Cancer Development. Annu. Rev. Cancer Biol. 2017, 1, 163–184. [Google Scholar] [CrossRef]
Figure 2.
Pathogenic mutations of the formins causing monogenic disorders. Depending on the specific mutation, some formins produce different disorders. In these cases, we used the colors, as indicated, to refer to each of the diseases and the corresponding mutations. *, stop codon. The mutations without reported familial studies are indicated in italics.
Figure 2.
Pathogenic mutations of the formins causing monogenic disorders. Depending on the specific mutation, some formins produce different disorders. In these cases, we used the colors, as indicated, to refer to each of the diseases and the corresponding mutations. *, stop codon. The mutations without reported familial studies are indicated in italics.

Figure 3.
Some of the organs and cell types affected by formin alterations. (A) The formins involved in human disorders and the affected organs and systems are indicated in the schematic of the human body. Those causing monogenic disorders are highlighted in bold. (B) Some of the affected cell types. Their most characteristic structures are indicated.
Figure 3.
Some of the organs and cell types affected by formin alterations. (A) The formins involved in human disorders and the affected organs and systems are indicated in the schematic of the human body. Those causing monogenic disorders are highlighted in bold. (B) Some of the affected cell types. Their most characteristic structures are indicated.

Figure 4.
Schematic of a migrating cancer cell. The processes and cell functions that can be altered by aberrant expression of formins in cancer cells are indicated. Cells, such as cancer-associated fibroblasts and immune system cells, in the surroundings of the cancerous cell influence cancer progression.
Figure 4.
Schematic of a migrating cancer cell. The processes and cell functions that can be altered by aberrant expression of formins in cancer cells are indicated. Cells, such as cancer-associated fibroblasts and immune system cells, in the surroundings of the cancerous cell influence cancer progression.


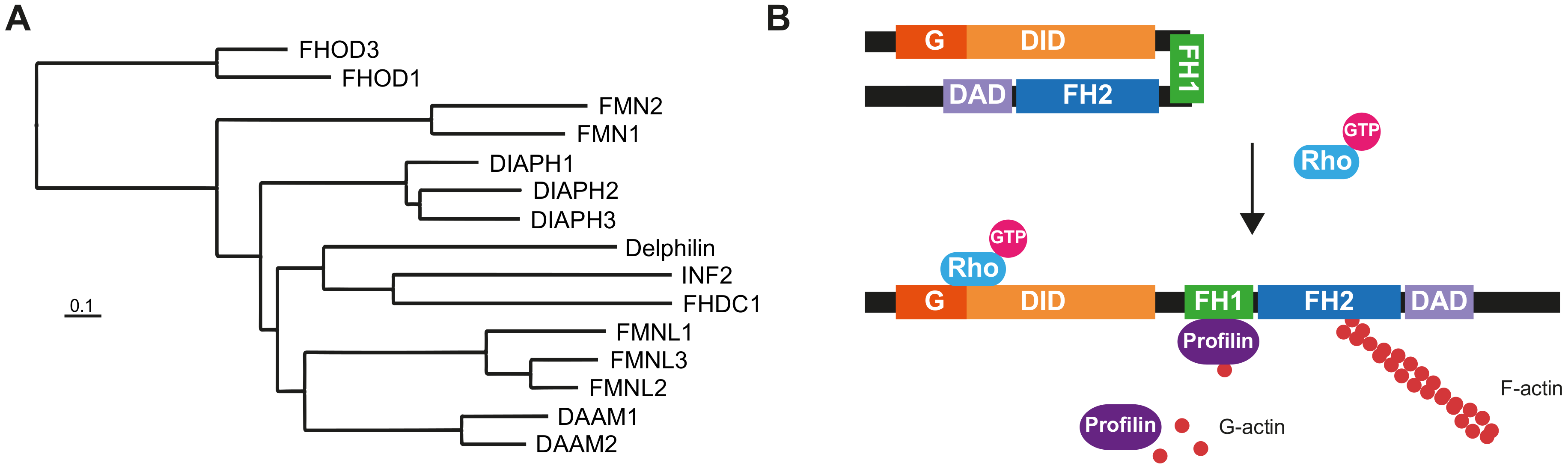{kind=link}
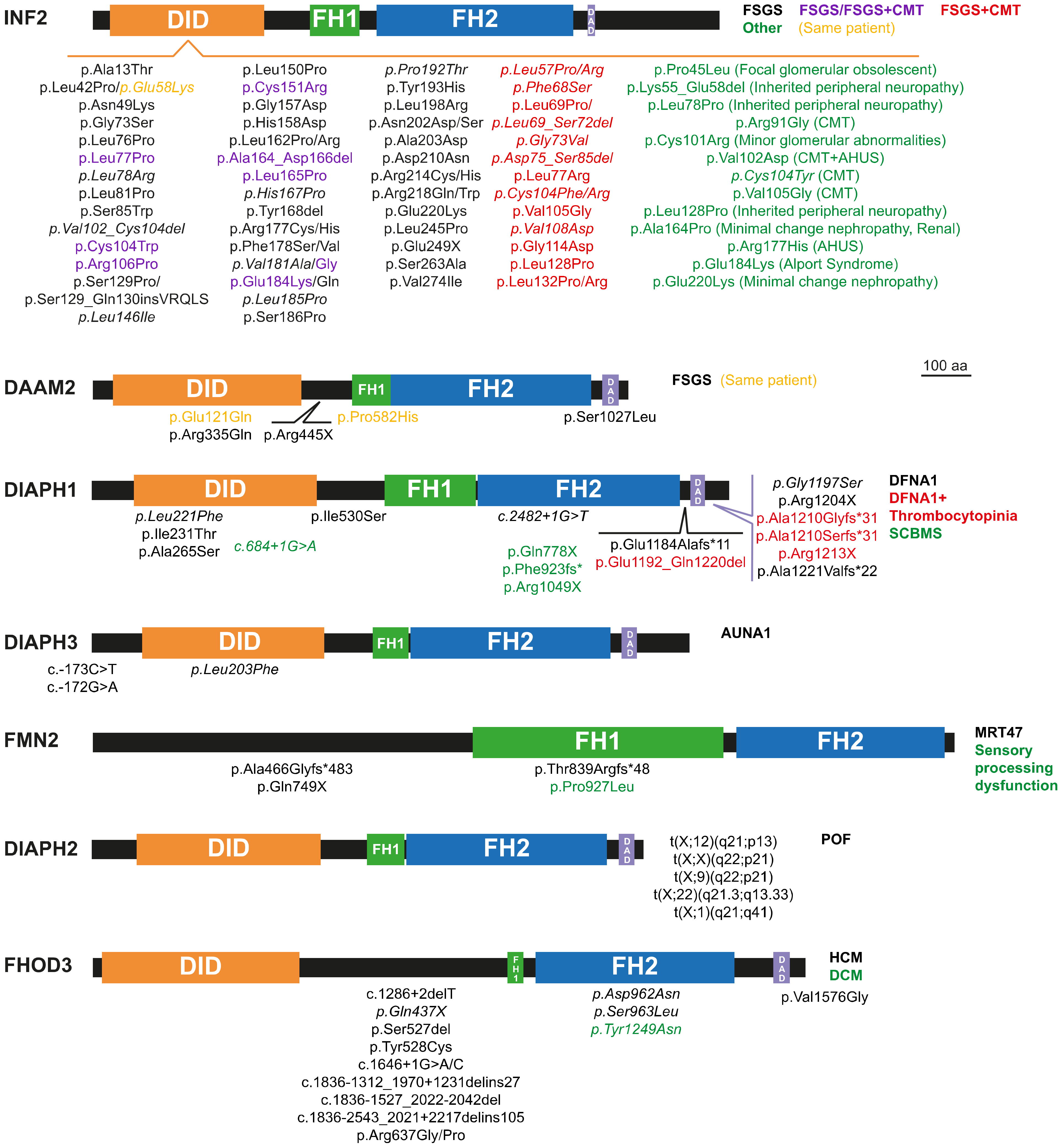{kind=link}
{kind=link}
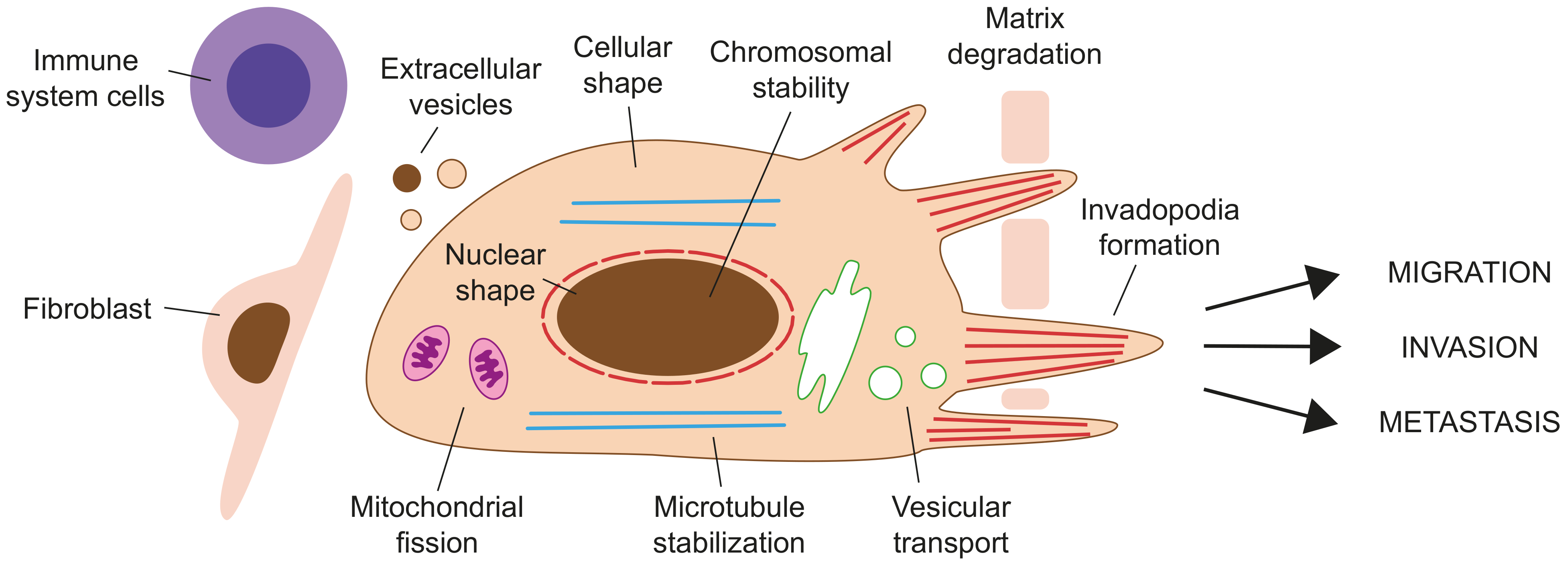{kind=link}
Table 1.
Summary of formin expression in different types of cancer. The in vitro and in vivo experiments providing evidence of the involvement of formins in cancers and the use of formin expression as prognostic biomarkers are also summarized.
Table 1.
Summary of formin expression in different types of cancer. The in vitro and in vivo experiments providing evidence of the involvement of formins in cancers and the use of formin expression as prognostic biomarkers are also summarized.
| Cancer | Formin | Expression Levels (Technique) or Mutations | In Vitro | In Vivo (Nude Mice) | Prognosis | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Matrix Degradation | Invadopodia Formation | Migration | Invasion | Metastasis | Tumor size/Weight | |||||
| Brain | DIAPH1 | ↑ (IHC/T) | ↑ | ↑ | ↑ | ↑ | ↑ | [228,229,230] | ||
| DIAPH1 | ↑ | [231] | ||||||||
| DIAPH3 | [232] | |||||||||
| DAAM1 | ↑ | [233] | ||||||||
| DAAM1 | ↑ (Mass spectrometry) | [234] | ||||||||
| DAAM2 | ↑ (ISH/T) | ↑ | [235] | |||||||
| FMNL1 | ↑ (IHC/T) | ↑ | ↑ | ↑ | Poor | [236] | ||||
| FMNL2 FMN2 | Mutants | [237] | ||||||||
| FHOD3 | ↑ (WB) | ↑ | [238] | |||||||
| FHOD3 | ↑ (IHC/T) | ↑ | ↑ | Poor | [239] | |||||
| FHOD1 INF2 | ↑ (IHC/T) | ↑ | Not clear | [240] | ||||||
| Breast | All | ↑ or ↓ depending on the specific formin | [241,242] | |||||||
| DIAPH1/2/3 | ↑ | ↑ | ↑ | [243] | ||||||
| DIAPH1 | ↑ (T/WB) | ↑ | ↑ | [244] | ||||||
| DIAPH3 | Deletions | ↑ | ↑ | [232] | ||||||
| DIAPH3 | ↓ (IHC/T/WB) | ↑ | ↑ | ↑ | [245] | |||||
| DIAPH3 | [246] | |||||||||
| DAAM1 | ↑ (IHC) | ↑ | ↑ | ↑ | No effect | Poor | [247,248,249,250] | |||
| FHOD1 INF2 | ↑ (IHC/T) | ↑ | ↑ | No effect | [251] | |||||
| Bone | DAAM1 | ↑ | [252] | |||||||
| FMNL1 | ↑ (T) | ↑ Lung metastasis | [253] | |||||||
| Cervix | DAAM2 | ↑ (T) | Poor | [254] | ||||||
| Colorectal | DIAPH1 | ↑ (IHC) | ↑ | ↑ | ↑ | ↑ | [255] | |||
| DIAPH1 | [256] | |||||||||
| DIAPH2 | Mutants | Poor | [257] | |||||||
| DIAPH2 | [258] | |||||||||
| DIAPH3 | ↑ (T) Mutants | Poor | [259] | |||||||
| DAAM1/2 | ↑ (T) | [260] | ||||||||
| DAAM2 | ↑ (IHC/T/WB) | ↑ | ↑ | Poor | [261] | |||||
| FMNL2 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | [262,263,264,265,266] | ||
| FMNL3 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | ↑ | ↑ | Poor | [267,268] | ||
| FMN2 | ↓ (T) | No effect | [269] | |||||||
| FMN2 | Mutants | [270] | ||||||||
| Esophagus | DIAPH2 | ↓ (T) | [271] | |||||||
| DAAM1 | ↑ | [272] | ||||||||
| Gallbladder | FMNL2 | ↑ (IHC) | Poor | [273] | ||||||
| Head & neck | DIAPH1*/2/3 | ↑* (IHC/T/WB) Mutants | Poor | [274] | ||||||
| DIAPH2*/3 | ↓* (T) Mutants in DIAPH2 SNP in DIAPH3 | [275] | ||||||||
| DIAPH2 | SNP | Protective | [276] | |||||||
| FMNL1 | ↑ (IHC/ISH/WB) Amplifications | ↑ | ↑ | ↑ | ↑ | Poor | [277] | |||
| FMNL3 | ↑ (IHC/T/WB) | ↑ | ↑ | Poor | [278] | |||||
| FMNL3 | ↑ (IHC) | ↑ | ↑ | No effect | No effect | [279] | ||||
| FHOD1 | ↑ (IHC/T) | ↑ | ↑ | ↑ | ↑ | [280] | ||||
| Kidney | FMNL1 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | Poor | [281] | |||
| DAAM2 | SNP | [282] | ||||||||
| Leukemia | DIAPH1 | ↑ | ↑ | [283] | ||||||
| FMNL1 | ↑ (T/WB) | ↑ | ↑ | [284,285] | ||||||
| FHOD3 | Mutants | [286] | ||||||||
| Lymphoma | FMNL1 | ↑ (IHC/WB) | [287] | |||||||
| Liver | DIAPH3 | ↑ (IHC/T/WB) | ↑ | ↑ | [288] | |||||
| DIAPH3 | Deletions | [232] | ||||||||
| DAAM2 | ↑ (IHC/T/WB) | ↑ | Poor | [289] | ||||||
| FMNL2 | ↓ (IHC/T/WB) | ↑ | ↑ | Poor | [290] | |||||
| Lung | DIAPH3 | ↑ (IHC/T/WB) | ↑ | Poor | [291] | |||||
| DAAM2 | SNP | Protective | [292] | |||||||
| FMNL1 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | ↑ | Poor | [293] | |||
| FHOD1 | ↑ | ↑ | Poor | [294] | ||||||
| Ovary | DIAPH1 | ↑ (IHC/T/WB) | Protective | [295] | ||||||
| DIAPH3 | ↑ | ↓ | [296] | |||||||
| FMNL1/2/3 FHOD3 | ↑ | ↑ (in vivo zebrafish) | [297] | |||||||
| Pancreas | DIAPH3 | ↑ (IHC/T/WB) | ↑ | ↑ | Poor | [298] | ||||
| FMNL1/3 FMN2 | Mutants | [299] | ||||||||
| FMN1 | SNP | Higher risk | [300] | |||||||
| FMN2 | ↑ (T) | Protective | [301] | |||||||
| Prostate | DIAPH3 | Deletions (IHC/ISH) | ↑ | ↑ | ↑ | ↑ | [232,302] | |||
| DIAPH3 | Knockdown | ↑ | ↑ | [246] | ||||||
| FMNL3 | ↑ | [303] | ||||||||
| FMN1 | SNP | Higher risk | [304] | |||||||
| INF2 | ↑ | ↑ | [305] | |||||||
| Skin | DIAPH1 | ↑ | [241] | |||||||
| DAAM1 | ↑ (IHC) | ↑ | ↑ | ↑ | [306] | |||||
| FMNL2/3 | ↑ (IHC/WB) | ↑ | Poor (FMNL2) | [307] | ||||||
| FMNL2 | ↑ (T/WB) | ↑ | ↑ | [308] | ||||||
| FHOD1 | ↑ | [309] | ||||||||
| FHOD1 | ↑ (IHC/WB) | ↑ | ↑ | ↑ | [310] | |||||
| Stomach | FMNL1/2/3 | ↑ (T) | Poor (FMNL1/3) No effect (FMNL2) | [311] | ||||||
| FMNL2 | ↑ (WB) | ↑ | ↑ | [312] | ||||||
| FMNL1 FHOD1 | [313] | |||||||||
| FHOD1 | ↑ (T) | [314] | ||||||||
| Thyroid | FHOD3 | ↑ (T) Copy number variations | [315] | |||||||
IHC, immunohistochemistry; ISH, in situ hybridization; T, transcript levels; WB, western blot; ↑/↓, High/Low. The asteisks (*) label a pair of a given formin and its corresponding expression levels.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Labat-de-Hoz, L.; Alonso, M.A. Formins in Human Disease. Cells 2021, 10, 2554. https://doi.org/10.3390/cells10102554
AMA Style
Labat-de-Hoz L, Alonso MA. Formins in Human Disease. Cells. 2021; 10(10):2554. https://doi.org/10.3390/cells10102554
Chicago/Turabian StyleLabat-de-Hoz, Leticia, and Miguel A. Alonso. 2021. "Formins in Human Disease" Cells 10, no. 10: 2554. https://doi.org/10.3390/cells10102554
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.