
Integrated miRNA and mRNA Transcriptome Analysis Reveals Eggplant’s (Solanum melongena L.) Responses to Waterlogging Stress


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Expression Profiling (Transcriptome) Database Sequencing Process
2.3. RNA Information Analysis Program
2.3.1. Assembly and Annotation of Transcriptome Sequencing Data
- Inchworm: We constructed a k-mer library with a k-mer size of 25. Low-frequency k-mers were filtered out, and the most frequent k-mer, excluding complexity and single k-mers, was selected as the seed. The k-1 length overlap between k-mers was used to extend the seeds until a linear contig was formed.
- Chrysalis: Contig sets that potentially contained variable splicing and other parallel genes were identified and defined as components. De Bruijn graphs were constructed for each component, and read validation was performed to determine the reads supporting each component.
- Butterfly: A linear path with continuous nodes was merged to form longer sequences in the de Bruijn graph. Bifurcations, likely caused by sequencing errors and rarely supported by reads, were eliminated to ensure even edges. The following steps were carried out: scoring with a dynamic programming algorithm, identifying the path supported by reads and read pairs, and eliminating the path supported by reads. The resulting Trinity assembly was referred to as Unigene.
2.3.2. Quantitative, Differential and Functional Enrichment Analysis of Sequencing Data of Expression Profiles
2.3.3. Small RNA Information Analysis Scheme
2.3.4. Differential Expression Analysis
2.4. qRT-PCR Assay
2.5. Statistical Calculations
3. Results
3.1. Morphological and Physiological Differences in Eggplants Grown under Different Waterlogging Conditions
3.2. Analysis of Differential Gene Expression under Different Waterlogging Conditions
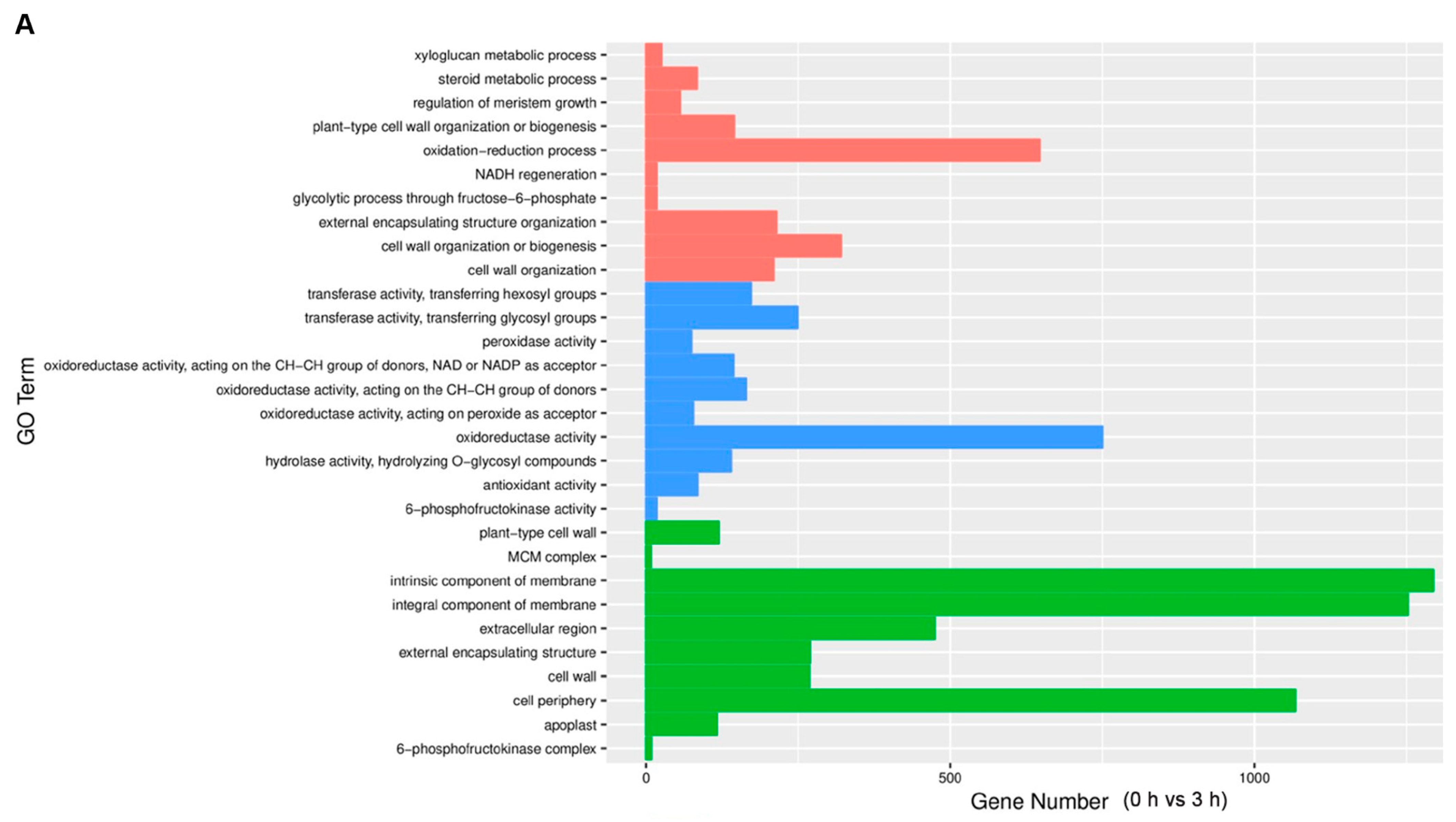
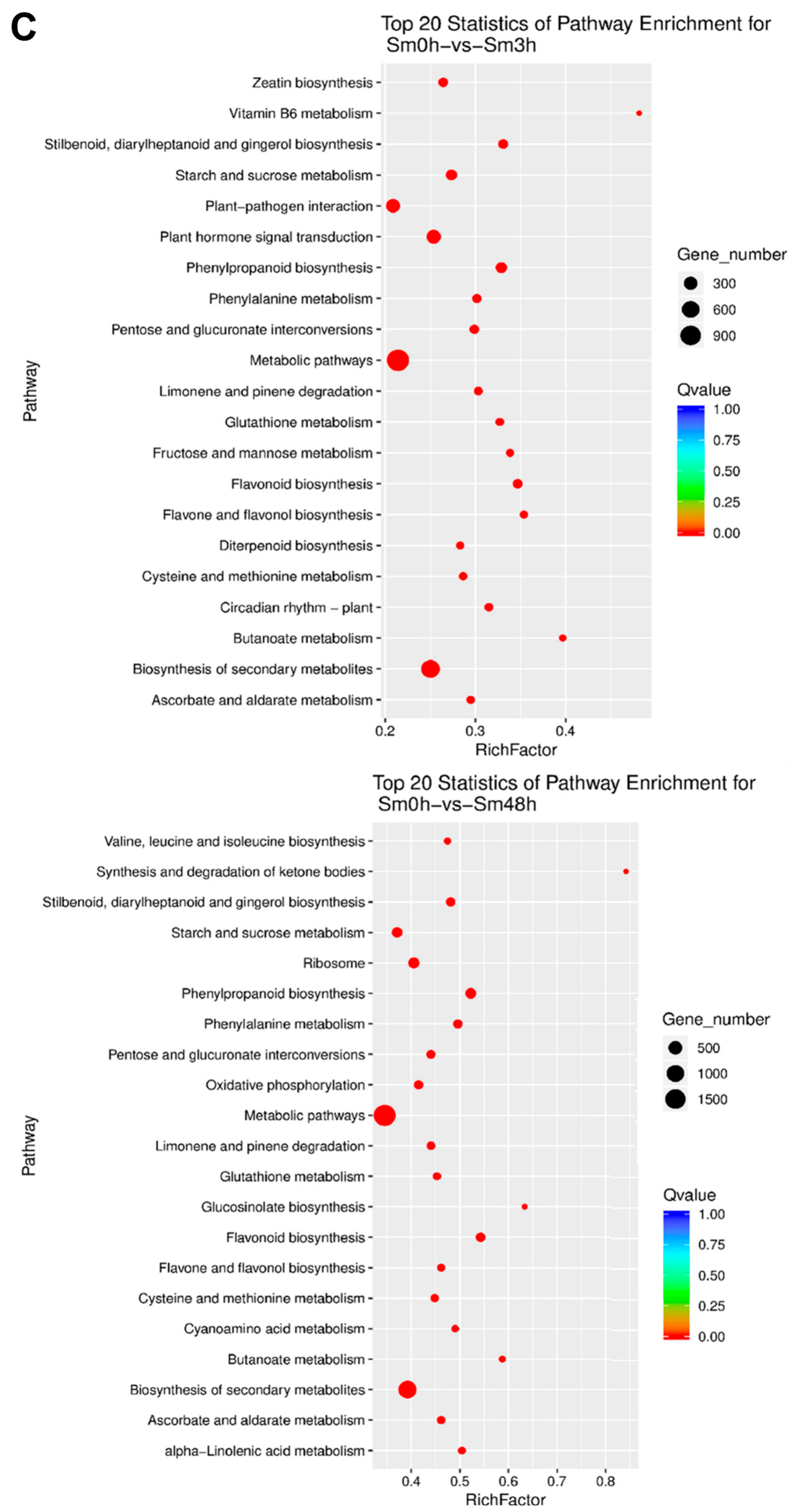
3.3. Functional Analysis of Differentially Expressed Genes under Different Waterlogging Conditions (GO and KEGG Pathway)
3.4. Classification Annotation of Small RNAs (sRNAs)
3.5. Differential Expression Analysis of miRNAs under Different Waterlogging Conditions
3.6. Prediction of Target Gene Loci
3.7. GO Enrichment Analysis of miRNA Target Genes
3.8. The KEGG Metabolic Pathway Analysis of miRNA-Targeted mRNA
3.9. Prediction of the miRNA–Target Gene Interaction Network
3.10. qRT-PCR Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakshi, A.; Gilroy, S. Analysis of plant flooding response. Methods Enzym. 2023, 680, 461–491. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, F.; Meng, Y.; Chandrasekaran, U.; Luo, X.; Yang, W.; Shu, K. Plant waterlogging/flooding stress responses: From seed germination to maturation. Plant Physiol. Biochem. 2020, 148, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Sairam, R.K.; Kumutha, D.; Ezhilmathi, K.; Deshmukh, P.S.; Srivastava, G.C. Physiology and biochemistry of waterlogging tolerance in plants. Biol. Plant. 2008, 52, 401–412. [Google Scholar] [CrossRef]
- Luo, Q.; Xie, H.; Chen, Z.; Ma, Y.; Yang, H.; Yang, B.; Ma, Y. Morphology, photosynthetic physiology and biochemistry of nine herbaceous plants under water stress. Front. Plant Sci. 2023, 14, 1147208. [Google Scholar] [CrossRef]
- Niki, T.; Gladish, D.K. Changes in growth and structure of pea primary roots (Pisum sativum L. cv. Alaska) as a result of sudden flooding. Plant Cell Physiol. 2001, 42, 694–702. [Google Scholar] [CrossRef]
- Wei, W.; Li, D.; Wang, L.; Ding, X.; Zhang, Y.; Gao, Y.; Zhang, X. Morpho-anatomical and physiological responses to waterlogging of sesame (Sesamum indicum L.). Plant Sci. 2013, 208, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Last, L.; Widmer, F.; Fjellstad, W.; Stoyanova, S.; Kolliker, R. Genetic diversity of natural orchardgrass (Dactylis glomerata L.) populations in three regions in Europe. BMC Genet. 2013, 14, 102. [Google Scholar] [CrossRef]
- De la Rosa, C.; Covarrubias, A.A.; Reyes, J.L. A dicistronic precursor encoding miR398 and the legume-specific miR2119 coregulates CSD1 and ADH1 mRNAs in response to water deficit. Plant Cell Environ. 2019, 42, 133–144. [Google Scholar] [CrossRef]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.J.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2010, 61, 165–177. [Google Scholar] [CrossRef]
- Jin, Q.; Xu, Y.; Mattson, N.; Li, X.; Wang, B.; Zhang, X.; Jiang, H.; Liu, X.; Wang, Y.; Yao, D. Identification of Submergence-Responsive MicroRNAs and Their Targets Reveals Complex MiRNA-Mediated Regulatory Networks in Lotus (Nelumbo nucifera Gaertn). Front. Plant Sci. 2017, 8, 6. [Google Scholar] [CrossRef]
- Liu, Z.; Kumari, S.; Zhang, L.; Zheng, Y.; Ware, D. Characterization of miRNAs in response to short-term waterlogging in three inbred lines of Zea mays. PLoS ONE 2012, 7, e39786. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Sharif, R.; Xu, X.; Chen, X. Mechanisms of Waterlogging Tolerance in Plants: Research Progress and Prospects. Front. Plant Sci. 2020, 11, 627331. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, F.; Zhang, Y.; Wang, L.; Cheng, Y.F. Cold-responsive miRNAs and their target genes in the wild eggplant species Solanum aculeatissimum. BMC Genom. 2017, 18, 1000. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Talla, A.; Qiu, W. Small RNA profiling of virus-infected grapevines: Evidences for virus infection-associated and variety-specific miRNAs. Funct. Integr. Genom. 2012, 12, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Tian, X.; Li, Y.J.; Wu, C.A.; Zheng, C.C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. Rna 2008, 14, 836–843. [Google Scholar] [CrossRef]
- Wan, F.; Pan, Y.; Li, J.; Chen, X.; Pan, Y.; Wang, Y.; Tian, S.; Zhang, X. Heterologous expression of Arabidopsis C-repeat binding factor 3 (AtCBF3) and cold-regulated 15A (AtCOR15A) enhanced chilling tolerance in transgenic eggplant (Solanum melongena L.). Plant Cell Rep. 2014, 33, 1951–1961. [Google Scholar] [CrossRef]
- Waseem, M.; Ahmad, F.; Habib, S.; Li, Z. Genome-wide identification of the auxin/indole-3-acetic acid (Aux/IAA) gene family in pepper, its characterisation, and comprehensive expression profiling under environmental and phytohormones stress. Sci. Rep. 2018, 8, 12008. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Sun, J.; Li, M.; Li, Z.; Xue, J.; Lan, X.; Zhang, C.; Lei, C.; Chen, H. Identification and profiling of conserved and novel microRNAs from Chinese Qinchuan bovine longissimus thoracis. BMC Genom. 2013, 14, 42. [Google Scholar] [CrossRef]
- Yang, X.; Wang, L.; Yuan, D.; Lindsey, K.; Zhang, X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J. Exp. Bot. 2013, 64, 1521–1536. [Google Scholar] [CrossRef]
- Carnavale Bottino, M.; Rosario, S.; Grativol, C.; Thiebaut, F.; Rojas, C.A.; Farrineli, L.; Hemerly, A.S.; Ferreira, P.C. High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE 2013, 8, e59423. [Google Scholar] [CrossRef] [PubMed]
- Khraiwesh, B.; Pugalenthi, G.; Fedoroff, N.V. Identification and analysis of red sea mangrove (Avicennia marina) microRNAs by high-throughput sequencing and their association with stress responses. PLoS ONE 2013, 8, e60774. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lai, Z. Comparative analysis reveals dynamic changes in miRNAs and their targets and expression during somatic embryogenesis in longan (Dimocarpus longan Lour.). PLoS ONE 2013, 8, e60337. [Google Scholar] [CrossRef]
- Jiang, Z.; Shen, L.; He, J.; Du, L.; Xia, X.; Zhang, L.; Yang, X. Functional Analysis of SmMYB39 in Salt Stress Tolerance of Eggplant (Solanum melongena L.). Horticulturae 2023, 9, 848. [Google Scholar] [CrossRef]
- Zhang, X.; Zou, Z.; Gong, P.; Zhang, J.; Ziaf, K.; Li, H.; Xiao, F.; Ye, Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 2011, 33, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Zhou, X.H.; Liu, J. Conserved miRNAs and Their Response to Salt Stress in Wild Eggplant Solanum linnaeanum Roots. Int. J. Mol. Sci. 2014, 15, 839–849. [Google Scholar] [CrossRef]
- Li, S.H.; Ge, Z.M.; Xie, L.N.; Chen, W.; Yuan, L.; Wang, D.Q.; Li, X.Z.; Zhang, L.Q. Ecophysiological response of native and exotic salt marsh vegetation to waterlogging and salinity: Implications for the effects of sea-level rise. Sci. Rep. 2018, 8, 2441. [Google Scholar] [CrossRef]
- Ma, M.H.; Wang, H.X.; Jia, P.F.; Liu, R.H.; Hong, Z.; Labriola, L.G.; Hong, Y.; Miao, L.J. Investigation of inducements and defenses of flash floods and urban waterlogging in Fuzhou, China, from 1950 to 2010. Nat. Hazards 2018, 91, 803–818. [Google Scholar] [CrossRef]
- Guo, H.; Tian, H.; Wang, Y.Q.; Xiong, X.W.; Tian, H.Z.; Zhang, S.Q.; Geng, G.D. Comparative analysis of growth, physiological and transcriptomic response revealed mechanisms of waterlogging tolerance of hot pepper (Capsicum annuum var. conoides). Plant Breed. 2022, 141, 807–819. [Google Scholar] [CrossRef]
- de Souza, W.R.; Mitchell, R.A.C.; Cesarino, I. Editorial: The plant cell wall: Advances and current perspectives. Front. Plant Sci. 2023, 14, 1235749. [Google Scholar] [CrossRef]
- Gu, L.; Wu, Y.; Li, M.; Wang, F.; Li, Z.; Yuan, F.; Zhang, Z. Over-immunity mediated abnormal deposition of lignin arrests the normal enlargement of the root tubers of Rehmannia glutinosa under consecutive monoculture stress. Plant Physiol. Biochem. 2021, 165, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.S.; Qiu, H.J.; Peng, D.X.; Chen, J.H.; Wang, Y.F.; Stybayev, G.; Baitelenova, A.; Kipshakpayeva, G.; Begalina, A.; Wu, Z.M. The Mechanisms Underlying Physiological and Molecular Responses to Waterlogging in Flax. J. Nat. Fibers 2023, 20, 2198275. [Google Scholar] [CrossRef]
- Kharel, B.; Rusalepp, L.; Bhattarai, B.; Kaasik, A.; Kupper, P.; Lutter, R.; Mand, P.; Rohula-Okunev, G.; Rosenvald, K.; Tullus, A. Effects of air humidity and soil moisture on secondary metabolites in the leaves and roots of Betula pendula of different competitive status. Oecologia 2023, 202, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Liu, Z.; Zou, X.; Jiang, Y.; Qiu, F.; Zheng, Y.; Zhang, Z. Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol. Plant. 2013, 147, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef]
- Xu, X.; Wang, K.; Pan, J.; Chen, X. Small RNA sequencing identifies cucumber miRNA roles in waterlogging-triggered adventitious root primordia formation. Mol. Biol. Rep. 2019, 46, 6381–6389. [Google Scholar] [CrossRef]
- Phukan, U.J.; Mishra, S.; Shukla, R.K. Waterlogging and submergence stress: Affects and acclimation. Crit. Rev. Biotechnol. 2016, 36, 956–966. [Google Scholar] [CrossRef]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef]
- Visentin, I.; Pagliarani, C.; Deva, E.; Caracci, A.; Tureckova, V.; Novak, O.; Lovisolo, C.; Schubert, A.; Cardinale, F. A novel strigolactone-miR156 module controls stomatal behaviour during drought recovery. Plant Cell Environ. 2020, 43, 1613–1624. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Fukuda, H. HD-zip III homeobox genes that include a novel member, ZeHB-13 (Zinnia)/ATHB-15 (Arabidopsis), are involved in procambium and xylem cell differentiation. Plant Cell Physiol. 2003, 44, 1350–1358. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, L.; Zou, X.; Tao, Y.; Liu, Z.; Zheng, Y. Submergence-responsive MicroRNAs are potentially involved in the regulation of morphological and metabolic adaptations in maize root cells. Ann. Bot. 2008, 102, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Bussell, J.D.; Pcurar, D.I.; Schwambach, J.; Pcurar, M.; Bellini, C. Phenotypic Plasticity of Adventitious Rooting in Arabidopsis Is Controlled by Complex Regulation of AUXIN RESPONSE FACTOR Transcripts and MicroRNA Abundance. Plant Cell 2009, 21, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chiou, T.J.; Lin, S.I.; Aung, K.; Zhu, J.K. A miRNA involved in phosphate-starvation response in Arabidopsis. Curr. Biol. 2005, 15, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Datt Pant, B.; Stitt, M.; Scheible, W.R. PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rRNA | snoRNA | snRNA | rRNAetc | miRNA | Unann | Known_miRNA | tRNA | |
|---|---|---|---|---|---|---|---|---|
| 0 h | 1,688,348 | 15,383 | 9833 | 85,835 | 37,019 | 6,755,726 | 610,121 | 185,323 |
| 3 h | 1,759,930 | 20,098 | 12,877 | 98,196 | 44,790 | 8,754,388 | 753,614 | 276,079 |
| 6 h | 1,718,499 | 19,998 | 18,725 | 94,072 | 84,233 | 8,724,408 | 644,141 | 213,054 |
| 12 h | 2,259,877 | 18,544 | 19,832 | 86,215 | 58,605 | 6,605,543 | 466,736 | 141,748 |
| 24 h | 4,551,603 | 23,651 | 29,375 | 103,455 | 69,857 | 6,683,368 | 580,503 | 171,313 |
| 48 h | 6,225,970 | 9722 | 13,560 | 85,372 | 24,048 | 4,594,547 | 390,776 | 158,796 |
| rRNA | snoRNA | snRNA | rRNAetc | miRNA | Unann | Known_miRNA | tRNA | |
|---|---|---|---|---|---|---|---|---|
| 0 h | 248,404 | 5113 | 3771 | 20,294 | 318 | 4,784,562 | 3267 | 26,426 |
| 3 h | 236,165 | 6000 | 4522 | 21,284 | 361 | 5,929,137 | 3613 | 26,890 |
| 6 h | 199,923 | 5482 | 4668 | 19,392 | 354 | 5,767,460 | 3143 | 30,098 |
| 12 h | 216,642 | 5260 | 4968 | 18,901 | 298 | 4,675,733 | 3072 | 24,818 |
| 24 h | 334,239 | 6225 | 5699 | 20,748 | 294 | 4,561,252 | 3109 | 32,369 |
| 48 h | 373,456 | 3329 | 3486 | 16,620 | 245 | 3,225,708 | 2492 | 33,930 |
| miRNA_Number | Target_Gene_Number | miRNA::Target_Number | |
|---|---|---|---|
| Sm 0 h-vs.-Sm 3 h | 29 | 85 | 237 |
| Sm 0 h-vs.-Sm 6 h | 31 | 239 | 358 |
| Sm 0 h-vs.-Sm 12 h | 22 | 222 | 310 |
| Sm 0 h-vs.-Sm 24 h | 51 | 314 | 607 |
| Sm 0 h-vs.-Sm 48 h | 89 | 436 | 850 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Z.; Xia, X.; Liu, Y.; He, J.; Yang, X. Integrated miRNA and mRNA Transcriptome Analysis Reveals Eggplant’s (Solanum melongena L.) Responses to Waterlogging Stress. Agronomy 2023, 13, 2215. https://doi.org/10.3390/agronomy13092215
Jiang Z, Xia X, Liu Y, He J, Yang X. Integrated miRNA and mRNA Transcriptome Analysis Reveals Eggplant’s (Solanum melongena L.) Responses to Waterlogging Stress. Agronomy. 2023; 13(9):2215. https://doi.org/10.3390/agronomy13092215
Chicago/Turabian StyleJiang, Zheng, Xin Xia, Yiyong Liu, Jie He, and Xu Yang. 2023. "Integrated miRNA and mRNA Transcriptome Analysis Reveals Eggplant’s (Solanum melongena L.) Responses to Waterlogging Stress" Agronomy 13, no. 9: 2215. https://doi.org/10.3390/agronomy13092215