
Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Mitogenome Assembly and Annotation
2.3. Mitochondrial Gene Annotation and Analysis
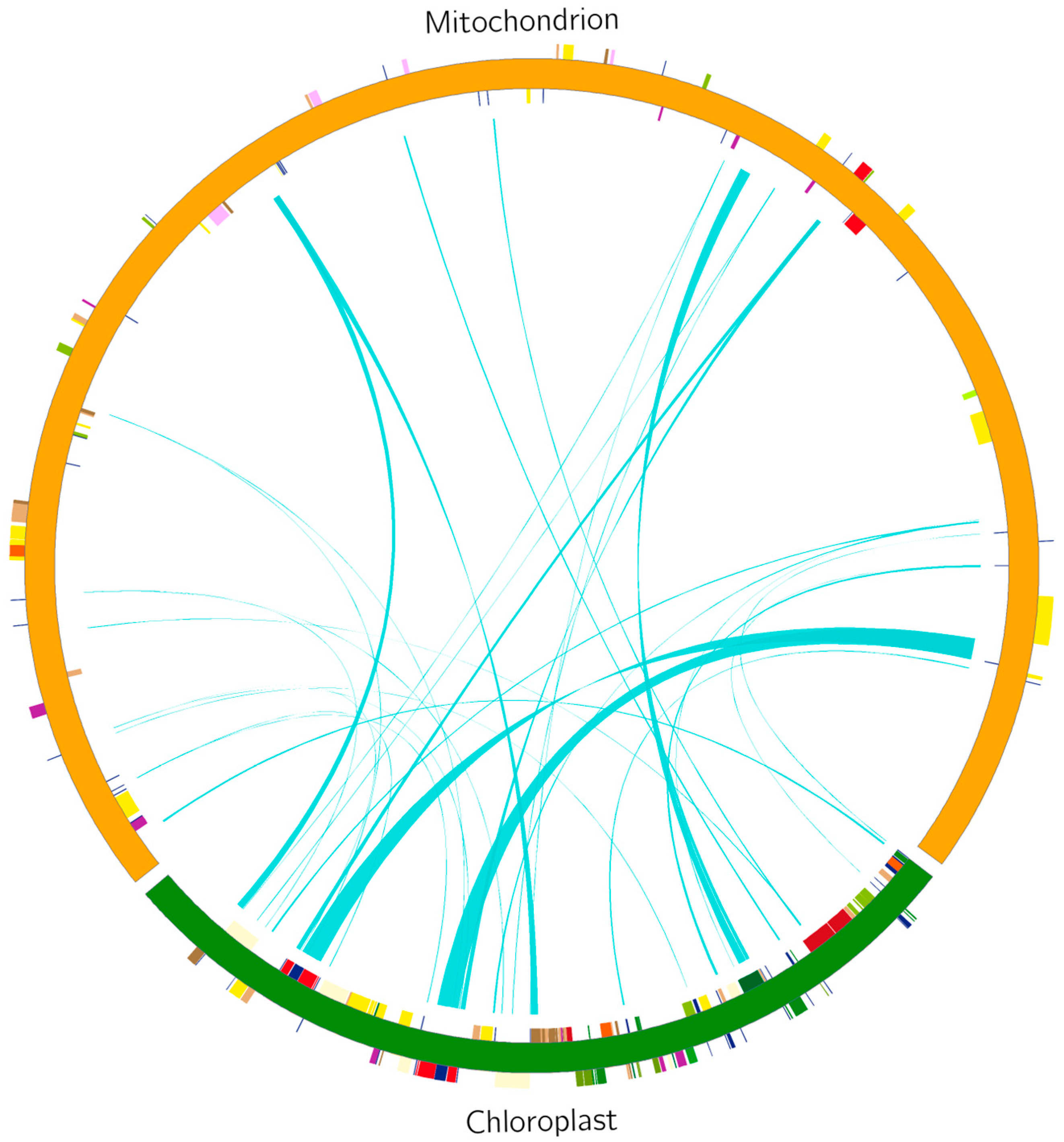
2.4. Repeat Sequence Analysis and Chloroplast-to-Mitochondrial DNA Transfer Analysis
2.5. Mitochondrial Genome Feature Analysis
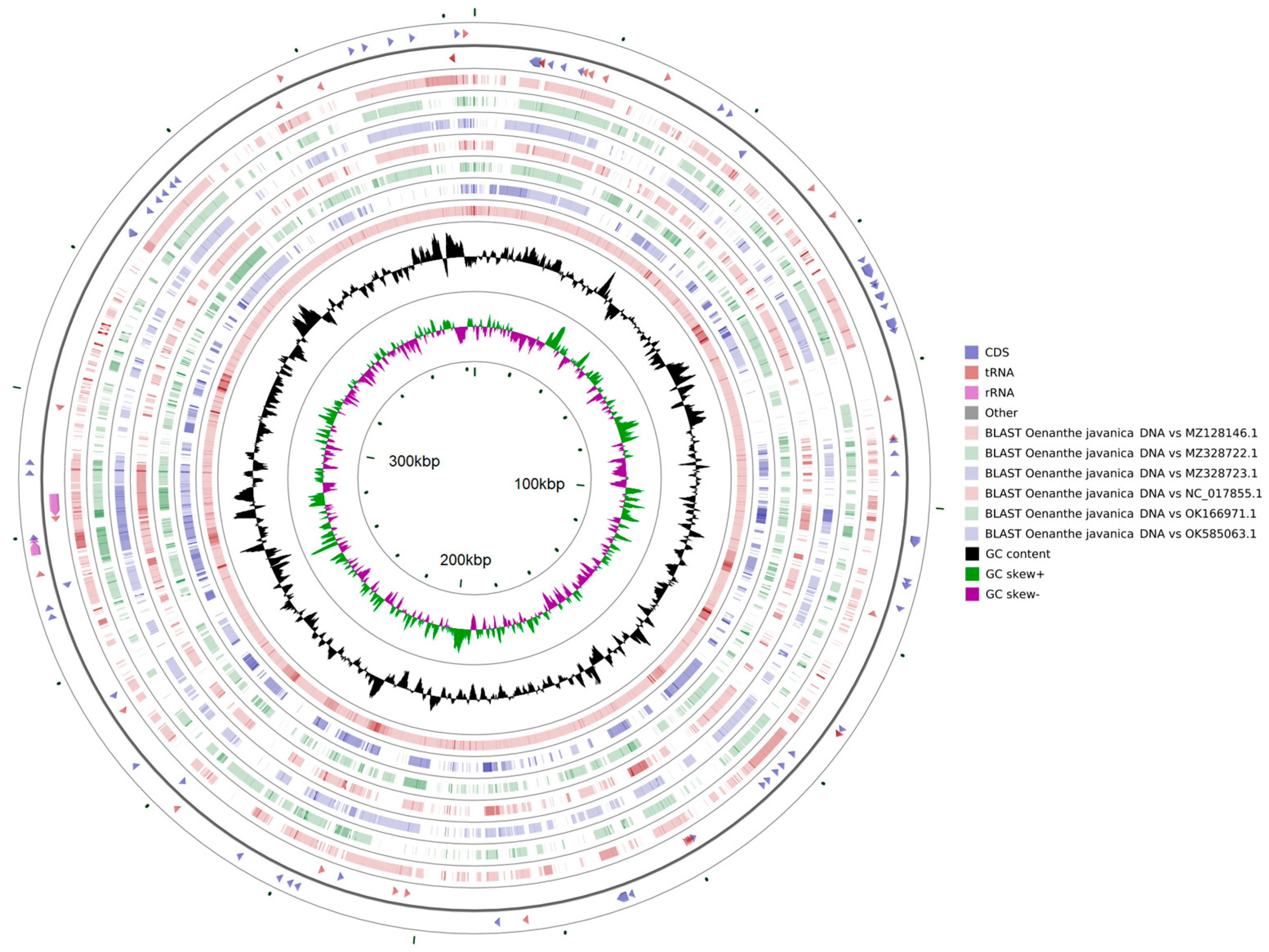
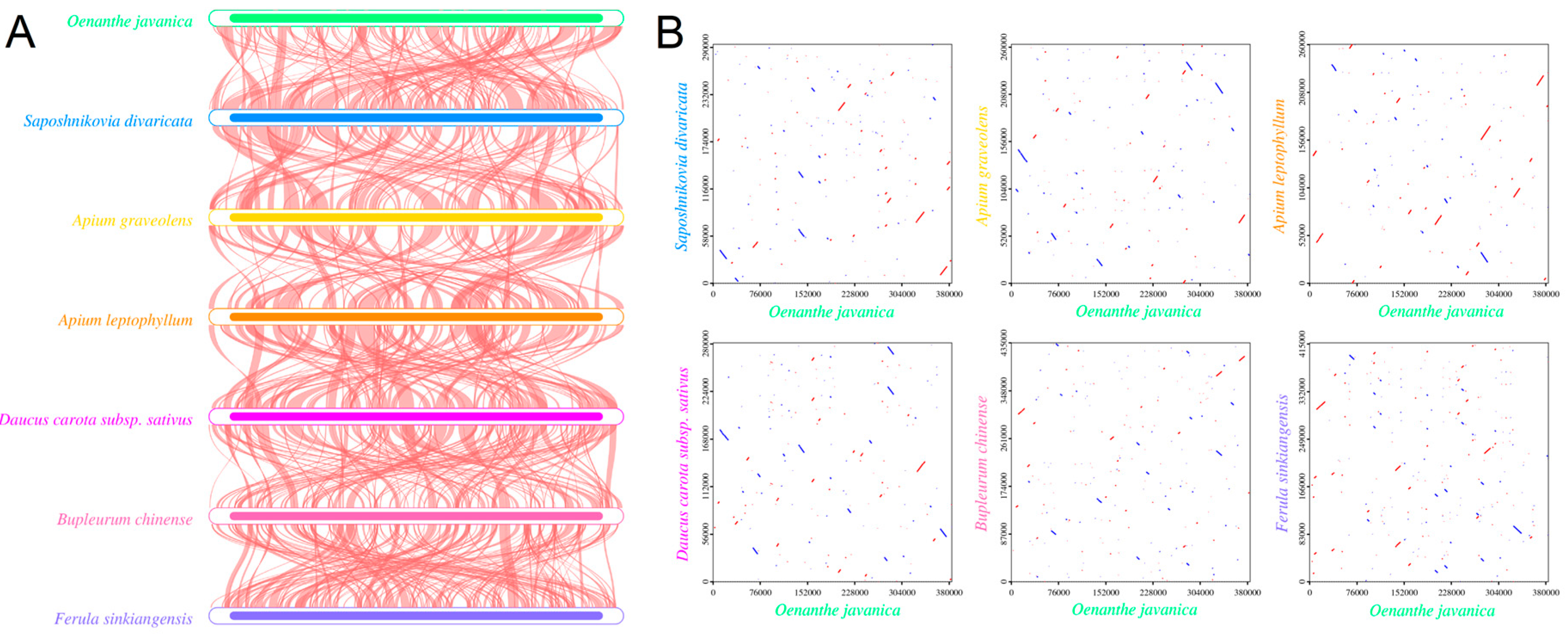
2.6. Genome Comparative Analysis
2.7. Phylogenetic Analysis
3. Results
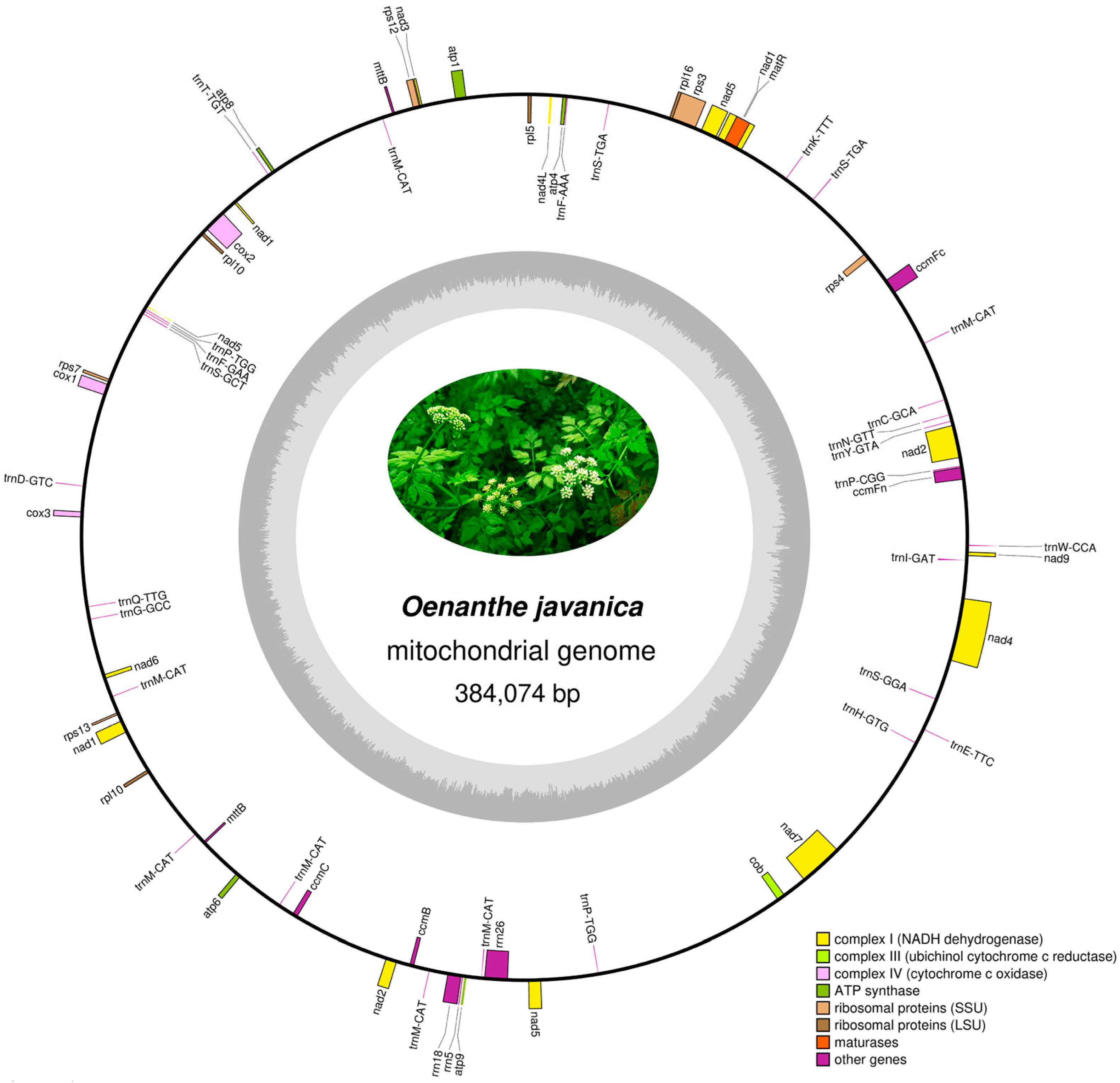
3.1. Assembly and Basic Features of the O. javanica Mitochondrial Genome
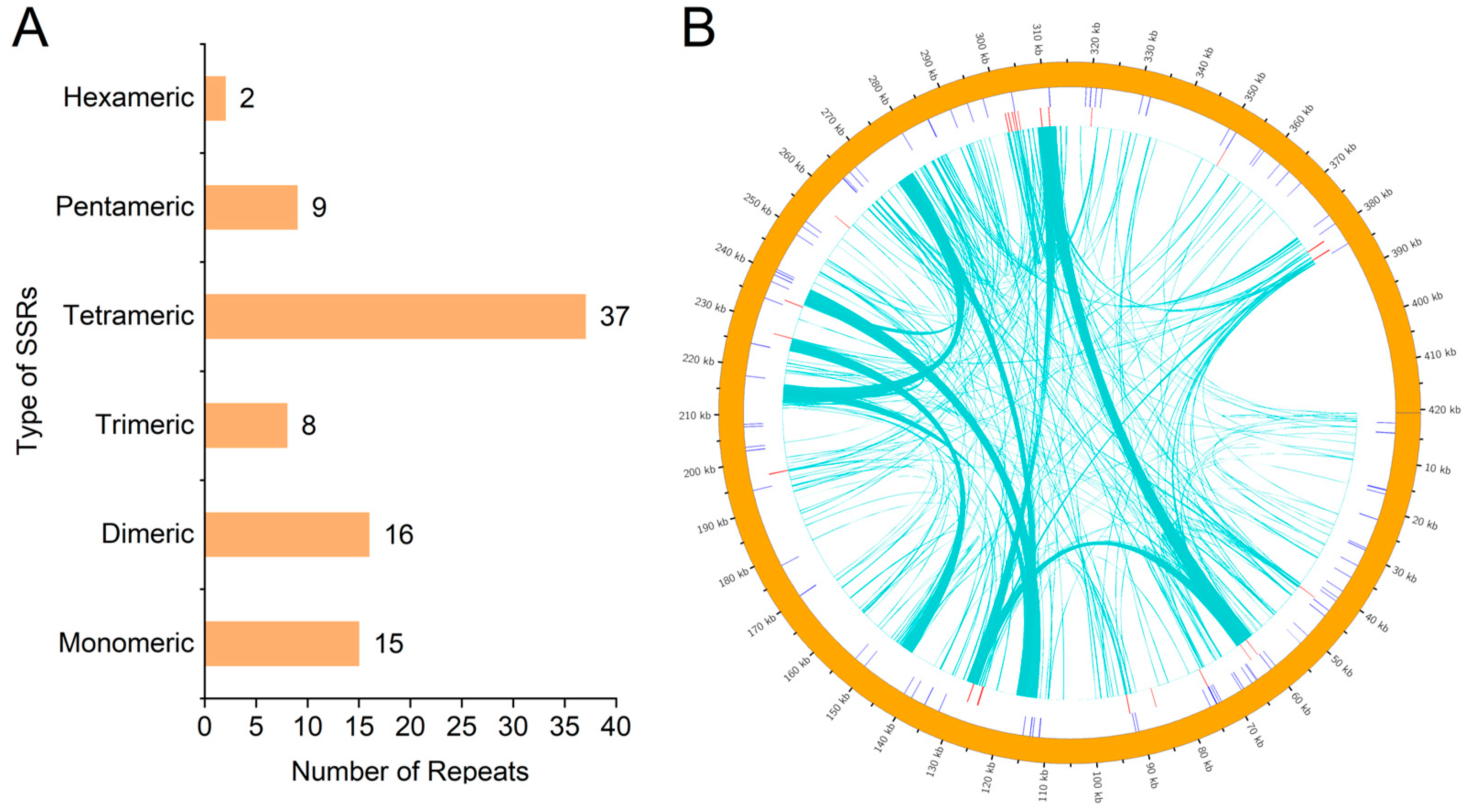
3.2. Repeat Sequence Analysis
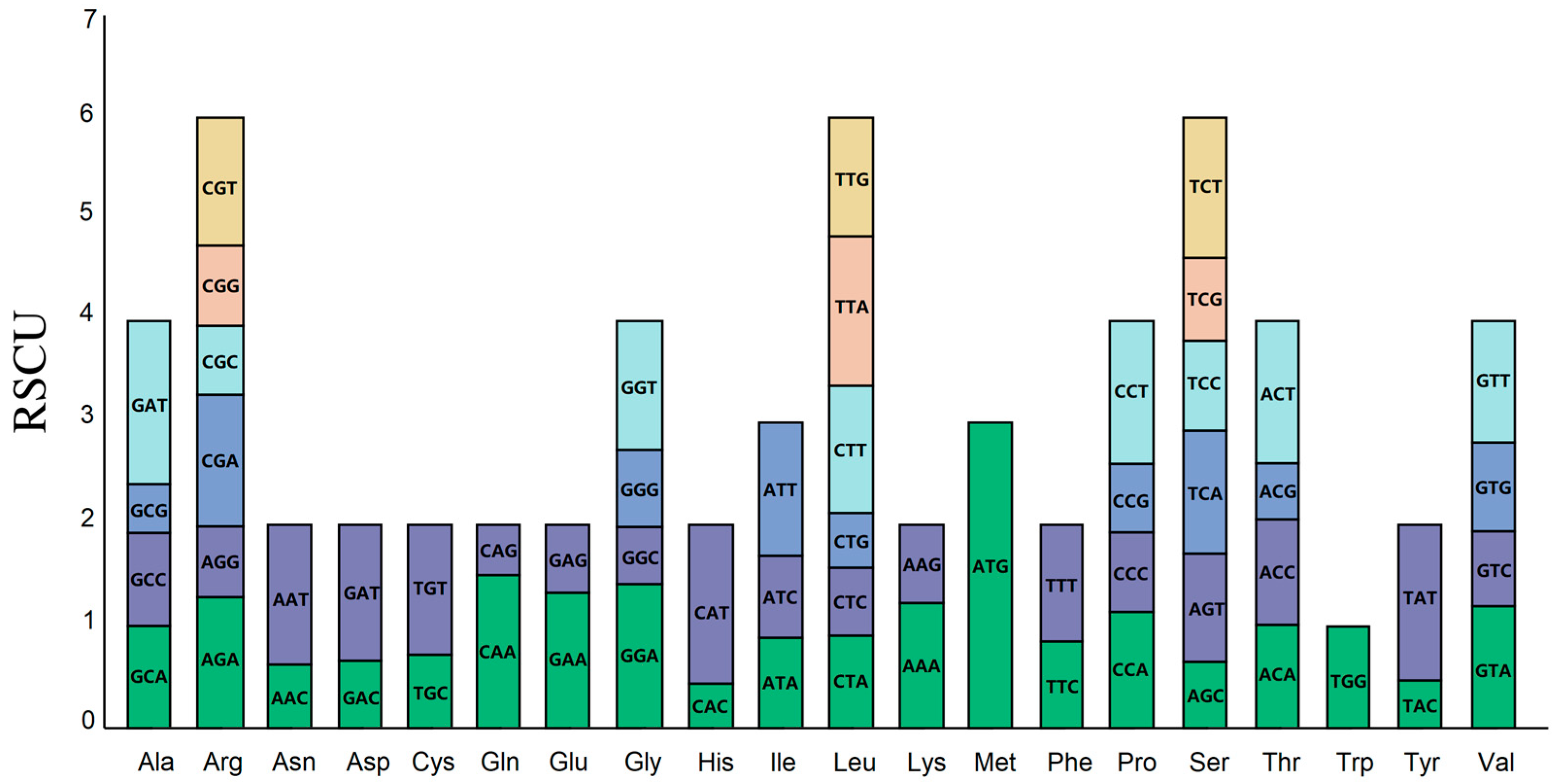
3.3. Analysis of Codon Usage Bias
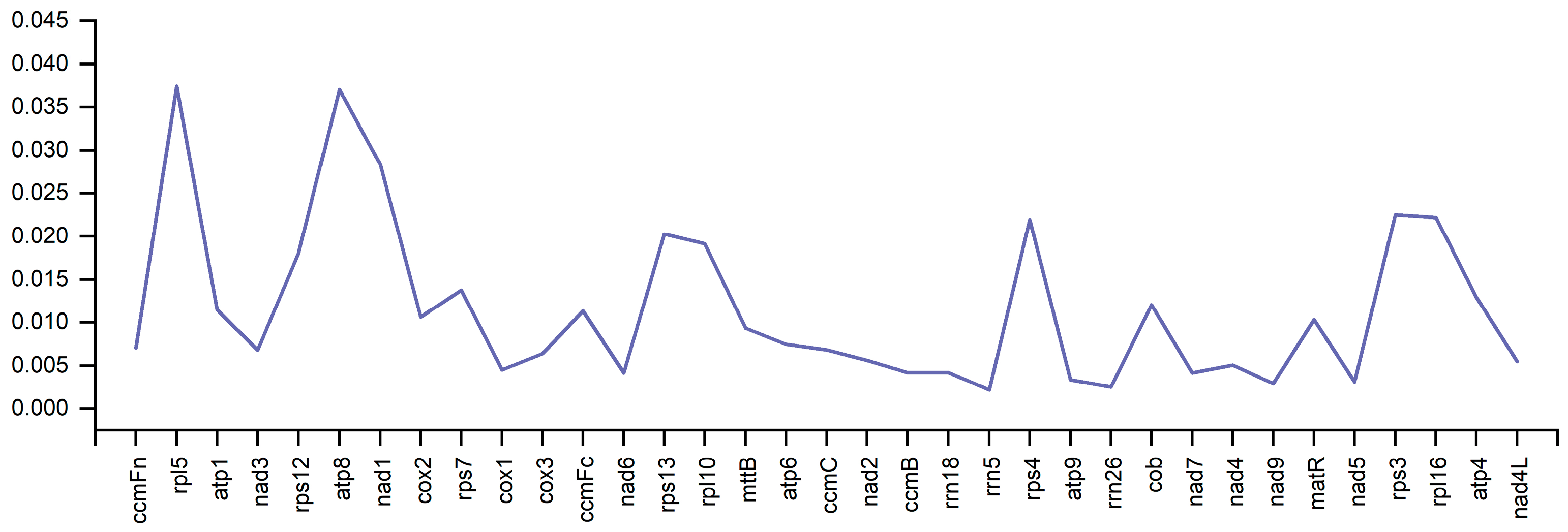
3.4. Analysis of Pi Nucleotide Diversity
3.5. Analysis of Chloroplast-to-Mitochondria Genomic Transfers in Watercress
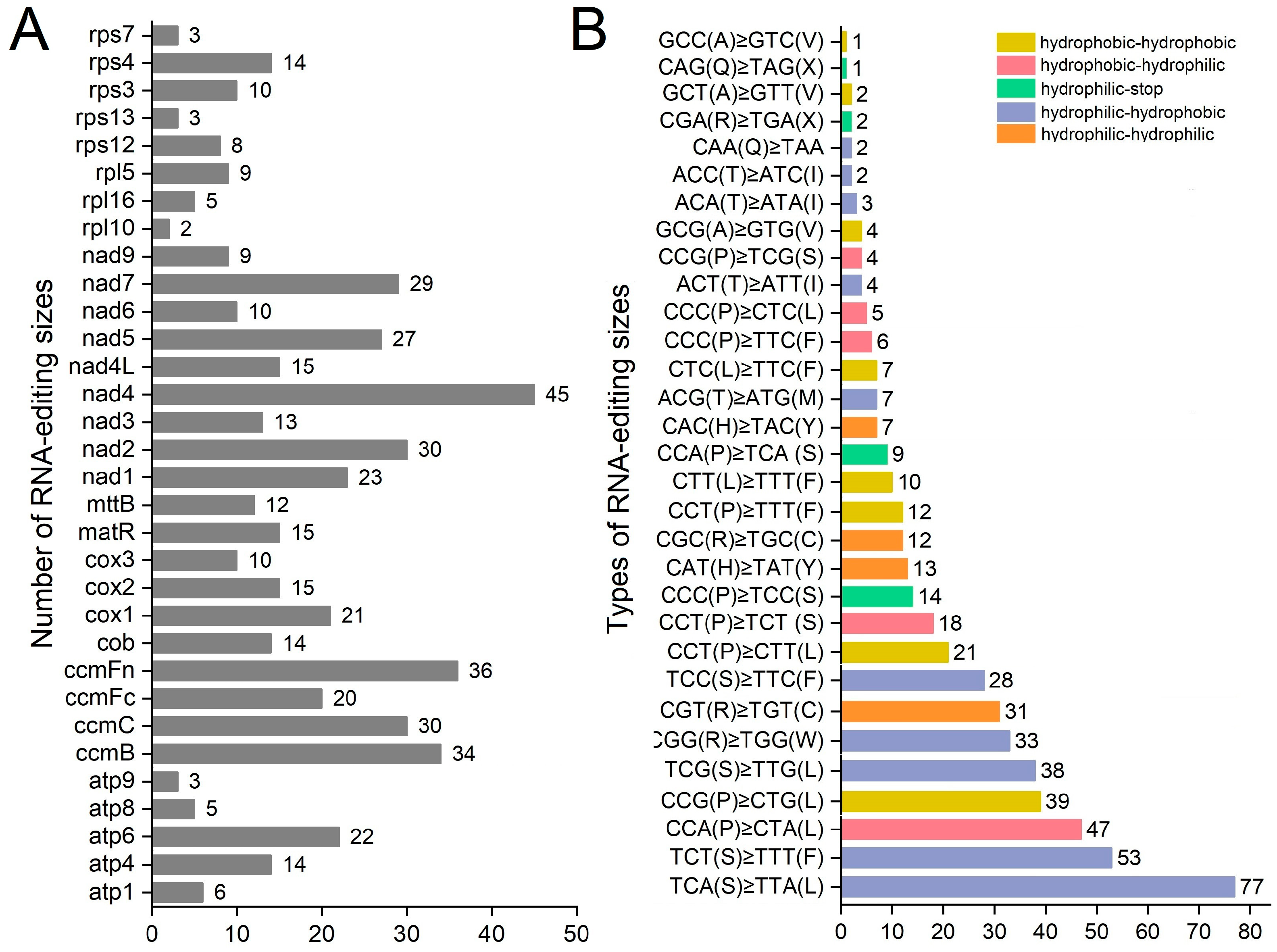
3.6. RNA Editing Site Analysis
3.7. Comparative Analysis of Mitochondrial Structures
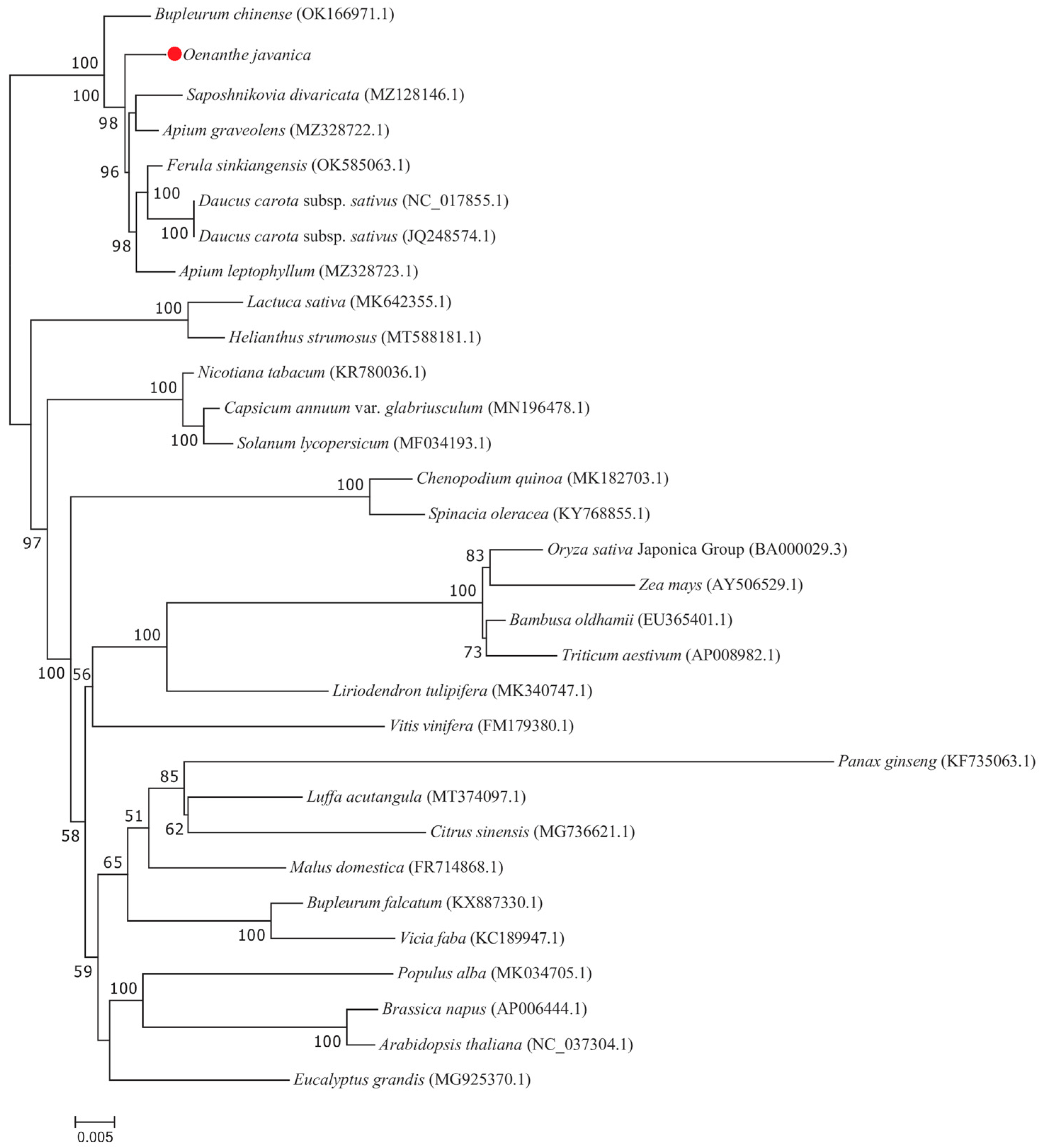
3.8. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grosser, M.R.; Sites, S.K.; Murata, M.M.; Lopez, Y.; Chamusco, K.C.; Love Harriage, K.; Grosser, J.W.; Graham, J.H.; Gmitter, F.; Chase, C.D. Plant mitochondrial introns as genetic markers-conservation and variation. Front. Plant Sci. 2023, 14, 1116851. [Google Scholar]
- Jang, W.; Lee, H.O.; Kim, J.-U.; Lee, J.-W.; Hong, C.-E.; Bang, K.-H.; Chung, J.-W.; Jo, I.-H. Complete mitochondrial genome and a set of 10 novel kompetitive allele-specific PCR markers in ginseng (Panax ginseng C. A. Mey.) . Agronomy 2020, 10, 1868. [Google Scholar]
- Xiong, Y.; Yu, Q.; Xiong, Y.; Zhao, J.; Lei, X.; Liu, L.; Liu, W.; Peng, Y.; Zhang, J.; Li, D.; et al. The complete mitogenome of Elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front. Plant Sci. 2022, 12, 802321. [Google Scholar]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar]
- Adams, K.L.; Palmer, J.D. Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 2003, 29, 380–395. [Google Scholar]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar]
- Palmer, J.D.; Adams, K.L.; Cho, Y.; Parkinson, C.L.; Qiu, Y.L.; Song, K. Dynamic evolution of plant mitochondrial genomes: Mobile genes and introns and highly variable mutation rates. Proc. Natl. Acad. Sci. USA 2000, 97, 6960–6966. [Google Scholar]
- Cheng, N.; Lo, Y.S.; Ansari, M.I.; Ho, K.C.; Jeng, S.T.; Lin, N.S.; Dai, H. Correlation between mtDNA complexity and mtDNA replication mode in developing cotyledon mitochondria during mung bean seed germination. New Phytol. 2017, 213, 751–763. [Google Scholar]
- Varré, J.-S.; D’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M.; et al. Complete Sequence, Multichromosomal Architecture and Transcriptome Analysis of the Solanum tuberosum Mitochondrial Genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. 2016, 2016, 5040598. [Google Scholar]
- Mustafa, S.I.; Heslop-Harrison, J.S.; Schwarzacher, T. The complete mitochondrial genome from Iraqi Meriz goats and the maternal lineage using whole genome sequencing data. Iran. J. Appl. Anim. Sci. 2021, 12, 321–328. [Google Scholar]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2017, 27, 1436–1448. [Google Scholar]
- Zhong, F.; Ke, W.; Li, Y.; Chen, X.; Zhou, T.; Xu, B.; Qi, L.; Yan, Z.; Ma, Y. Comprehensive analysis of the complete mitochondrial genomes of three Coptis species (C. chinensis, C. deltoidea and C. omeiensis): The important medicinal plants in China. Front. Plant Sci. 2023, 29, 1166420. [Google Scholar]
- Wang, B.; Xue, J.; Li, L.; Liu, Y.; Qiu, Y.L. The complete mitochondrial genome sequence of the liverwort Pleurozia purpurea reveals extremely conservative mitochondrial genome evolution in liverworts. Curr. Genet. 2009, 55, 601–609. [Google Scholar]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar]
- Yu, R.; Chen, X.; Long, L.; Jost, M.; Zhao, R.; Liu, L.; Mower, J.P.; de Pamphilis, C.W.; Wanke, S.; Jiao, Y. De novo assembly and comparative analyses of mitochondrial genomes in Piperales. Genom. Biol. Evol. 2023, 15, evad041. [Google Scholar]
- Ren, W.; Wang, L.; Feng, G.; Tao, C.; Liu, Y.; Yang, J. High-quality assembly and comparative analysis of Actinidia latifolia and A. valvata mitogenomes. Genes 2023, 14, 863. [Google Scholar]
- Cheng, Q.; Wang, P.; Li, T.; Liu, J.; Zhang, Y.; Wang, Y.; Sun, L.; Shen, H. Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in celery (Apium graveolens L.). Int. J. Mol. Sci. 2021, 22, 8584. [Google Scholar]
- Chen, C.M.; Chen, I.C.; Chen, Y.L.; Lin, T.H.; Chen, W.L.; Chao, C.Y.; Wu, Y.R.; Lu, Y.T.; Lee, C.Y.; Chien, H.C.; et al. Medicinal herbs Oenanthe javanica (Blume) DC., Casuarina equisetifolia L. and Sorghum bicolor (L.) Moench protect human cells from MPP+ damage via inducing FBXO7 expression. Phytomedicine 2016, 23, 1422–1433. [Google Scholar]
- Lu, C.L.; Li, X.F. A review of Oenanthe javanica (Blume) DC. as traditional medicinal plant and its therapeutic potential. Evid. Based Complement. Altern. Med. 2019, 2019, 6495819. [Google Scholar]
- Feng, K.; Kan, X.-Y.; Liu, Q.; Yan, Y.-J.; Sun, N.; Yang, Z.-Y.; Zhao, S.-P.; Wu, P.; Li, L.-J. Metabolomics Analysis Reveals Metabolites and Metabolic Pathways Involved in the Growth and Quality of Water Dropwort [Oenanthe javanica (Blume) DC.] under Nutrient Solution Culture. Plants 2023, 12, 1459. [Google Scholar] [CrossRef]
- Kumar, S.; Huang, X.; Ji, Q.; Qayyum, A.; Zhou, K.; Ke, W.; Zhu, H.; Zhu, G. Influence of Blanching on the Gene Expression Profile of Phenylpropanoid, Flavonoid and Vitamin Biosynthesis, and Their Accumulation in Oenanthe javanica. Antioxidants 2022, 11, 470. [Google Scholar] [CrossRef]
- Kumar, S.; Li, G.; Yang, J.; Huang, X.; Ji, Q.; Liu, Z.; Ke, W.; Hou, H. Effect of salt stress on growth, physiological parameters, and ionic concentration of water dropwort (Oenanthe javanica) cultivars. Front. Plant Sci. 2021, 12, 660409. [Google Scholar]
- Seo, W.H.; Baek, H.H. Identification of characteristic aroma-active compounds from water dropwort (Oenanthe javanica DC.). J. Agric. Food Chem. 2005, 53, 6766–6770. [Google Scholar]
- Zhang, Z.; Dong, H.; Yuan, M.; Yu, Y. The complete chloroplast genome of Oenanthe javanica. Mitochondrial DNA B Resour. 2020, 5, 3151–3153. [Google Scholar]
- Jiang, Q.; Wang, F.; Tan, H.W.; Li, M.Y.; Xu, Z.S.; Tan, G.F.; Xiong, A.S. De novo transcriptome assembly, gene annotation, marker development, and miRNA potential target genes validation under abiotic stresses in Oenanthe javanica. Mol. Genet. Genom. 2015, 290, 671–683. [Google Scholar]
- Yao, J.; Zhao, F.; Xu, Y.; Zhao, K.; Quan, H.; Su, Y.; Hao, P.; Liu, J.; Yu, B.; Yao, M.; et al. Complete chloroplast genome sequencing and phylogenetic analysis of two Dracocephalum plants. BioMed Res. Int. 2020, 2020, 4374801. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genom. Res. 2017, 27, 722–736. [Google Scholar]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar]
- Li, M.Y.; Zhang, R.; Li, R.; Zheng, K.M.; Xiao, J.C.; Zheng, Y.X. Analyses of chloroplast genome of Eutrema japonicum provide new Insights into the evolution of Eutrema Species. Agronomy 2021, 11, 2546. [Google Scholar]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar]
- Avvaru, A.K.; Saxena, S.; Sowpati, D.T.; Mishra, R.K. MSDB: A comprehensive database of simple sequence repeats. Genom. Biol. Evol. 2017, 9, 1797–1802. [Google Scholar]
- Gu, C.; Dong, B.; Xu, L.; Tembrock, L.R.; Zheng, S.; Wu, Z. The complete chloroplast genome of Heimia myrtifolia and comparative analysis within myrtales. Molecules 2018, 23, 846. [Google Scholar]
- Dong, S.; Chen, L.; Liu, Y.; Wang, Y.; Zhang, S.; Yang, L.; Lang, X.; Zhang, S. The draft mitochondrial genome of Magnolia biondii and mitochondrial phylogenomics of angiosperms. PLoS ONE 2020, 15, e0231020. [Google Scholar]
- Shi, Y.; Liu, Y.; Zhang, S.; Zou, R.; Tang, J.; Mu, W.; Peng, Y.; Dong, S. Assembly and comparative analysis of the complete mitochondrial genome sequence of Sophora japonica ‘JinhuaiJ2’. PLoS ONE 2018, 13, e0202485. [Google Scholar]
- Sun, Y.; Li, M.Y.; Ma, J.Y.; He, M.L.; Zheng, Y.X. Complete chloroplast genome sequence of a new variety of Brasenia schreberi: Genome characteristics, comparative analysis, and phylogenetic relationships. Agronomy 2022, 12, 2972. [Google Scholar]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar]
- Wynn, E.L.; Christensen, A.C. Repeats of unusual size in plant mitochondrial genomes: Identification, incidence and evolution. G3 2019, 9, 549–559. [Google Scholar]
- Ni, Y.; Li, J.; Chen, H.; Yue, J.; Chen, P.; Liu, C. Comparative analysis of the chloroplast and mitochondrial genomes of Saposhnikovia divaricata revealed the possible transfer of plastome repeat regions into the mitogenome. BMC Genom. 2022, 23, 570. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bai, D.; Zhong, Y.; Abid, M.; Qi, X.; Hu, C.; Fang, J. Physiological responses of two contrasting kiwifruit (Actinidia spp.) rootstocks against waterlogging stress. Plants 2021, 10, 2586. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar]
- Tang, D.; Huang, S.; Quan, C.; Huang, Y.; Miao, J.; Wei, F. Mitochondrial genome characteristics and phylogenetic analysis of the medicinal and edible plant Mesona chinensis Benth. Front. Genet. 2023, 13, 1056389. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Dang, Y.; Zhou, Z.; Wu, C.; Zhao, F.; Sachs, M.S.; Liu, Y. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol. Cell 2015, 59, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Xiao, G.; Liu, H.; Zhang, L.; Zhao, L.; Tang, M.; Huang, S.; An, Y.; Yu, J. Two pivotal RNA editing sites in the mitochondrial atp1 mRNA are required for ATP synthase to produce sufficient ATP for cotton fiber cell elongation. New Phytol. 2018, 218, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Tripp, E.A. The draft genome of Ruellia speciosa (Beautiful Wild Petunia: Acanthaceae). DNA Res. 2017, 24, 179–192. [Google Scholar]
- Feng, Y.; Xiang, X.; Akhter, D.; Pan, R.; Fu, Z.; Jin, X. Mitochondrial phylogenomics of fagales provides insights into plant mitogenome mosaic evolution. Front. Plant Sci. 2021, 12, 762195. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Group of Genes | Name of Genes |
|---|---|---|
| Protein coding genes (PCGs) | ATP synthase | atp1, atp4, atp6, atp8, atp9 |
| NADH dehydrogenase | nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 | |
| Cytochrome c biogenesis | cob | |
| Ubiquinol cytochrome c reductase | ccmB, ccmC, ccmFC, ccmFN | |
| Cytochrome c oxidase | cox1, cox2, cox3 | |
| Maturases | matR | |
| Transport membrane protein | mttB(×2) | |
| Ribosomal proteins (LSU) | rpl5, rpl10(×2), rpl16 | |
| Ribosomal proteins (SSU) | rps3, rps4, rps7, rps12, rps13 | |
| Ribosomal RNAs | rrn5, rrn18, rrn26 | |
| Transfer RNAs | trnC-GCA, trnD-GTC, trnE-TTC, trnF-AAA, trnF-GAA, trnG-GCC, trnH-GTG, trnI-GAT *, trnK-TTT, trnM-CAT(×7), trnN-GTT, trnP-CGG *, trnP-TGG(×2), trnQ-TTG, trnS-GCT, trnS-GGA, trnS-TGA(×2), trnT-TGT *, trnW-CCA, trnY-GTA | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Han, Q.; Li, M.; Luo, Q.; Zhu, S.; Zheng, Y.; Tan, G. Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica. Agronomy 2023, 13, 2103. https://doi.org/10.3390/agronomy13082103
Li X, Han Q, Li M, Luo Q, Zhu S, Zheng Y, Tan G. Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica. Agronomy. 2023; 13(8):2103. https://doi.org/10.3390/agronomy13082103
Chicago/Turabian StyleLi, Xiaoyan, Qiuju Han, Mengyao Li, Qing Luo, Shunhua Zhu, Yangxia Zheng, and Guofei Tan. 2023. "Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica" Agronomy 13, no. 8: 2103. https://doi.org/10.3390/agronomy13082103