
Genetic Diversity Analysis of Non-Heading Chinese Cabbage of Resistance to Clubroot Disease Based on SNP Molecular Markers
 , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Materials
2.2. Resequencing and Data Quality Control
2.3. Sequence Alignment and SNP/Indel Detection
2.4. Group Evolution Analysis
2.5. Linkage Disequilibrium Analysis
3. Results
3.1. Data Statistics and Evaluation
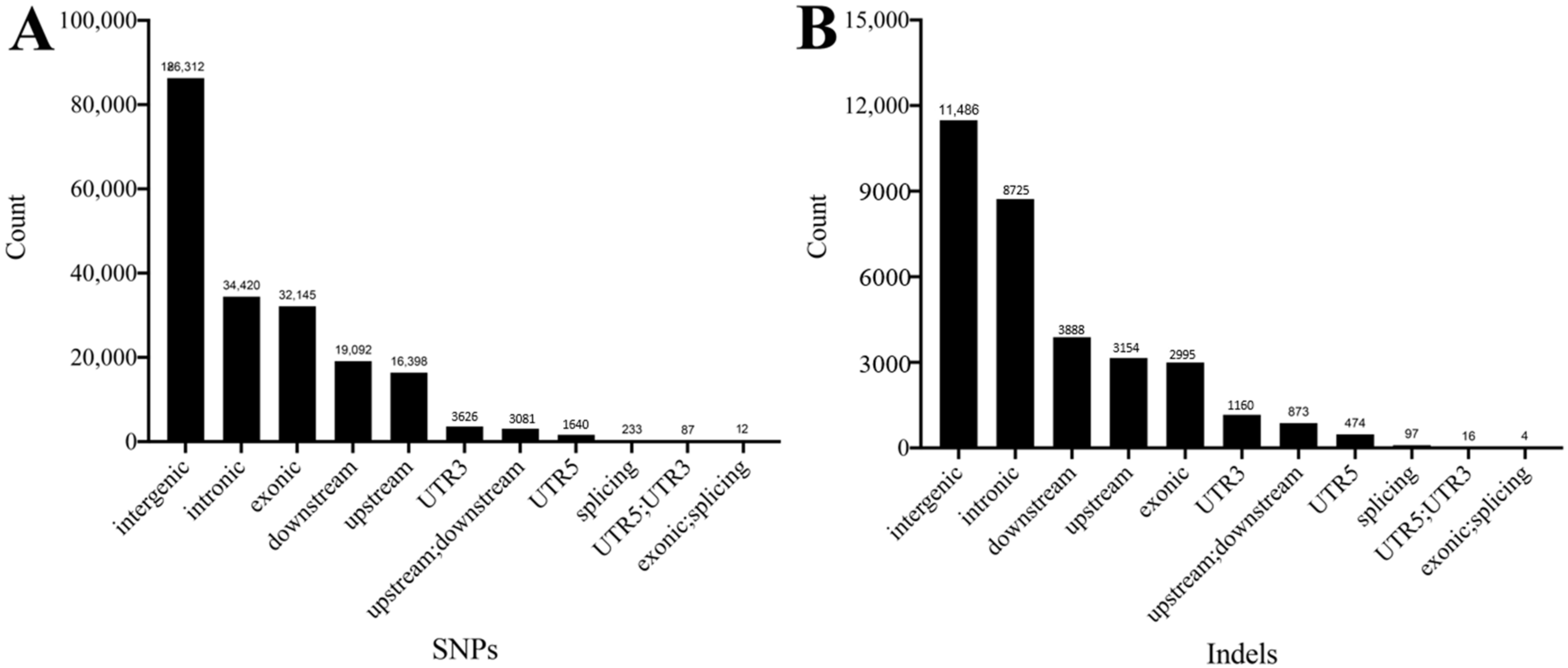
3.2. SNP and Indel Detection and Analysis
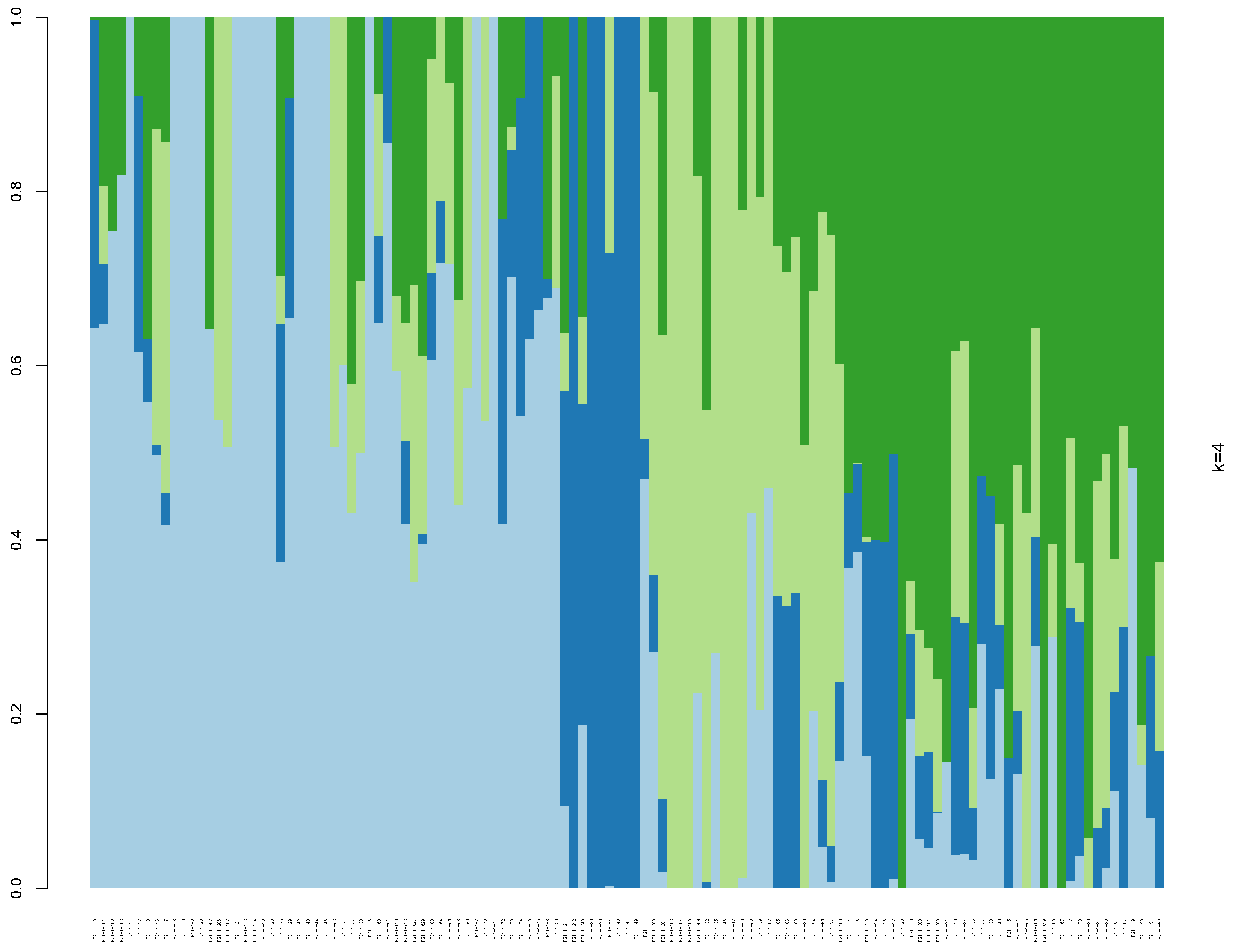
3.3. Population Genetic Structure and Phylogenetic Tree Analysis
3.4. Analysis of Population Genetic Diversity
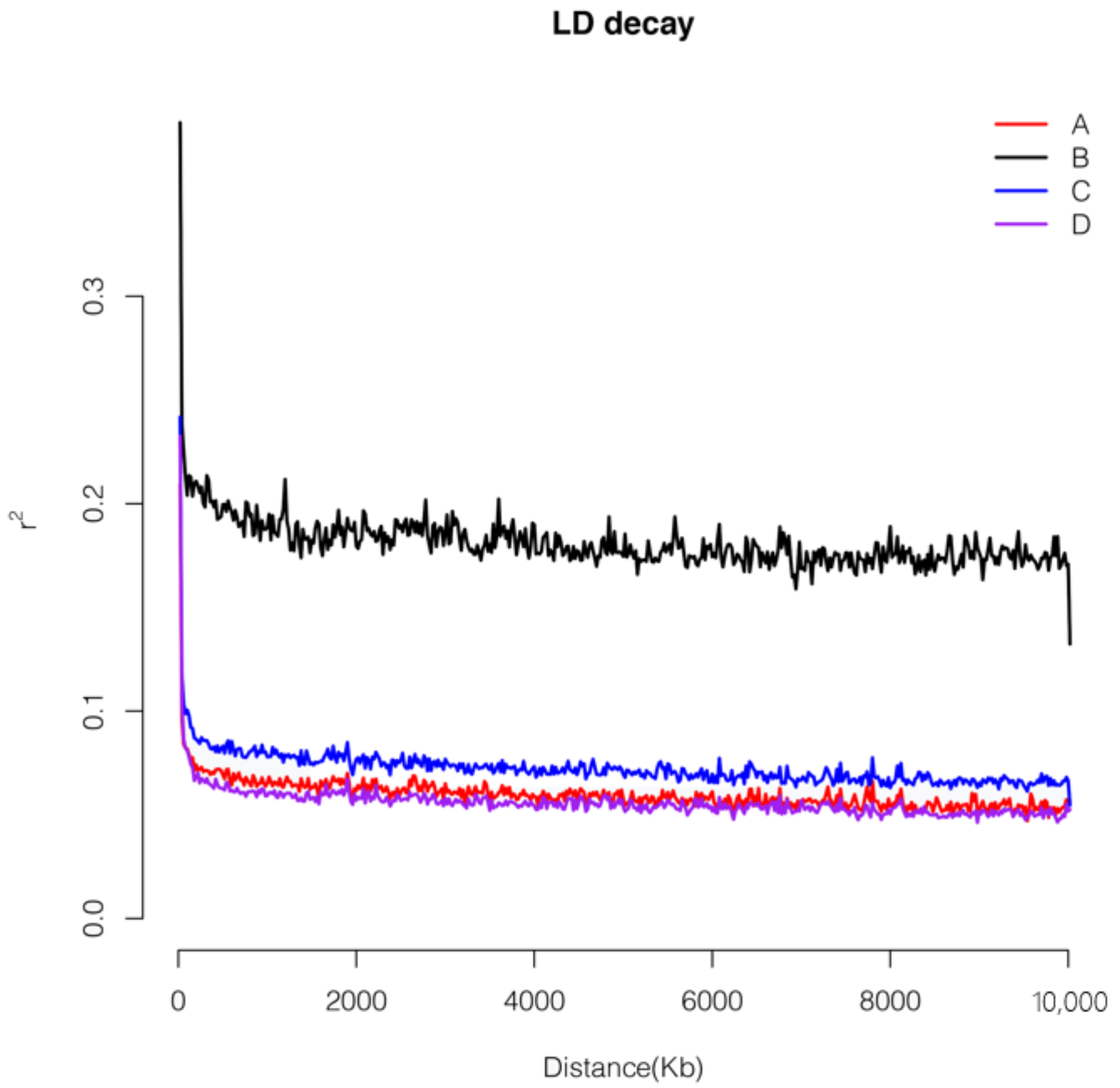
3.5. Linkage Disequilibrium Analysis
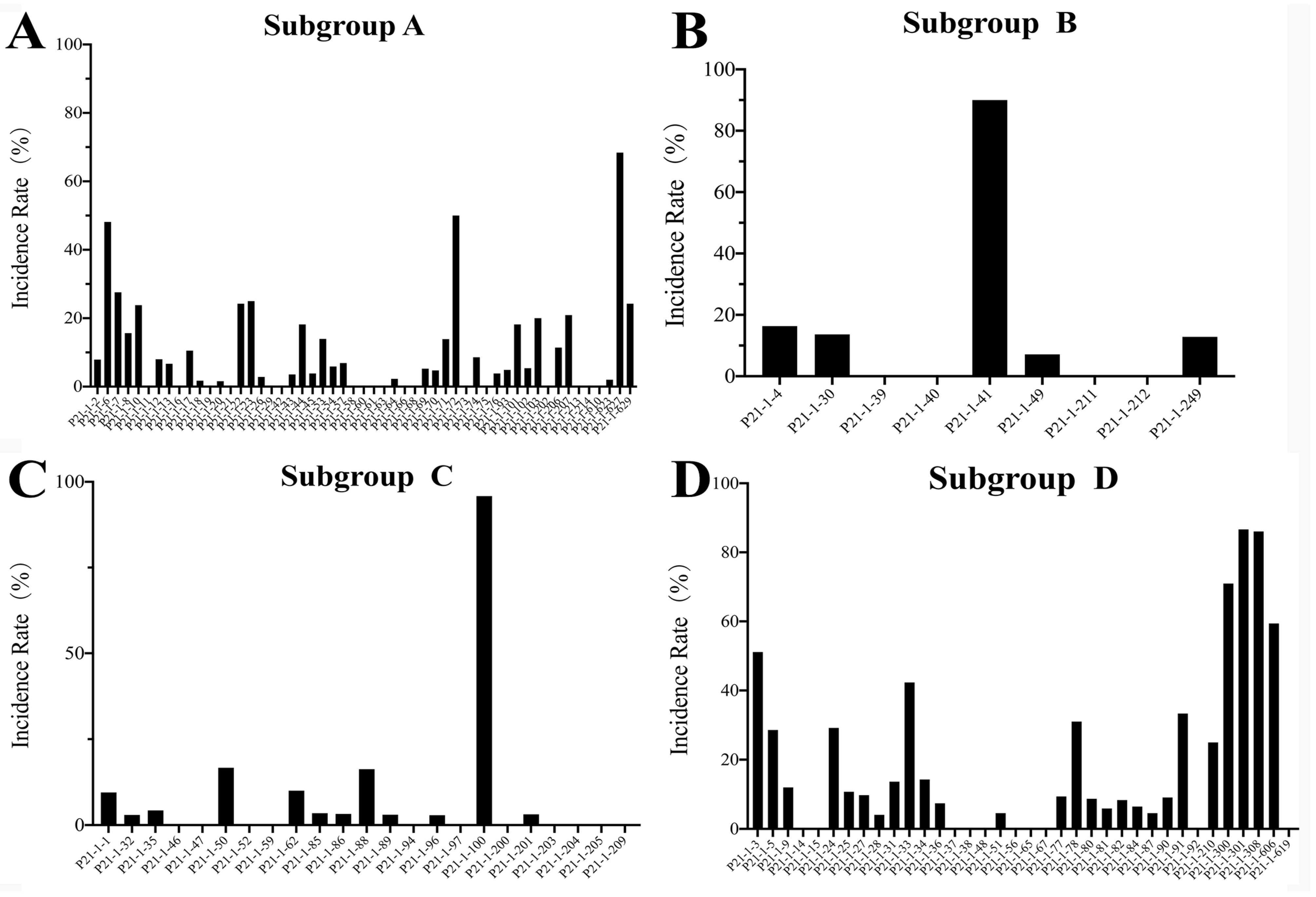
3.6. Selective Deletion Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piao, Z.; Ramchiary, N.; Lim, Y.P. Genetics of clubroot resistance in Brassica species. J. Plant Growth Regul. 2009, 28, 252–264. [Google Scholar] [CrossRef]
- Dixon, G.R. The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. J. Plant Growth Regul. 2009, 28, 194–202. [Google Scholar] [CrossRef]
- Hwang, S.-F.; Strelkov, S.E.; Feng, J.; Gossen, B.D.; Howard, R.J. Plasmodiophora Brassicae: A review of an emerging pathogen of the Canadian canola (Brassica napus) crop: Progress on canola clubroot research. Mol. Plant Pathol. 2012, 13, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Matsumoto, E.; Aruga, D.; Kitagawa, S.; Matsumura, H.; Hayashida, N. Molecular characterization of the CRa gene conferring clubroot resistance in Brassica rapa. Plant Mol. Biol. 2012, 80, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, K.; Suwabe, K.; Tomita, R.; Kato, T.; Nunome, T.; Fukuoka, H.; Matsumoto, S. Identification and characterization of Crr1a, a gene for resistance to clubroot disease (Plasmodiophora brassicae woronin) in Brassica rapa L. PLoS ONE 2013, 8, e54745. [Google Scholar] [CrossRef]
- Wallenhammar, A. Prevalence of Plasmodiophora brassicae in a spring oilseed rape growing area in central sweden and factors influencing soil infestation levels. Plant Pathol. 1996, 45, 710–719. [Google Scholar] [CrossRef]
- Diederichsen, E.; Frauen, M.; Linders, E.G.A.; Hatakeyama, K.; Hirai, M. Status and perspectives of clubroot resistance breeding in crucifer crops. J. Plant Growth Regul. 2009, 28, 265–281. [Google Scholar] [CrossRef]
- Wilson, E.O. The Biological Diversity Crisis: Despite unprecedented extinction rates, the extent of biological diversity remains unmeasured. BioScience 1985, 35, 700–706. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.; Skolnick, M.; Davis, R. Construction of a genetic-linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Williams, J.G.K.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [Green Version]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Vandelee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M.; et al. AFLP—A new technique for DNA-fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic Distribution, Putative Functions and Mutational Mechanisms: A Review: Microsatellite evolution. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Reyes, V.P.; Angeles-Shim, R.B.; Mendioro, M.S.; Manuel, M.C.C.; Lapis, R.S.; Shim, J.; Sunohara, H.; Nishiuchi, S.; Kikuta, M.; Makihara, D.; et al. Marker-assisted introgression and stacking of major QTLs controlling grain number (Gn1a) and number of primary branching (WFP) to NERICA cultivars. Plants 2021, 10, 844. [Google Scholar] [CrossRef]
- Kitony, J.K.; Sunohara, H.; Tasaki, M.; Mori, J.-I.; Shimazu, A.; Reyes, V.P.; Yasui, H.; Yamagata, Y.; Yoshimura, A.; Yamasaki, M.; et al. Development of an Aus-derived nested association mapping (Aus-NAM) population in rice. Plants 2021, 10, 1255. [Google Scholar] [CrossRef]
- Reyes, V.P.; Kitony, J.K.; Nishiuchi, S.; Makihara, D.; Doi, K. Utilization of genotyping-by-sequencing (GBS) for rice pre-breeding and improvement: A review. Life 2022, 12, 1752. [Google Scholar] [CrossRef]
- Song, L.; Cao, B.; Zhang, Y.; Meinhardt, L.W.; Zhang, D. Mining Single Nucleotide Polymorphism (SNP) markers for accurate genotype identification and diversity analysis of chinese jujube (Ziziphus Jujuba Mill.) germplasm. Agronomy 2021, 11, 2303. [Google Scholar] [CrossRef]
- Yu, Z.; Fredua-Agyeman, R.; Hwang, S.-F.; Strelkov, S.E. Molecular genetic diversity and population structure analyses of rutabaga accessions from nordic countries as revealed by single nucleotide polymorphism markers. BMC Genomics 2021, 22, 442. [Google Scholar] [CrossRef]
- Abdel-Haleem, H.; Luo, Z.; Szczepanek, A. Genetic diversity and population structure of the USDA collection of Brassica juncea L. Ind. Crops Prod. 2022, 187, 115379. [Google Scholar] [CrossRef]
- Tanhuanpää, P.; Erkkilä, M.; Tenhola-Roininen, T.; Tanskanen, J.; Manninen, O. SNP diversity within and among Brassica rapa accessions reveals no geographic differentiation. Genome 2016, 59, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, Y.; Han, F.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H.; Wang, Y.; Ji, J.; et al. Genetic diversity and population structure analysis of 161 broccoli cultivars based on SNP markers. Hortic. Plant J. 2021, 7, 423–433. [Google Scholar] [CrossRef]
- Yousef, E.A.A.; Müller, T.; Börner, A.; Schmid, K.J. Comparative analysis of genetic diversity and differentiation of cauliflower (Brassica oleracea var. botrytis) accessions from two ex situ genebanks. PLoS ONE 2018, 13, e0192062. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and WANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helyar, S.J.; Hemmer-Hansen, J.; Bekkevold, D.; Taylor, M.I.; Ogden, R.; Limborg, M.T.; Cariani, A.; Maes, G.E.; Diopere, E.; Carvalho, G.R.; et al. Application of SNPs for population genetics of nonmodel organisms: New opportunities and challenges. Mol. Ecol. Resour. 2011, 11, 123–136. [Google Scholar] [CrossRef]
- Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-sequencing in plants. Biology 2012, 1, 460–483. [Google Scholar] [CrossRef] [Green Version]
- Stoeckel, S.; Grange, J.; Fernández-Manjarres, J.F.; Bilger, I.; Frascaria-Lacoste, N.; Mariette, S. Heterozygote excess in a self-incompatible and partially clonal forest tree species—Prunus Avium L. Mol. Ecol. 2006, 15, 2109–2118. [Google Scholar] [CrossRef]
- Navascués, M.; Stoeckel, S.; Mariette, S. Genetic diversity and fitness in small populations of partially asexual, self-incompatible plants. Heredity 2010, 104, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, Y.; Wan, Y.; Li, Z.; Ma, H. Genetic diversity of Phyllanthus emblica from two different climate type areas. Front. Plant Sci. 2020, 11, 580812. [Google Scholar] [CrossRef]
- González-Martínez, S.; Gerber, S.; Cervera, M.; Martínez-Zapater, J.; Gil, L.; Alía, R. Seed gene flow and fine-scale structure in a mediterranean pine (Pinus pinaster ait.) using nuclear microsatellite markers. Theor. Appl. Genet. 2002, 104, 1290–1297. [Google Scholar] [CrossRef]
- Fu, Q.; Lu, G.; Fu, Y.; Wang, Y. Genetic differentiation between two varieties of Oreocharis benthamii (Gesneriaceae) in sympatric and allopatric regions. Ecol. Evol. 2020, 10, 7792–7805. [Google Scholar] [CrossRef]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [Green Version]
- Gross, B.L.; Olsen, K.M. Genetic perspectives on crop domestication. Trends Plant Sci. 2010, 15, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Sezen, U.; Paterson, A.H. Domestication and plant genomes. Curr. Opin. Plant Biol. 2010, 13, 160–166. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Widmer, A.; Arntz, A.M.; Burke, J.M. Directional selection is the primary cause of phenotypic diversification. Proc. Natl. Acad. Sci. USA 2002, 99, 12242–12245. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, E.; Yasui, C.; Ohi, M.; Tsukada, M. Linkage analysis of RFLP markers for clubroot resistance and pigmentation in Chinese cabbage (Brassica rapa ssp. pekinensis). Euphytica 1998, 104, 79–86. [Google Scholar] [CrossRef]
- Hayashida, N.; Takabatake, Y.; Nakazawa, N.; Arugal, D.; Nakanishi, H.; Taguchi, G.; SakaMot, K.; Matsumoto, E. Construction of a practical SCAR marker linked to clubroot resistance in Chinese cabbage, with intensive analysis of HC352b genes. J. Jpn. Soc. Hortic. Sci. 2008, 77, 150–154. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Kubo, N.; Matsumoto, S.; Suwabe, K.; Tsukada, M.; Hirai, M. Fine mapping of the clubroot resistance gene, Crr3, in Brassica rapa. Theor. Appl. Genet. 2006, 114, 81–91. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, X.; Peng, G.; Falk, K.C.; Strelkov, S.E.; Gossen, B.D. Genotyping-by-sequencing reveals three QTL for clubroot resistance to six pathotypes of Plasmodiophora brassicae in Brassica rapa. Sci. Rep. 2017, 7, 4516. [Google Scholar] [CrossRef]
- Pang, W.; Fu, P.; Li, X.; Zhan, Z.; Yu, S.; Piao, Z. Identification and mapping of the clubroot resistance gene CRd in Chinese cabbage (Brassica rapa ssp. pekinensis). Front. Plant Sci. 2018, 9, 653. [Google Scholar] [CrossRef] [PubMed]
- Laila, R.; Park, J.-I.; Robin, A.H.K.; Natarajan, S.; Vijayakumar, H.; Shirasawa, K.; Isobe, S.; Kim, H.-T.; Nou, I.-S. Mapping of a novel clubroot resistance QTL using DdRAD-seq in Chinese cabbage (Brassica rapa L.). BMC Plant Biol. 2019, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.; Godiard, L.; Straube, E.; Ashfield, T.; Lewald, J.; Sattler, A.; Innes, R.; Dangl, J. Structure of the Arabidopsis RPM1 gene enabling dual-specificity disease resistance. Science 1995, 269, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.B.; Ji, W.Q. Cloning and characterization of disease resistance protein RPM1 genes against powdery mildew in wheat line N9134. Cereal Res. Commun. 2019, 47, 473–483. [Google Scholar] [CrossRef]
- Lapin, D.; Bhandari, D.; Parker, J. Origins and immunity networking functions of EDS1 family proteins. Annu. Rev. Phytopathol. 2020, 58, 253–276. [Google Scholar] [CrossRef]
- Dongus, J.A.; Parker, J.E. EDS1 signalling: At the nexus of intracellular and surface receptor immunity. Curr. Opin. Plant Biol. 2021, 62, 102039. [Google Scholar] [CrossRef]
- Feng, J.; Hwang, S.; Strelkov, S. Studies into primary and secondary infection processes by Plasmodiophora brassicae on canola. Plant Pathol. 2013, 62, 177–183. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
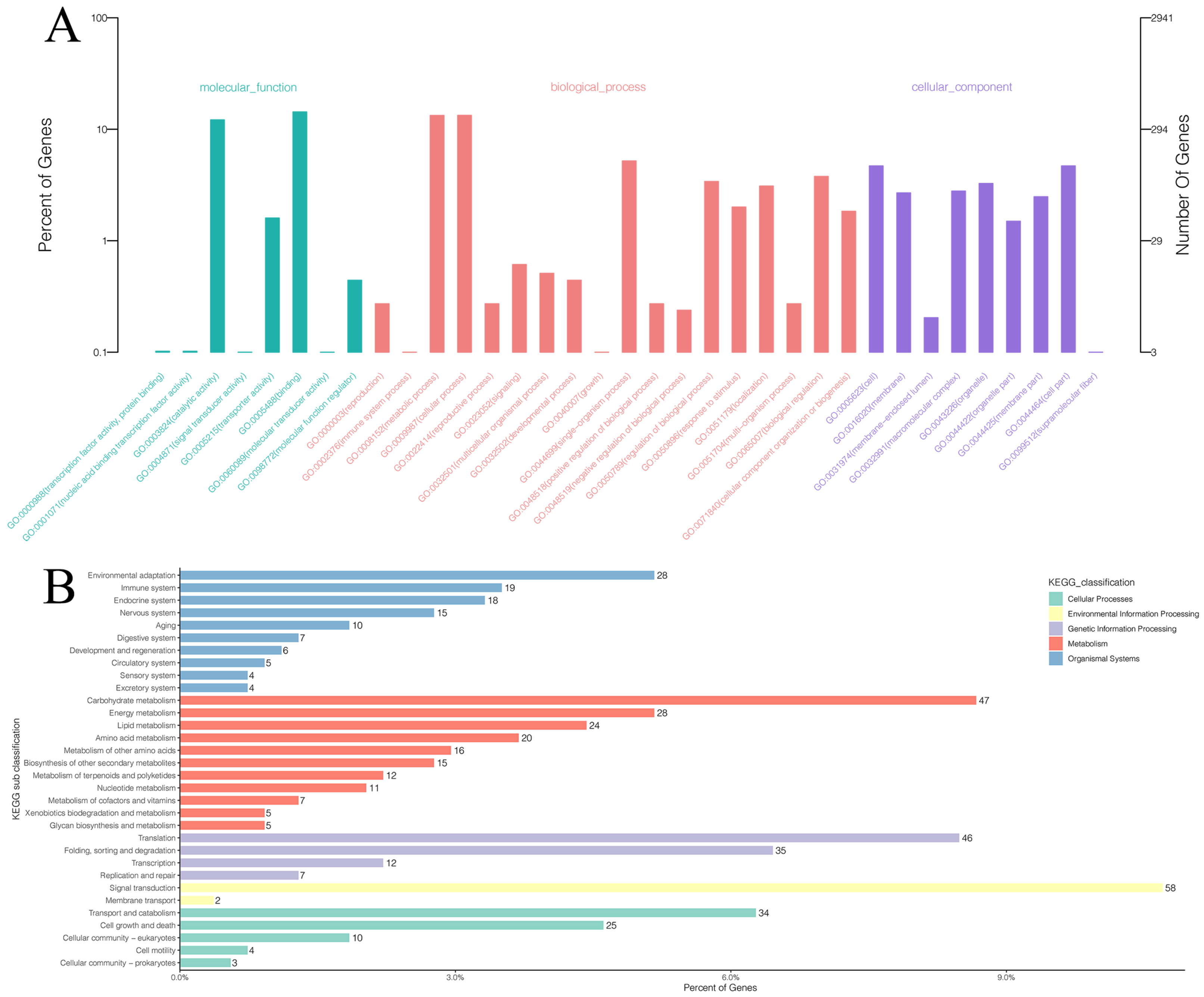{kind=link}
| Maximum Value | Minimum Value | Average Value | ||
|---|---|---|---|---|
| Reads statistic | Raw reads | 34,566,744 | 12,680,398 | 16,794,360 |
| Raw data bases | 5,185,011,600 | 1,902,059,700 | 2,519,154,000 | |
| Clean reads | 34,538,110 | 12,672,024 | 16,784,963 | |
| Clean data bases | 5,113,683,916 | 1,871,020,512 | 2,484,015,380 | |
| Clean data Q20 (%) | 97.95 | 94.79 | 96.17 | |
| Clean data Q30 (%) | 94.03 | 85.13 | 89.00 | |
| Clean data GC (%) | 39.43 | 37.34 | 38.19 | |
| Mapped reads statistic | Total Reads | 35,124,117 | 12,871,646 | 17,038,713 |
| Map Reads | 34,762,929 | 12,649,523 | 16,750,465 | |
| Map Rate | 99.12% | 91.31% | 98.31% | |
| UniqMap Rate | 95.96% | 87.69% | 94.54% | |
| Sequencing depth | Coverage (%) | 90.53 | 77.02 | 83.27 |
| Coverage 5X (%) | 71.81 | 21.60 | 37.15 | |
| Coverage 10X (%) | 34.65 | 3.60 | 7.74 | |
| Depth | 13.85 | 4.92 | 6.75 | |
| Coverage Base | 319,702,365 | 271,985,352 | 294,071,230 |
| Sample | Maximum Value | Sample | Minimum Value | Average Value | ||
|---|---|---|---|---|---|---|
| SNP | Ref | P21-1-88 | 132,447 | P21-1-5 | 40,558 | 99,270 |
| Alt (Homo) | P21-1-80 | 18,230 | P21-1-86 | 6610 | 13,847 | |
| Het | P21-1-249 | 51,452 | P21-1-5 | 10,189 | 28,723 | |
| Miss | P21-1-5 | 118,656 | P21-1-249 | 3013 | 11,801 | |
| Total | P21-1-5 | 177,601 | P21-1-249 | 136,896 | 153,641 | |
| INDEL | Ref | P21-1-88 | 23,499 | P21-1-5 | 7875 | 18,460 |
| Alt (Homo) | P21-1-80 | 1841 | P21-1-86 | 778 | 1410 | |
| Het | P21-1-249 | 5288 | P21-1-5 | 1005 | 2692 | |
| Miss | P21-1-5 | 19,125 | P21-1-249 | 350 | 1657 | |
| Total | P21-1-5 | 28,865 | P21-1-249 | 20,525 | 24,219 |
| Subgroup | Num_Indv | Obs_Het | Obs_Hom | Exp_Het | Exp_Hom | Pi | Fis |
|---|---|---|---|---|---|---|---|
| A | 50.77558 | 0.28931 | 0.71069 | 0.28849 | 0.71151 | 0.29138 | 0.07317 |
| B | 8.79188 | 0.2588 | 0.7412 | 0.22567 | 0.77433 | 0.23934 | −0.01984 |
| C | 22.03601 | 0.26015 | 0.73985 | 0.28823 | 0.71177 | 0.29497 | 0.12222 |
| D | 33.86832 | 0.26989 | 0.73011 | 0.31143 | 0.68857 | 0.31612 | 0.15415 |
| A | B | C | D | |
|---|---|---|---|---|
| A | 0.0455735 | 0.040152 | 0.0276534 | |
| B | 0.0744347 | 0.0447235 | ||
| C | 0.0321242 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, L.; Xi, D.; Li, X.; Zhang, C.; Gao, L.; Li, Y.; Zhu, Y.; Zhu, H. Genetic Diversity Analysis of Non-Heading Chinese Cabbage of Resistance to Clubroot Disease Based on SNP Molecular Markers. Agronomy 2023, 13, 245. https://doi.org/10.3390/agronomy13010245
Miao L, Xi D, Li X, Zhang C, Gao L, Li Y, Zhu Y, Zhu H. Genetic Diversity Analysis of Non-Heading Chinese Cabbage of Resistance to Clubroot Disease Based on SNP Molecular Markers. Agronomy. 2023; 13(1):245. https://doi.org/10.3390/agronomy13010245
Chicago/Turabian StyleMiao, Liming, Dandan Xi, Xiaofeng Li, Changwei Zhang, Lu Gao, Ying Li, Yuying Zhu, and Hongfang Zhu. 2023. "Genetic Diversity Analysis of Non-Heading Chinese Cabbage of Resistance to Clubroot Disease Based on SNP Molecular Markers" Agronomy 13, no. 1: 245. https://doi.org/10.3390/agronomy13010245