
Comparative Analysis of Transcriptome Profiles Reveals the Mechanisms in the Difference of Low Potassium Tolerance among Cultivated and Tibetan Wild Barleys

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Plant Growth
2.2. RNA-Seq Sample Collection and Preparation
2.3. DEG Identification, GO Annotation, and KEGG Pathway Analysis
2.4. Statistical Analysis
3. Results
3.1. Evaluation of RNA-Seq Reads and Mapping Results
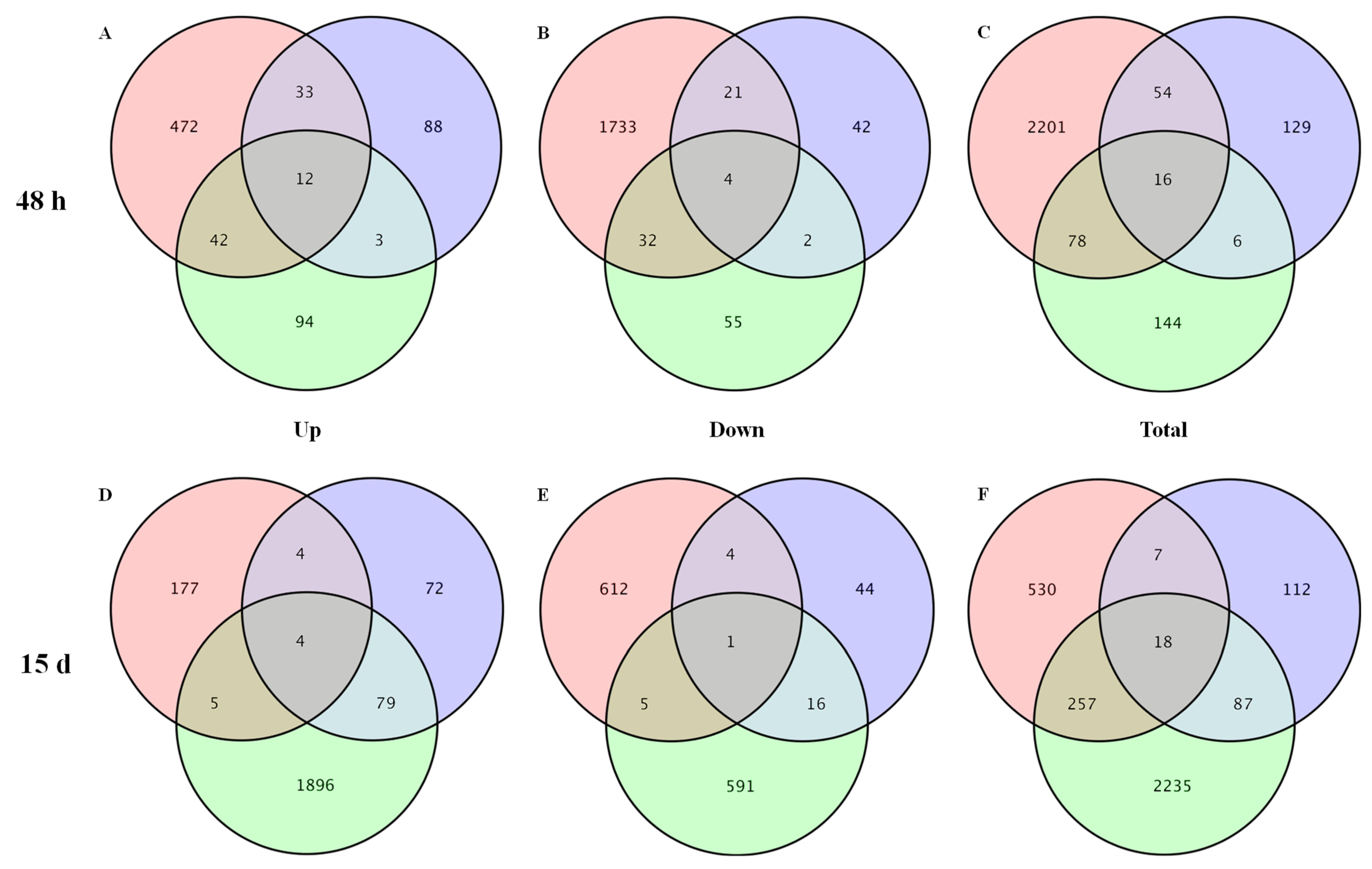
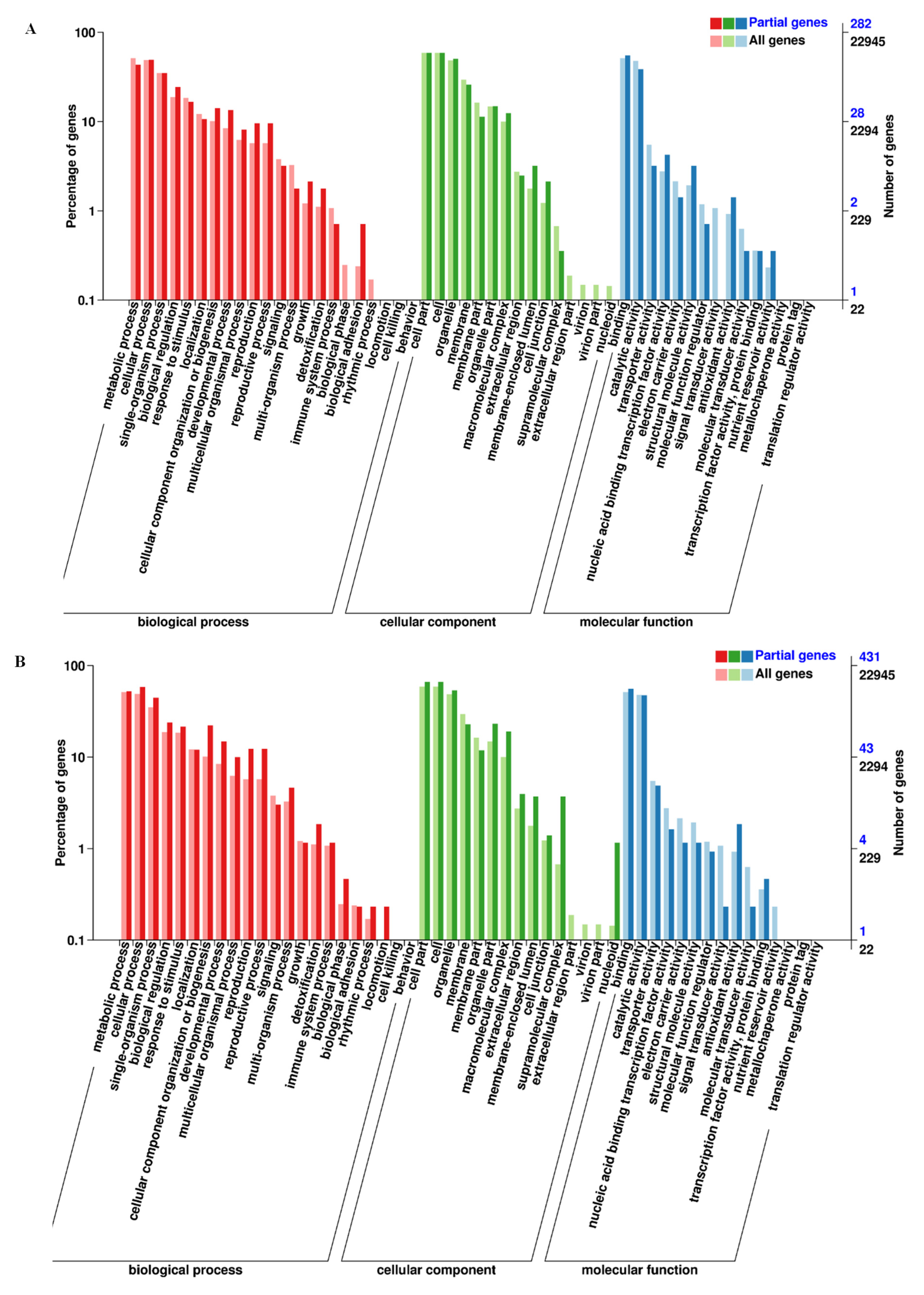
3.2. DEGs Identification, GO Function, and KEGG Analysis
3.3. DEGs Associated with Transcription Factors (TFs), Transporters, and Kinases
3.4. DEGs Involved in K Transposition and Ca2+ Signaling Pathway
3.5. DEGs Involved in the Ethylene Biosynthesis Process
3.6. The DEGs Involved in the Four Interrelated Metabolic Pathways
3.7. DEGs Associated with Photosynthesis
4. Discussion
4.1. K Channels and Ca2+ Signaling Pathway Contribute to LK Stress Response in XZ153
4.2. Ethylene Biosynthesis Process May Benefit to LK Stress Response
4.3. The TCA Cycle, Glycolysis, and PPP Are Closely Related to LK Tolerant Stress
4.4. The DEGs Involved in Photosynthesis Contribute to LK Tolerance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Wu, W.H. Genetic approaches for improvement of the crop potassium acquisition and utilization efficiency. Curr. Opin. Plant Biol. 2015, 25, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengel, Z.; Damon, P.M. Crops and genotypes differ in efficiency of potassium uptake and use. Physiol. Plant 2008, 133, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Nevo, E.; Wu, D.Z.; Comadran, J.; Zhou, M.X.; Qiu, L.; Chen, Z.H.; Beiles, A.; Chen, G.X.; Zhang, G.P. Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. USA 2012, 109, 16969–16973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.G.; Wu, D.Z.; Jabeen, Z.; Huang, Y.Q.; Huang, Y.C.; Zhang, G.P. Genome-wide association analysis of aluminum tolerance in cultivated and Tibetan wild barley. PLoS ONE 2013, 8, e69776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.Y.; Zeng, J.B.; Cao, F.B.; Ahmed, I.M.; Zhang, G.P.; Vincze, E.; Wu, F.B. HvEXPB7, a novel β-expansin gene revealed by the root hair transcriptome of Tibetan wild barley, improves root hair growth under drought stress. J. Exp. Bot. 2015, 66, 7405–7419. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.Z.; Shen, Q.F.; Cai, S.G.; Chen, Z.H.; Dai, F.; Zhang, G.P. Ionomic responses and correlations between elements and metabolites under salt stress in wild and cultivated barley. Plant Cell Physiol. 2013, 54, 1976–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.Z.; Sato, K.; Ma, J.F. Genome-wide association mapping of cadmium accumulation in different organs of barley. New Phytol. 2015, 208, 817–829. [Google Scholar] [CrossRef]
- Zhu, B.; Zeng, J.B.; Wu, D.Z.; Cai, S.G.; Yang, L.N.; Zhang, G.P. Identification and physiological characterization of low potassium tolerant germplasm in Tibetan Plateau annual wild barley. J. Zhejiang Univ. (Agric. Life Sci.) 2014, 40, 165–174. [Google Scholar]
- Ye, Z.L.; Zeng, J.B.; Li, X.; Zeng, F.R.; Zhang, G.P. Physiological characterizations of three barley genotypes in response to low potassium stress. Acta Physiol. Plant. 2017, 39, 232. [Google Scholar] [CrossRef]
- Zeng, J.B.; Quan, X.Y.; He, X.Y.; Cai, S.G.; Ye, Z.L.; Chen, G.; Zhang, G.P. Root and leaf metabolite profiles analysis reveals the adaptive strategies to low potassium stress in barley. BMC Plant Biol. 2018, 18, 187. [Google Scholar] [CrossRef]
- Ye, Z.L.; Zeng, J.B.; Ma, X.Y.; Long, L.Z.; Zhang, G.P. Transcriptome profilling analysis reveals involvement of SAM cycle and methionine pathway in low potassium tolerance in barley. Curr. Plant Biol. 2021, 25, 100190. [Google Scholar] [CrossRef]
- Zeng, J.B.; He, X.Y.; Wu, D.Z.; Zhu, B.; Cai, S.G.; Nadira, U.A.; Jabeen, Z.; Zhang, G.P. Comparative transcriptome profiling of two Tibetan wild barley genotypes in responses to low potassium. PLoS ONE 2014, 9, e100567. [Google Scholar] [CrossRef] [Green Version]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-Seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, O.A.; Shao, J.; Nemchinov, L.G. Analysis of the Alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol. 2013, 54, 1041–1055. [Google Scholar] [CrossRef]
- Hübner, S.; Korol, A.B.; Schmid, K.J. RNA-Seq analysis identifies genes associated with differential reproductive success under drought-stress in accessions of wild barley Hordeum spontaneum. BMC Plant Biol. 2015, 15, 134. [Google Scholar] [CrossRef] [Green Version]
- Gleason, L.U.; Burton, R.S. RNA-seq reveals regional differences in transcriptome response to heat stress in the marine snail Chlorostoma funebralis. Mol. Ecol. 2015, 24, 610–627. [Google Scholar] [CrossRef]
- Wu, L.Y.; Yu, J.H.; Shen, Q.F.; Huang, L.; Wu, D.Z.; Zhang, G.P. Identification of microRNAs in response to aluminum stress in the roots of Tibetan wild barley and cultivated barley. BMC Genom. 2018, 19, 560. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Yadav, B.S.; Raj, U.; Freilich, S.; Varadwaj, P.K. Transcriptomic analysis of soil Grown T. aestivum cv. root to reveal the changes in expression of genes in response to multiple nutrients deficiency. Front. Plant Sci. 2017, 8, 1025. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.C.; Yang, X.Y.; Yu, H.J.; Jiang, W.J.; Sun, N.; Liu, X.R.; Liu, X.L.; Zhang, X.M.; Wang, Y.; Gu, X.F. RNA-Seq-based transcriptome profiling of early nitrogen deficiency response in cucumber seedlings provides new insight into the putative nitrogen regulatory network. Plant Cell Physiol. 2015, 56, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Long, L.Z.; Ma, X.Y.; Ye, L.Z.; Zeng, J.B.; Chen, G.; Zhang, G.P. Root plasticity and Pi recycling within plants contribute to low-P tolerance in Tibetan wild barley. BMC Plant Biol. 2019, 19, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Yang, S.S.; Miller, S.S.; Bucciarelli, B.; Liu, J.Q.; Rydeen, A.; Bozsoki, Z.; Uhde-Stone, C.; Tu, Z.J.; Allan, D.; et al. An RNASeq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol. 2013, 161, 705–724. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, W.H. Potassium transport and signaling in higher plants. Annu. Rev. Plant Biol. 2013, 64, 451–476. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.P.; Takeshi, Y.; Kondou, Y.; Schachtman, D.P.; Matsui, M.; Shin, R. Identification and characterization of transcription factors regulating Arabidopsis HAK5. Plant Cell Physiol. 2013, 54, 1478–1490. [Google Scholar] [CrossRef] [Green Version]
- Kwak, J.M.; Murata, Y.; Baizabal-Aguirre, V.M.; Merrill, J.; Wang, M.; Kemper, A.; Hawke, S.D.; Tallman, G.; Schroeder, J.I. Dominant negative guard cell K+ channel mutants reduce inward-rectifying K+ currents and light-induced stomatal opening in Arabidopsis. Plant Physiol. 2001, 27, 473–485. [Google Scholar] [CrossRef]
- Szyroki, A.; Ivashikina, N.; Dietrich, P.; Roelfsema, M.R.G.; Ache, P.; Reintanz, B.; Deeken, R.; Godde, M.; Felle, H.; Steinmeyer, R.; et al. KAT1 is not essential for stomatal opening. Proc. Natl. Acad. Sci. USA 2001, 98, 2917–2921. [Google Scholar] [CrossRef] [Green Version]
- Ache, P.; Becker, D.; Ivashikina, N.; Dietrich, P.; Roelfsema, M.R.G.; Hedrich, R. GORK, a delayed outward rectifier expressed in guard cells of Arabidopsis thaliana, is a K+-selective, K+-sensing ion channel. FEBS Lett. 2000, 486, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Hosy, E.; Vavasseur, A.; Mouline, K.; Dreyer, I.; Gaymard, F.; Poree, F.; Boucherez, J.; Lebaudy, A.; Bouchez, D.; Very, A.A.; et al. The Arabidopsis outward K+ channel GORK is involved in regulation of stomatal movements and plant transpiration. Proc. Natl. Acad. Sci. USA 2003, 100, 5549–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signaling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bowler, C.; Fluhr, R. The role of calcium and activated oxygens as signals for controlling cross-tolerance. Trends Plant Sci. 2000, 5, 241–246. [Google Scholar] [CrossRef]
- Valentina, L.V.; Paola, D.; Alessandra, A. Towards understanding plant calcium signaling through calmodulin-like proteins: A biochemical and structural perspective. Int. J. Mol. Sci. 2018, 19, 1331. [Google Scholar]
- Cheong, Y.H.; Pandey, G.K.; Grant, J.J.; Batistic, O.; Li, L.; Kim, B.G.; Lee, S.C.; Kudla, J.; Luan, S. Two calcineurin B-like calcium sensors, interacting with protein kinase CIPK23, regulate leaf transpiration and root potassium uptake in Arabidopsis. Plant J. 2007, 52, 223–239. [Google Scholar] [CrossRef]
- Zhu, X.; Dunand, C.; Snedden, W.; Galaud, J.P. CaM and CML emergence in the green lineage. Trends Plant Sci. 2015, 20, 483–489. [Google Scholar] [CrossRef]
- Merchante, C.; Alonso, J.M.; Stepanova, A.N. Ethylene signaling: Simple ligand, complex regulation. Curr. Opin. Plant Biol. 2013, 16, 554–560. [Google Scholar] [CrossRef]
- Shin, R.; Schachtman, D.P. Hydrogen peroxide mediates plant root cell response to nutrient deprivation. Proc. Natl. Acad. Sci. USA 2004, 101, 8827–8832. [Google Scholar] [CrossRef] [Green Version]
- Giovanelli, J.; Mudd, S.H.; Datko, A.H. Quantitative analysis of pathways of methionine metabolism and their regulation in Lemna. Plant Physiol. 1985, 78, 555–560. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Ruzicka, D.; Shin, R.; Schachtman, D.P. The Arabidopsis AP2/ERF transcription factor RAP2.11 modulates plant response to low-potassium conditions. Mol. Plant 2012, 5, 1042–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.L.; Zeng, J.B.; Long, L.Z.; Ye, L.Z.; Zhang, G.P. Identification of microRNAs in response to low potassium stress in the shoots of Tibetan wild barley and cultivated barley. Curr. Plant Biol. 2020, 25, 100193. [Google Scholar] [CrossRef]
- Ye, Z.L.; Zeng, J.B.; Ye, L.Z.; Long, L.Z.; Zhang, G.P. Genome-wide association analysis of potassium uptake and translocation rates under low K stress in Tibetan wild barley. Euphytica 2020, 216, 35. [Google Scholar] [CrossRef]
- Schnarrenberger, C.; Martin, W. Evolution of the enzymes of the citric acid cycle and the glyoxylate cycle of higher plants. Eur. J. Biochem. 2002, 269, 868–883. [Google Scholar] [CrossRef] [Green Version]
- Millar, A.H.; Knorpp, C.; Leaver, C.J.; Hill, S.A. Plant mitochondrial pyruvate dehydrogenase complex: Purification and identification of catalytic components in potato. Biochem. J. 1998, 334, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Timm, S.; Wittmiss, M.; Gamlien, S.; Ewald, R.; Florian, A.; Frank, M.; Wirtz, M.; Hell, R.; Fernie, A.R.; Bauwea, H. Mitochondrial Dihydrolipoyl Dehydrogenase Activity Shapes Photosynthesis and Photorespiration of Arabidopsis thaliana. Plant Cell 2015, 27, 1968–1984. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.; Harada, Y.; Yamatoya, K.; Kawano, N.; Kanai, S.; Miyamoto, Y.; Nakamura, A.; Miyado, M.; Hayashi, Y.; Kuroki, Y.; et al. Correction, Extra-mitochondrial citrate synthase initiates calcium oscillation and suppresses age-dependent sperm dysfunction. Lab. Investig. 2020, 100, 665. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.M.; Yang, X.; Bie, J.T.; Wang, Z.; Liu, M.M.; Li, Y.T.; Shao, G.Z.; Luo, J.Y. Citrate synthase desuccinylation by SIRT5 promotes colon cancer cell proliferation and migration. Biol. Chem. 2020, 401, 1031–1039. [Google Scholar] [CrossRef]
- Anoop, V.M.; Basu, U.; McCammon, M.T.; McAlister-Henn, L.; Taylor, G.J. Modulation of citrate metabolism alters aluminum tolerance in yeast and transgenic canola overexpressing a mitochondrial citrate synthase. Plant Physiol. 2003, 132, 2205–2217. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Guan, C.F.; Wang, M.K.; Zhang, Y.F.; Ruan, X.F.; Zhang, Q.L.; Luo, Z.R.; Yang, Y. DkWRKY interacts with pyruvate kinase gene DkPK1 and promotes natural deastringency in C-PCNA persimmon. Plant Sci. 2020, 290, 110285. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, C.; Johann, P.; Nehls, U.; Hampp, R. Evidence for an up-regulation of the host and a down-regulation of the fungal phosphofructokinase activity in ectomycorrhizas of Norway spruce and fly agaric. New Phytol. 1996, 134, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, A.; Stock, J.; Hess, N.; Aldous, S.; Dreilich, A.; Grimm, B. Characterization of the phosphofructokinase gene family in rice and its expression under oxygen deficiency stress. Front. Plant Sci. 2013, 4, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwell, J.; Gill, A.; Nimmo, G.A.; Wilkins, M.B.; Jenkins, G.L.; Nimmo, H.G. Phosphoenolpyruvate carboxylase kinase is a novel protein kinase regulated at the level of expression. Plant J. 1999, 20, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; DeFraia, C.; Williams, D.; Zhang, X.D.; Mou, Z.L. Characterization of Arabidopsis 6-phosphogluconolactonase T-DNA insertion mutants reveal an essential role for the oxidative section of the plastidic pentose phosphate pathway in plant growth and development. Plant Cell Physiol. 2009, 50, 1277–1291. [Google Scholar] [CrossRef]
- Lebaudy, A.; Vavasseur, A.; Hosy, E.; Hosy, E.; Dreyer, I.; Leonhardt, N.; Thibaud, J.B.; Sentenac, H. Plant adaptation to fluctuating environment and biomass production are strongly dependent on guard cell potassium channels. Proc. Natl. Acad. Sci. USA 2008, 105, 5271–5276. [Google Scholar] [CrossRef] [Green Version]
- Martin, W.F.; Bryant, D.A.; Beatty, J.T. A physiological perspective on the origin and evolution of photosynthesis. FEMS Microbiol. Rev. 2018, 42, 205–231. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Du, Q.; Zhao, Y.; Wang, H.; Li, Y.; Wang, X.; Yu, H. Effects of different potassium stress on leaf photosynthesis and chlorophyll fluorescence in Maize (Zea mays L.) at seedling stage. Agric. Sci. 2016, 7, 44–53. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Groups | Gene Id | Log2 (Fold Change) | Seq Description | |||||
|---|---|---|---|---|---|---|---|---|---|
| XZ153 | XZ141 | ZD9 | |||||||
| 48 h | K channel | MLOC_56043 | 1.08 | up | 1.01 | normal | 0.84 | normal | Potassium channel GORK |
| Ca2+ signaling pathway | MLOC_13902 | 1.09 | up | 0.90 | normal | 0.19 | normal | Probable calcium-binding protein CML29 | |
| 15 d | K channel | MLOC_60763 | 1.47 | up | -- | -- | −0.99 | normal | Potassium channel KAT3 |
| Ca2+ signaling pathway | MLOC_75452 | 2.08 | up | 0.29 | normal | 0.58 | normal | Calmodulin-like protein 1 CML1 | |
| MLOC_6391 | −0.21 | normal | −0.17 | normal | −1.13 | down | Calcium-dependent protein kinase 1 CDPK1 | ||
| Gene Id | Log2 (Fold Change) | Seq Description | |||||
|---|---|---|---|---|---|---|---|
| XZ153 | XZ141 | ZD9 | |||||
| MLOC_70078 | −1.00 | normal | −2.05 | down | −0.91 | normal | 1-aminocyclopropane-1-carboxylate oxidase 1 |
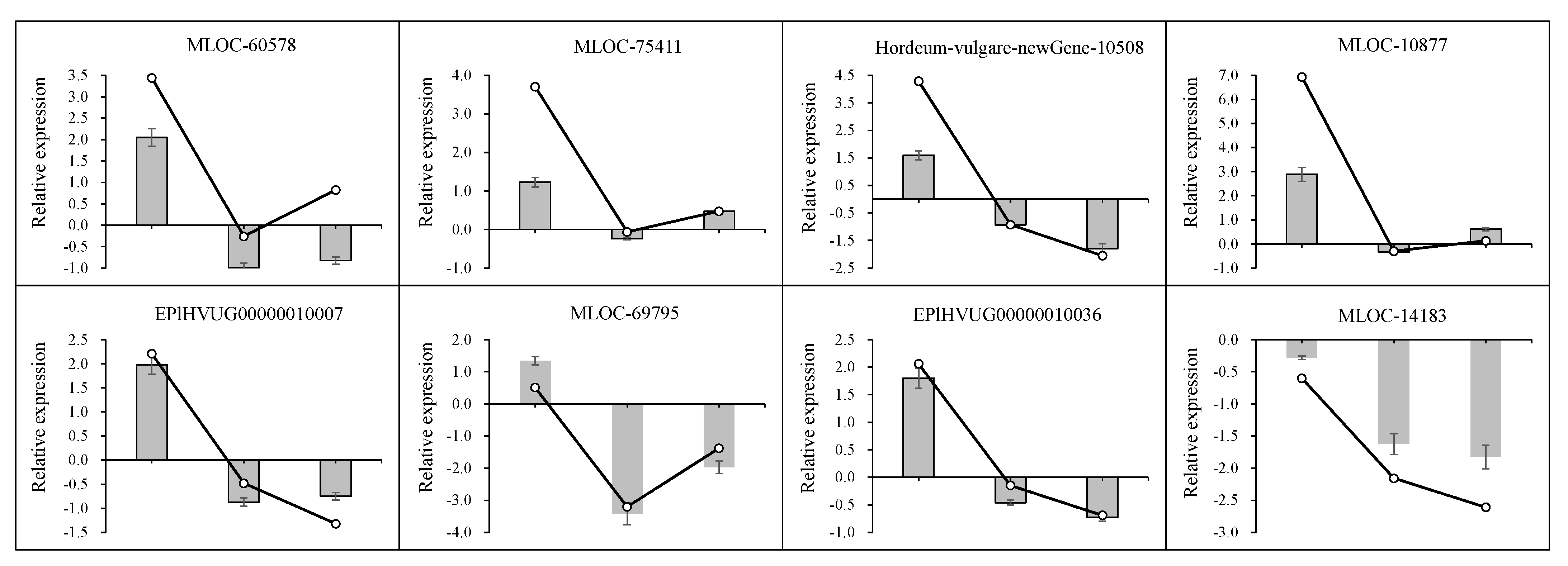
| MLOC_69795 | 0.51 | normal | −3.21 | down | −1.38 | down | S-adenosylmethionine decarboxylase beta chain |
| MLOC_61904 | 0.31 | normal | −0.87 | normal | −1.04 | down | DNA (cytosine-5)-methyltransferase 1B |
| MLOC_11952 | 0.09 | normal | −0.63 | normal | −1.14 | down | DNA (cytosine-5)-methyltransferase 3 |
| Time | Gene Id | Log2 (Fold Change) | Seq Description | |||||
|---|---|---|---|---|---|---|---|---|
| XZ153 | XZ141 | ZD9 | ||||||
| 48 h | NewGene_28417 | 1.61 | up | 0.74 | normal | 2.10 | normal | AP2-like ethylene-responsive transcription factor ANT |
| NewGene_2076 | 1.38 | up | 0.44 | normal | 0.95 | normal | AP2-like ethylene-responsive transcription factor AIL5 | |
| 15 d | NewGene_2076 | 0.30 | normal | −0.54 | normal | −1.02 | down | AP2-like ethylene-responsive transcription factor AIL5 |
| MLOC_10221 | 0.34 | normal | −0.86 | normal | −1.82 | down | AP2-like ethylene-responsive transcription factor ANT | |
| MLOC_63425 | −0.29 | normal | −0.26 | normal | −1.14 | down | AP2-like ethylene-responsive transcription factor ANT | |
| NewGene_28417 | −0.37 | normal | −0.93 | normal | −1.35 | down | AP2-like ethylene-responsive transcription factor ANT | |
| Time | Groups | Gene Id | Log2 (Fold Change) | Seq Description | |||||
|---|---|---|---|---|---|---|---|---|---|
| XZ153 | XZ141 | ZD9 | |||||||
| 48 h | TCA cycle | newGene_13796 | 1.01 | up | 0.15 | normal | −0.24 | normal | ATP-citrate synthase |
| MLOC_10877 | 6.93 | up | −0.30 | normal | 0.13 | normal | Dihydrolipoyl dehydrogenase 1 | ||
| MLOC_7202 | -- | -- | -- | -- | -inf | down | Pyruvate decarboxylase 2 PDC2 | ||
| The PPP | newGene_20892 | 1.20 | up | 0.17 | normal | 0.44 | normal | Phosphogluconate dehydrogenase | |
| 15 d | Glycolysis | newGene_1192 | 1.31 | normal | −0.21 | normal | −3.64 | down | Pyruvate kinase isozyme A |
| newGene_9744 | 0.52 | normal | −0.35 | normal | −1.10 | down | Phosphoglycerate kinase | ||
| MLOC_65676 | 0.23 | normal | −0.09 | normal | −1.29 | down | 6-phosphofructokinase complex | ||
| Gluconeogenesis | MLOC_13152 | −0.41 | normal | −0.95 | normal | −1.07 | down | Phosphoenolpyruvate carboxylase kinase 1 PPCK1 | |
| Time | Gene Id | Log2 (Fold Change) | Seq Description | |||||
|---|---|---|---|---|---|---|---|---|
| XZ153 | XZ141 | ZD9 | ||||||
| 48 h | MLOC_58581 | −0.03 | normal | −7.59 | down | 0.77 | normal | ATP synthase subunit |
| MLOC_38677 | -- | -- | −1.60 | down | -- | -- | Probable NAD(P)H dehydrogenase(quinone) FQR1-like 1 | |
| MLOC_27318 | 2.42 | up | -- | -- | 0.21 | normal | Probable NAD(P)H dehydrogenase(quinone) FQR1-like 2 | |
| MLOC_9033 | 0.03 | normal | −3.10 | down | −0.43 | normal | Light-harvesting complex II chlorophyll a/b binding protein 1 | |
| 15 d | EPlHVUG00000010007 | 2.21 | normal | −0.48 | normal | −1.32 | down | ATP synthase CF1 beta subunit |
| EPlHVUG00000010006 | 1.29 | up | 0.01 | normal | 0.13 | normal | NADH-plastoquinone oxidoreductase subunit 5 | |
| EPlHVUG00000010045 | 1.51 | up | −0.10 | normal | −0.33 | normal | Photosystem II reaction center protein K | |
| EPlHVUG00000010046 | 1.13 | up | 0.19 | normal | −0.29 | normal | Photosystem II reaction center protein I | |
| EPlHVUG00000010036 | 2.06 | up | −0.15 | normal | −0.69 | normal | Photosystem II reaction center protein Z | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; He, X.; Liu, C. Comparative Analysis of Transcriptome Profiles Reveals the Mechanisms in the Difference of Low Potassium Tolerance among Cultivated and Tibetan Wild Barleys. Agronomy 2022, 12, 1094. https://doi.org/10.3390/agronomy12051094
Ye Z, He X, Liu C. Comparative Analysis of Transcriptome Profiles Reveals the Mechanisms in the Difference of Low Potassium Tolerance among Cultivated and Tibetan Wild Barleys. Agronomy. 2022; 12(5):1094. https://doi.org/10.3390/agronomy12051094
Chicago/Turabian StyleYe, Zhilan, Xinyi He, and Chaorui Liu. 2022. "Comparative Analysis of Transcriptome Profiles Reveals the Mechanisms in the Difference of Low Potassium Tolerance among Cultivated and Tibetan Wild Barleys" Agronomy 12, no. 5: 1094. https://doi.org/10.3390/agronomy12051094