

Detection of Hub QTLs Underlying the Genetic Basis of Three Modules Covering Nine Agronomic Traits in an F2 Soybean Population
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Mapping
2.2. Trait Evaluation and Statistical Analysis
2.3. Molecular Marker Identification and Genetic Map Construction
2.4. QTL Analysis
2.5. Prediction of Candidate Genes
3. Results
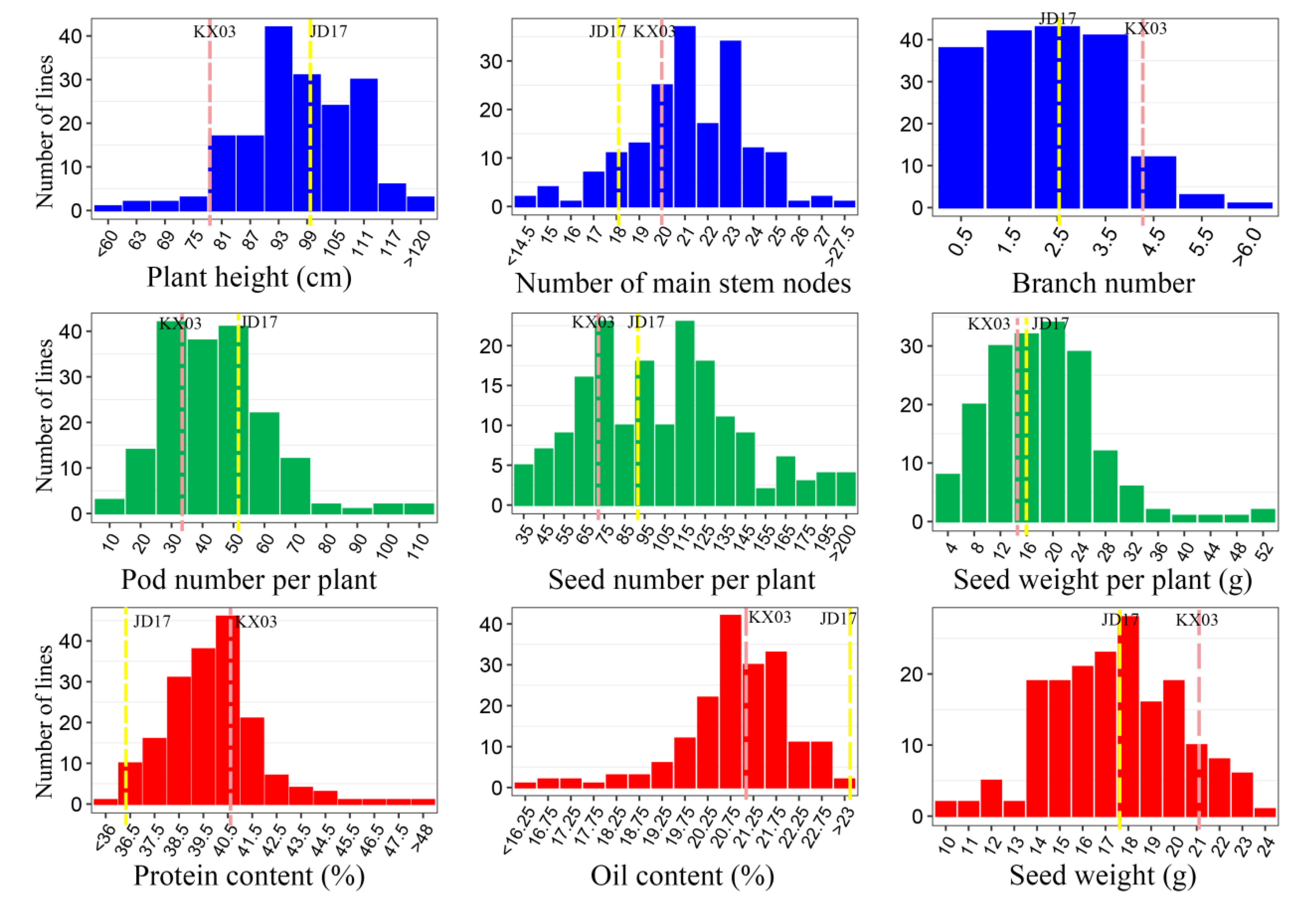
3.1. Variation of Three Trait Modules from Nine Agronomic Traits in the F2 Population
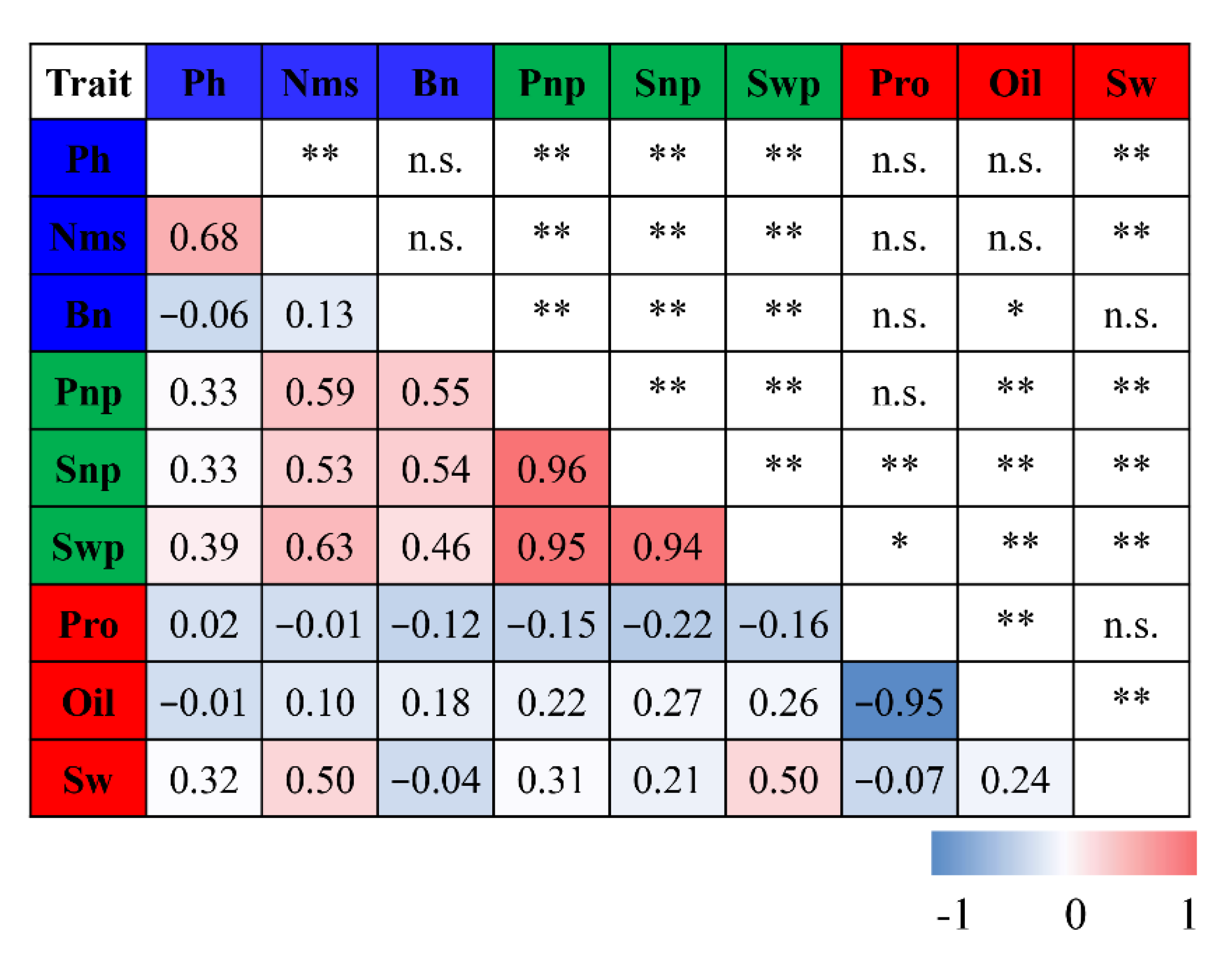
3.2. Correlation Analysis between Different Agronomic Traits
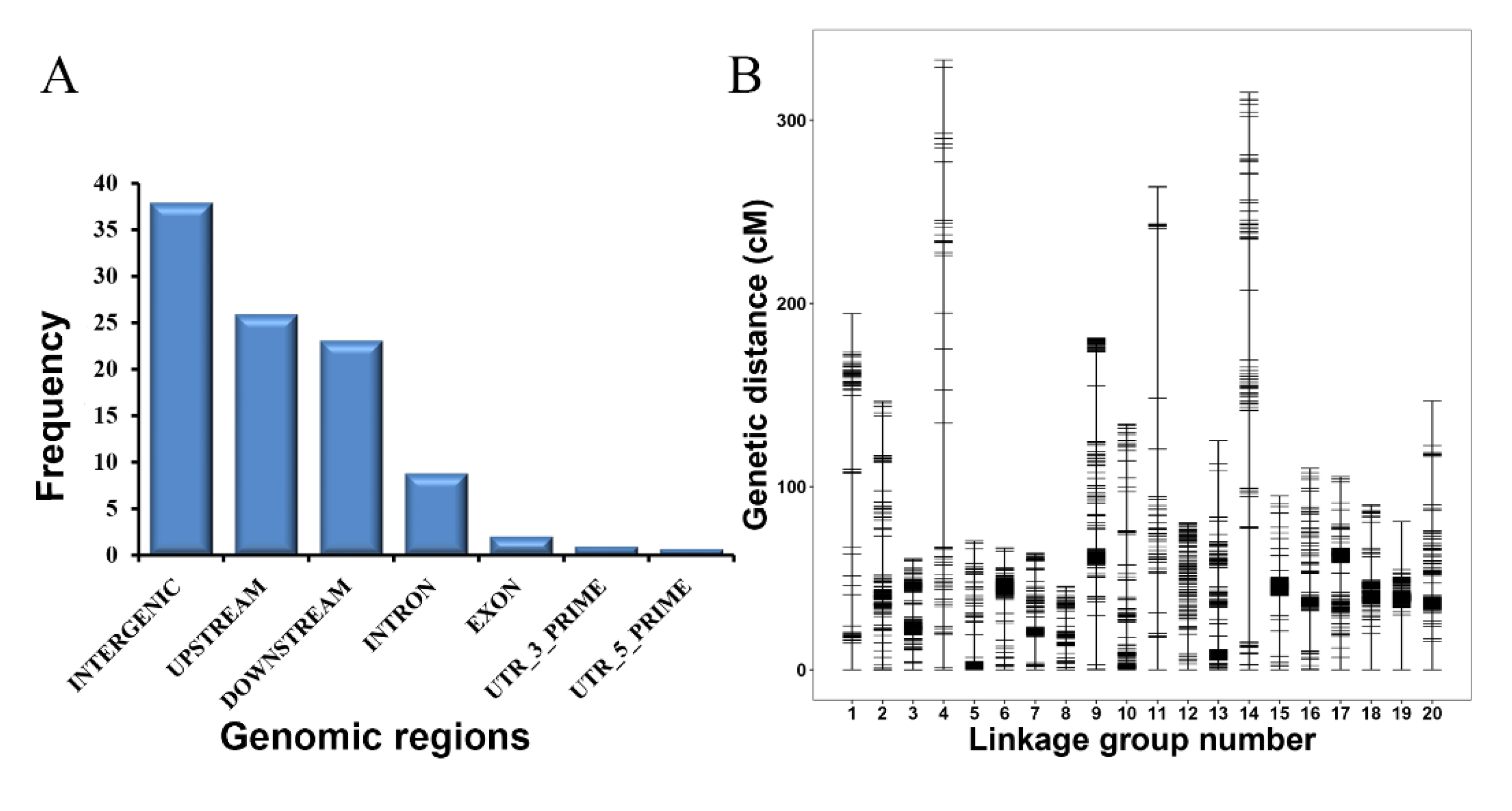
3.3. High-Quality SNP Linkage Map Construction for the KJ F2 Population
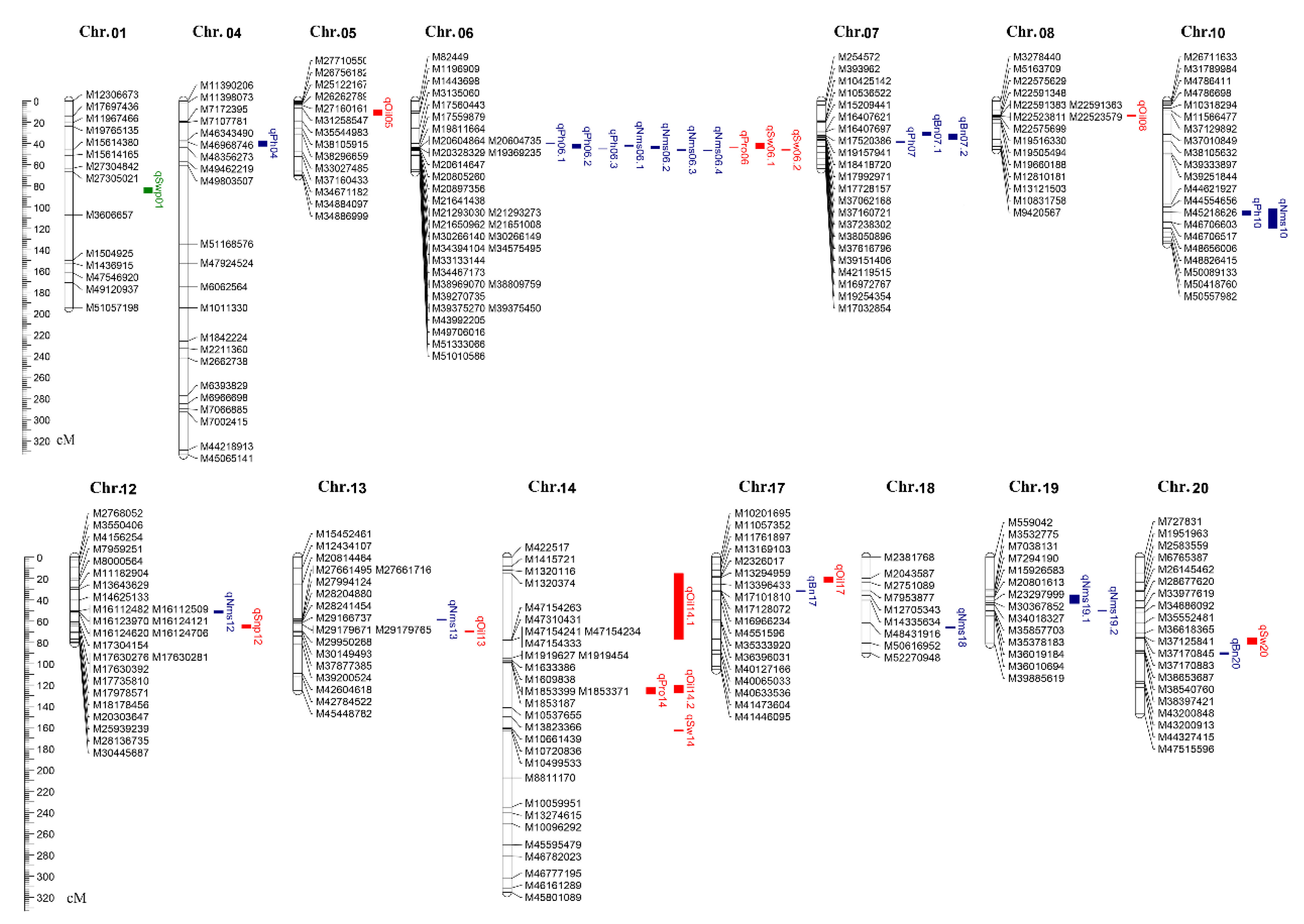
3.4. Identification of QTLs over Multiple Agronomic Traits
3.5. Co-Localization of QTLs Detected from Different Methods
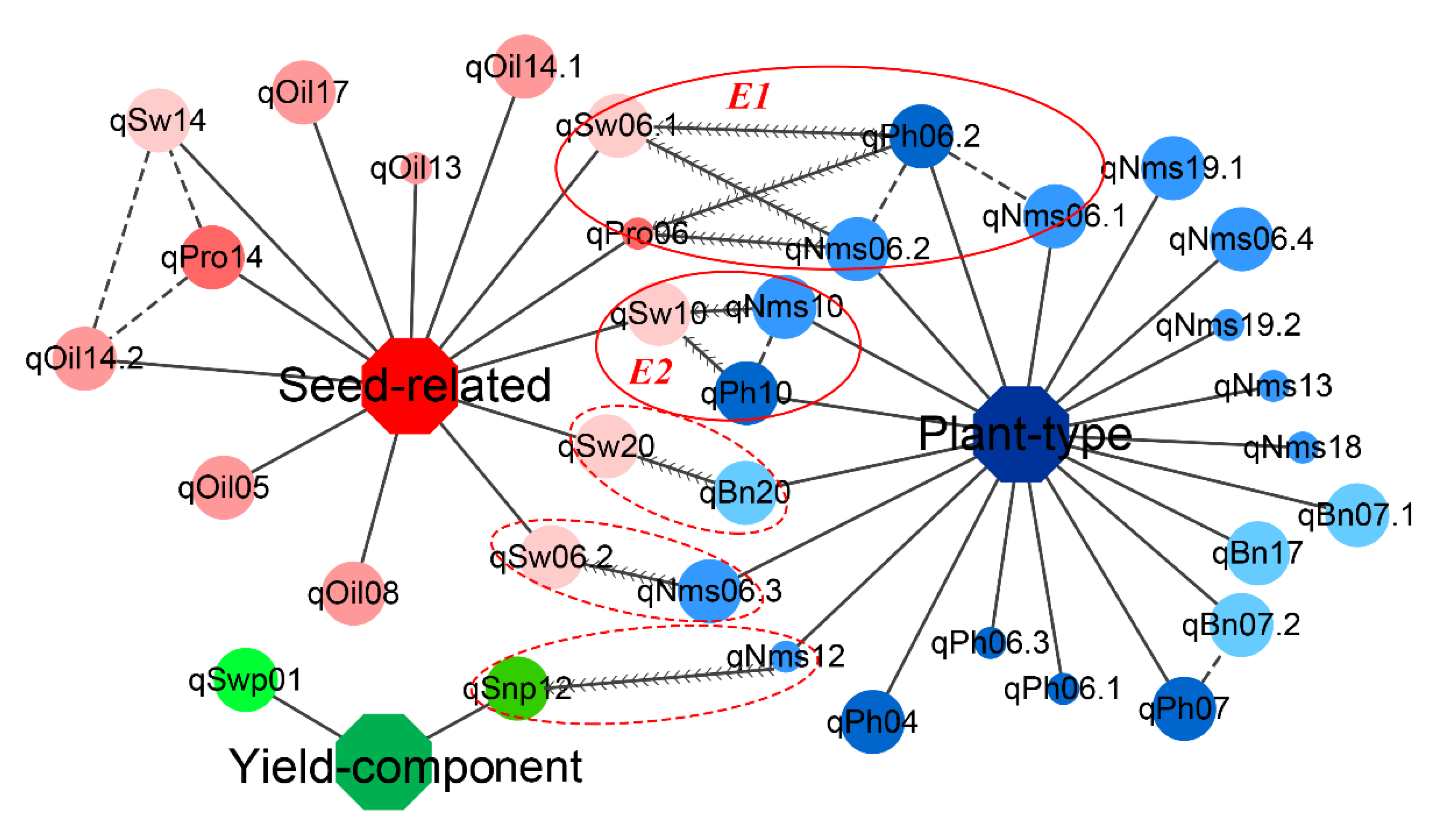
3.6. Exploration of Hub QTLs among Three Trait Modules in the KJ Population
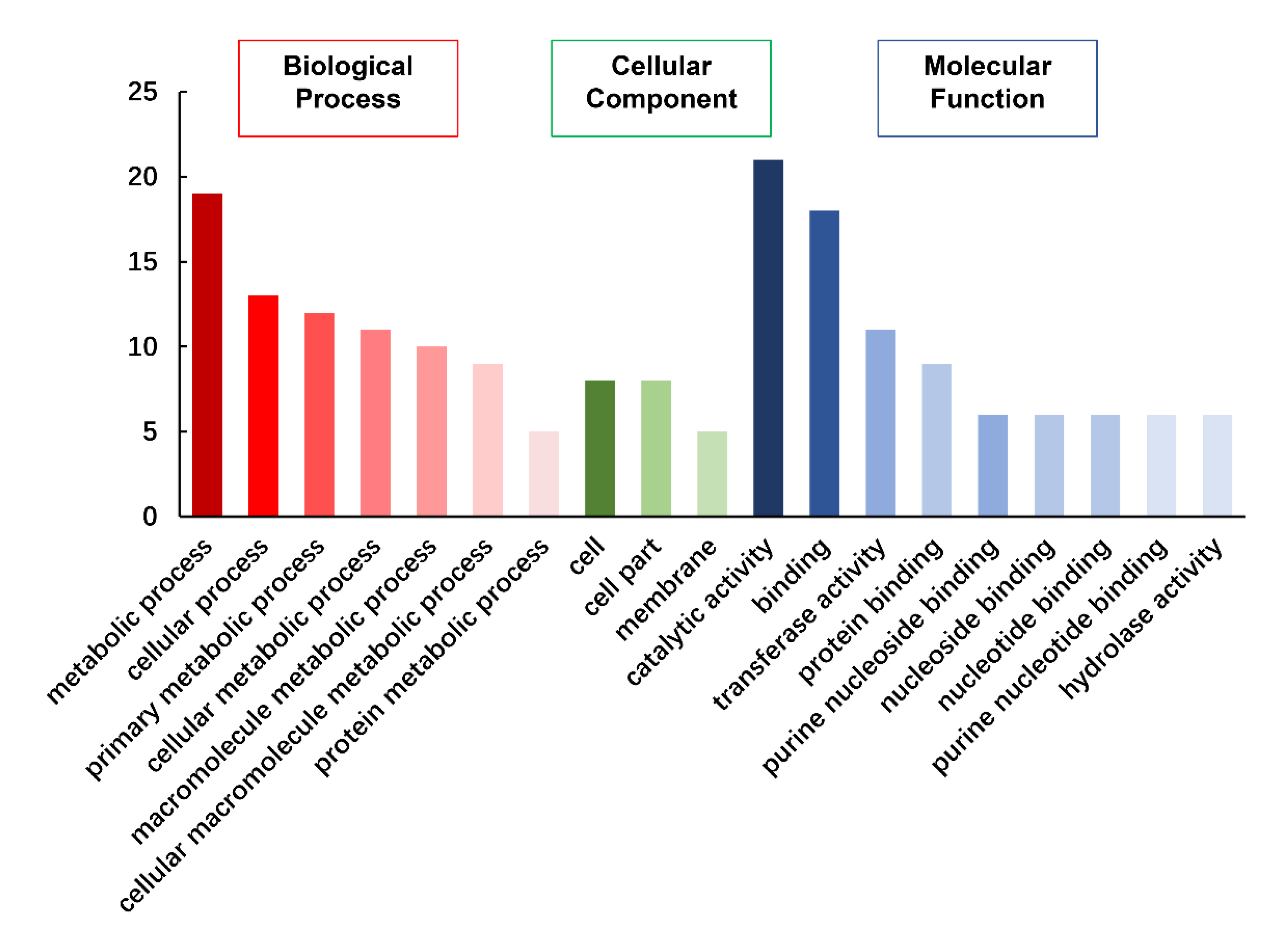
3.7. Exploration of Candidate Genes for Different Traits in the KJ Population
4. Discussion
4.1. The Factors Affecting QTL Mapping Analysis
4.2. The Novel QTL Loci and the Exploration of Candidate Genes from Hub QTLs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fu, M.; Wang, Y.; Ren, H.; Du, W.; Wang, D.; Bao, R.; Yang, X.; Tian, Z.; Fu, L.; Cheng, Y.; et al. Genetic dynamics of earlier maturity group emergence in south-to-north extension of Northeast China soybeans. Theor. Appl. Genet. 2020, 133, 1839–1857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hao, D.; Sitoe, H.M.; Yin, Z.; Hu, Z.; Zhang, G.; Yu, D.; Singh, R. Genetic dissection of the relationship between plant architecture and yield component traits in soybean (Glycine max) by association analysis across multiple environments. Plant Breed. 2015, 134, 564–572. [Google Scholar] [CrossRef]
- Chang, F.; Guo, C.; Sun, F.; Zhang, J.; Wang, Z.; Kong, J.; He, Q.; Sharmin, R.A.; Zhao, T. Genome-Wide Association Studies for Dynamic Plant Height and Number of Nodes on the Main Stem in Summer Sowing Soybeans. Front. Plant Sci. 2018, 9, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Liu, X.; Wang, G.; Mi, L.; Shen, Z.; Chen, X.; Herbert, S.J. Agronomic and physiological contributions to the yield improvement of soybean cultivars released from 1950 to 2006 in Northeast China. Field Crops Res. 2010, 115, 116–123. [Google Scholar] [CrossRef]
- Li, R.; Li, J.; Li, S.; Qin, G.; Novak, O.; Pencik, A.; Ljung, K.; Aoyama, T.; Liu, J.; Murphy, A.; et al. ADP1 Affects Plant Architecture by Regulating Local Auxin Biosynthesis. PLoS Genet. 2014, 10, e1003954. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-h.; Reif, J.C.; Hong, H.-l.; Li, H.-h.; Liu, Z.-x.; Ma, Y.-s.; Li, J.; Tian, Y.; Li, Y.-f.; Li, W.-b.; et al. Genome-wide association mapping of QTL underlying seed oil and protein contents of a diverse panel of soybean accessions. Plant Sci. 2018, 266, 95–101. [Google Scholar] [CrossRef]
- Sun, Y.-n.; Pan, J.-b.; Shi, X.-l.; Du, X.-y.; Wu, Q.; Qi, Z.-m.; Jiang, H.-w.; Xin, D.-w.; Liu, C.-y.; Hu, G.-h.; et al. Multi-environment mapping and meta-analysis of 100-seed weight in soybean. Mol. Biol. Rep. 2012, 39, 9435–9443. [Google Scholar] [CrossRef]
- Hartwig, E.E.; Hinson, K. Association between chemical composition of seed and seed yield of soybeans 1. Crop Sci. 1972, 12, 829–830. [Google Scholar] [CrossRef]
- Iqbal, Z.; Arshad, M.; Ashraf, M.; Naeem, R.; Malik, M.F.; Waheed, A. Genetic divergence and correlation studies of soybean (Glycine max (L.) Merrill.) genotypes. Pak. J. Bot. 2010, 42, 971–976. [Google Scholar]
- Mansur, L.M.; Orf, J.H.; Chase, K.; Jarvik, T.; Cregan, P.B.; Lark, K.G. Genetic mapping of agronomic traits using recombinant inbred lines of soybean. Crop Sci. 1996, 36, 1327–1336. [Google Scholar] [CrossRef]
- Panthee, D.; Pantalone, V.; West, D.; Saxton, A.; Sams, C. Quantitative trait loci for seed protein and oil concentration, and seed size in soybean. Crop Sci. 2005, 45, 2015–2022. [Google Scholar] [CrossRef]
- Ehrlich, P.R.; Harte, J. To feed the world in 2050 will require a global revolution. Proc. Natl. Acad. Sci. USA 2015, 112, 14743–14744. [Google Scholar] [CrossRef] [Green Version]
- Pandey, K.; Dangi, R.; Prajapati, U.; Kumar, S.; Maurya, N.K.; Singh, A.V.; Pandey, A.K.; Singh, J.; Rajan, R. Advance breeding and biotechnological approaches for crop improvement: A review. Int. J. Chem. Stud. 2019, 7, 837–841. [Google Scholar]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S. A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, L.; Shi, H.; Chern, M.; Yu, H.; Yi, H.; He, M.; Yin, J.; Zhu, X.; Li, Y. A single transcription factor promotes both yield and immunity in rice. Science 2018, 361, 1026–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, H. IPA1: A new “green revolution” gene? Mol. Plant 2017, 10, 779–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, H.; Ma, B.; Liu, G.; Wang, J.; Wang, J.; Gao, R.; Li, J.; Liu, J.; Xu, J. A natural tandem array alleviates epigenetic repression of IPA1 and leads to superior yielding rice. Nat. Commun. 2017, 8, 14789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; He, J.; Zhao, T.; Xing, G.; Wang, Y.; Yu, D.; Chen, S.; Gai, J. Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2018, 131, 2581–2599. [Google Scholar] [CrossRef]
- Li, S.; Cao, Y.; He, J.; Zhao, T.; Gai, J. Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2017, 130, 2297–2314. [Google Scholar] [CrossRef]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S.; et al. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Goettel, W.; Zhang, H.; Li, Y.; Qiao, Z.; Jiang, H.; Hou, D.; Song, Q.; Pantalone, V.R.; Song, B.-H.; Yu, D.; et al. POWR1 is a domestication gene pleiotropically regulating seed quality and yield in soybean. Nat. Commun. 2022, 13, 3051. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Loscalzo, J. The emerging paradigm of network medicine in the study of human disease. Circ. Res. 2012, 111, 359–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavarenne, J.; Guyomarc’h, S.; Sallaud, C.; Gantet, P.; Lucas, M. The spring of systems biology-driven breeding. Trends Plant Sci. 2018, 23, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, A.; Razzaq, A.; Ahmed, A.; Liu, A.; Qun, G.; Junwen, L.; Shi, Y.; Deng, X.; Zafar, M.M.; Ali, A. Identification of hub genes through co-expression network of major QTLs of fiber length and strength traits in multiple RIL populations of cotton. Genomics 2021, 113, 1325–1337. [Google Scholar] [CrossRef]
- Feng, L.; Ma, A.; Song, B.; Yu, S.; Qi, X.J.G. Mapping causal genes and genetic interactions for agronomic traits using a large F2 population in rice. G3 2021, 11, jkab318. [Google Scholar] [CrossRef]
- Lander, E.S.; Botstein, D. Mapping Mendelian Factors Underlying Quantitative Traits Using Rflp Linkage Maps. Genetics 1989, 121, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.B. Precision mapping of quantitative trait loci. Genetics 1994, 136, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ribaut, J.M.; Li, Z.; Wang, J. Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2008, 116, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, C.C.; Hu, H.; Yu, R.D.; Xia, Z.; Ye, X.Z.; Zhu, J. QTLNetwork: Mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 2008, 24, 721–723. [Google Scholar] [CrossRef]
- Li, H.; Hearne, S.; Banziger, M.; Li, Z.; Wang, J. Statistical properties of QTL linkage mapping in biparental genetic populations. Heredity 2010, 105, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.S.; Gore, M.; Buckler, E.S.; Yu, J.M. Status and Prospects of Association Mapping in Plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Tardivel, A.; Sonah, H.; Belzile, F.; O’Donoughue, L.S. Rapid identification of alleles at the soybean maturity gene E3 using genotyping by sequencing and a haplotype-based approach. Plant Genome 2014, 7, plantgenome2013-10. [Google Scholar] [CrossRef]
- St-Amour, V.T.B.; Mimee, B.; Torkamaneh, D.; Jean, M.; Belzile, F.; O’Donoughue, L.S. Characterizing resistance to soybean cyst nematode in PI 494182, an early maturing soybean accession. Crop Sci. 2020, 60, 2053–2069. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Wang, W.; Gong, S.; Zuo, J.; Li, S.; Xu, S. High Density Linkage Map Construction and Mapping of Yield Trait QTLs in Maize (Zea mays) Using the Genotyping-by-Sequencing (GBS) Technology. Front. Plant Sci. 2017, 8, 706. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Ooijen, J.W.V. JoinMap® 4.0: Software for the Calculation of Genetic Linkage Maps in Experimental Population; Kyazma BV: Wageningen, The Netherlands, 2006. [Google Scholar]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ye, G.; Wang, J. A Modified Algorithm for the Improvement of Composite Interval Mapping. Genetics 2006, 175, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Xia, Z.; Hideshima, R.; Tsubokura, Y.; Sato, S.; Yamanaka, N.; Takahashi, R.; Anai, T.; Tabata, S.; Kitamura, K.; et al. A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 2011, 188, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai, T.; Sato, S.; Yamazaki, T.; Lu, S.; et al. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2012, 109, E2155–E2164. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Zhai, H.; Zhang, Y.; Wang, Y.; Wang, L.; Xu, K.; Wu, H.; Zhu, J.; Jiao, S.; Wan, Z. QNE1 is a key flowering regulator determining the length of the vegetative period in soybean cultivars. Sci. China Life Sci. 2022, 65, 2472–2490. [Google Scholar] [CrossRef] [PubMed]
- Charmet, G. Power and accuracy of QTL detection: Simulation stusdies of one-QTL models. Agronomie 2000, 20, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Chuanzao, M.; Shihua, C. Analysis of accuracy and influence factor in QTL mapping about agronomic traits in rice (Oryza sativa L.). J. Agric. Biotechnol. 1999, 7, 386–394. [Google Scholar]
- Wang, Z.; Wang, J.; Peng, J.; Du, X.; Jiang, M.; Li, Y.; Han, F.; Du, G.; Yang, H.; Lian, S.; et al. QTL mapping for 11 agronomic traits based on a genome-wide Bin-map in a large F2 population of foxtail millet (Setaria italica (L.) P. Beauv). Mol. Breed. 2019, 39, 18. [Google Scholar] [CrossRef]
- Mackay, I.; Powell, W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007, 12, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yang, Z.; Tang, H.; Yu, Y.; Chen, Z.; Wei, S.; Sun, Q.; Peng, Z. High-density genetic map construction and mapping of the homologous transformation sterility gene (hts) in wheat using GBS markers. BMC Plant Biol. 2018, 18, 301. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, M.; Hu, X.; Ma, Q.; Mu, Y.; Tan, Z.; Xia, Q.; Zhang, G.; Nian, H. Construction of high-density genetic map and QTL mapping of yield-related and two quality traits in soybean RILs population by RAD-sequencing. BMC Genom. 2017, 18, 466. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-H.; Zhang, L.-Y.; Wang, J.-K. Analysis and Answers to Frequently Asked Questions in Quantitative Trait Locus Mapping. Acta Agron. Sin. 2010, 36, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.K.; Wang, Y.J.; Luo, G.Z.; Zhang, J.S.; He, C.Y.; Wu, X.L.; Gai, J.Y.; Chen, S.Y. QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theor. Appl. Genet. 2004, 108, 1131–1139. [Google Scholar] [CrossRef]
- Buzzell, R. Inheritance of a soybean flowering response to fluorescent-daylength conditions. Can. J. Genet. Cytol. 1971, 13, 703–707. [Google Scholar] [CrossRef]
- Brummer, E.; Graef, G.; Orf, J.; Wilcox, J.; Shoemaker, R. Mapping QTL for seed protein and oil content in eight soybean populations. Crop Sci. 1997, 37, 370–378. [Google Scholar] [CrossRef]
- Qi, Z.-m.; Wu, Q.; Han, X.; Sun, Y.-n.; Du, X.-y.; Liu, C.-y.; Jiang, H.-w.; Hu, G.-h.; Chen, Q.-s. Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 2011, 179, 499–514. [Google Scholar] [CrossRef]
- Iñigo, S.; Giraldez, A.N.; Chory, J.; Cerdán, P.D. Proteasome-mediated turnover of Arabidopsis MED25 is coupled to the activation of FLOWERING LOCUS T transcription. Plant Physiol. 2012, 160, 1662–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, Q.; Li, Z.; Staswick, P.E.; Wang, M.; Zhu, Y.; He, Z. Dual regulation role of GH3. 5 in salicylic acid and auxin signaling during Arabidopsis-Pseudomonas syringae interaction. Plant Physiol. 2007, 145, 450–464. [Google Scholar] [CrossRef] [Green Version]
- Hyten, D.L.; Pantalone, V.R.; Sams, C.E.; Saxton, A.M.; Landau-Ellis, D.; Stefaniak, T.R.; Schmidt, M.E. Seed quality QTL in a prominent soybean population. Theor. Appl. Genet. 2004, 109, 552–561. [Google Scholar] [CrossRef]
- Jiao, Y.; Vuong, T.D.; Liu, Y.; Meinhardt, C.; Liu, Y.; Joshi, T.; Cregan, P.B.; Xu, D.; Shannon, J.G.; Nguyen, H.T.J.T.; et al. Identification and evaluation of quantitative trait loci underlying resistance to multiple HG types of soybean cyst nematode in soybean PI 437655. Theor. Appl. Genet. 2015, 128, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; He, J.; Wang, Y.; Xing, G.; Zhao, J.; Li, Y.; Yang, S.; Palmer, R.G.; Zhao, T.; Gai, J. Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. J. Exp. Bot. 2015, 66, 6311–6325. [Google Scholar] [CrossRef] [Green Version]
- Oki, N.; Takagi, K.; Ishimoto, M.; Takahashi, M.; Takahashi, M. Evaluation of the resistance effect of QTLs derived from wild soybean (Glycine soja) to common cutworm (Spodoptera litura Fabricius). Breed. Sci. 2019, 69, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.; Wan, Z.; Jiao, S.; Zhou, J.; Xu, K.; Nan, H.; Liu, Y.; Xiong, S.; Fan, R.; Zhu, J.; et al. GmMDE genes bridge the maturity gene E1 and florigens in photoperiodic regulation of flowering in soybean. Plant Physiol. 2022, 189, 1021–1036. [Google Scholar] [CrossRef]
- Fahim, A.M.; Pan, L.; Li, C.; He, J.; Xing, G.; Wang, W.; Zhang, F.; Li, N.; Gai, J. QTL-allele system of main stem node number in recombinant inbred lines of soybean (Glycine max) using association versus linkage mapping. Plant Breed. 2021, 140, 870–883. [Google Scholar] [CrossRef]
- Thieme, C.J.; Rojas-Triana, M.; Stecyk, E.; Schudoma, C.; Zhang, W.; Yang, L.; Miñambres, M.; Walther, D.; Schulze, W.X.; Paz-Ares, J. Endogenous Arabidopsis messenger RNAs transported to distant tissues. Nat. Plants 2015, 1, 15025. [Google Scholar] [CrossRef]
- Huang, S.; Yu, J.; Li, Y.; Wang, J.; Wang, X.; Qi, H.; Xu, M.; Qin, H.; Yin, Z.; Mei, H. Identification of soybean genes related to soybean seed protein content based on quantitative trait loci collinearity analysis. J. Agric. Food Chem. 2018, 67, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, B.J.; Pearce, S.; van Bolderen-Veldkamp, R.; Marshall, A.; Widera, P.; Gilbert, J.; Drost, H.-G.; Bassel, G.W.; Müller, K.; King, J.R. Transcriptional dynamics of two seed compartments with opposing roles in Arabidopsis seed germination. Plant Physiol. 2013, 163, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, D.J.; Wei, Y.M.; Yu, Z.Q.; Dai, X.; Gao, X.-Q. Arabidopsis AtPRP17 functions in embryo development by regulating embryonic patterning. Planta 2021, 254, 58. [Google Scholar] [CrossRef] [PubMed]
- Bassel, G.W.; Lan, H.; Glaab, E.; Gibbs, D.J.; Gerjets, T.; Krasnogor, N.; Bonner, A.J.; Holdsworth, M.J.; Provart, N.J. Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc. Natl. Acad. Sci. USA 2011, 108, 9709–9714. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Trait | Parents | F2 Population | |||||
|---|---|---|---|---|---|---|---|---|
| KX03 | JD17 | Number | Mean | Min. | Max. | CV | ||
| Plant-type | Ph(cm) | 78.00 | 99.50 | 178 | 96.62 | 48.00 | 122.00 | 12.66 |
| Nms | 20.00 | 18.00 | 178 | 21.18 | 13.00 | 28.00 | 12.10 | |
| Bn | 4.20 | 2.50 | 180 | 2.71 | 0.00 | 8.00 | 54.09 | |
| Yield-component | Pnp | 34.20 | 51.70 | 179 | 44.33 | 12.00 | 113.00 | 39.53 |
| Snp | 73.80 | 91.20 | 178 | 104.12 | 30.00 | 271.00 | 39.93 | |
| Swp (g) | 15.50 | 15.96 | 178 | 18.34 | 3.87 | 51.36 | 45.52 | |
| Seed-related | Pro (%) | 40.76 | 36.36 | 181 | 39.91 | 35.86 | 48.36 | 5.02 |
| Oil (%) | 21.19 | 23.67 | 181 | 20.87 | 15.47 | 23.18 | 5.86 | |
| Sw (g) | 21.00 | 17.50 | 181 | 17.35 | 9.07 | 23.99 | 16.34 | |
| Linkage Group | Total Number of Markers | Total Size (cM) | Average Distance (cM) | Gap > 5 cM (%) |
|---|---|---|---|---|
| LG01 | 74 | 194.53 | 2.63 | 9.46 |
| LG02 | 136 | 146.6 | 1.08 | 2.94 |
| LG03 | 135 | 60.68 | 0.45 | 1.48 |
| LG04 | 79 | 332.88 | 4.21 | 16.46 |
| LG05 | 143 | 70.34 | 0.49 | 2.10 |
| LG06 | 225 | 66.61 | 0.30 | 1.33 |
| LG07 | 93 | 63.69 | 0.68 | 3.23 |
| LG08 | 97 | 45.58 | 0.47 | 0.00 |
| LG09 | 259 | 181.05 | 0.70 | 2.70 |
| LG10 | 134 | 134.18 | 1.00 | 5.22 |
| LG11 | 78 | 263.81 | 3.38 | 8.97 |
| LG12 | 147 | 80.32 | 0.55 | 0.68 |
| LG13 | 246 | 125.04 | 0.51 | 2.85 |
| LG14 | 110 | 315.20 | 2.87 | 8.18 |
| LG15 | 208 | 94.99 | 0.46 | 2.88 |
| LG16 | 136 | 109.93 | 0.81 | 2.21 |
| LG17 | 209 | 105.46 | 0.50 | 3.35 |
| LG18 | 209 | 90.01 | 0.43 | 1.44 |
| LG19 | 300 | 80.96 | 0.27 | 0.67 |
| LG20 | 170 | 146.77 | 0.86 | 3.53 |
| Total | 3188 | 2708.63 | 0.85 | 3.98 |
| Raw QTL | Chr. | Marker interval | Genetic Distance (cM) | LOD | Additive Effect | Dominant Effect | PVE (%) |
|---|---|---|---|---|---|---|---|
| q-c-Ph-06-1 | 6 | M5316720-M5344739 | 39.67–39.80 | 8.92 | −6.34 | 5.98 | 1.95 |
| q-c-Ph-06-2 | 6 | M17853562-M17944755 | 40.94–41.08 | 8.27 | 6.01 | 8.72 | 2.08 |
| q-c-Ph-06-3 | 6 | M18032241-M18032483 | 41.75–41.76 | 11.59 | 5.79 | 7.62 | 2.10 |
| q-c-Ph-06-4 | 6 | M18905749-M19276029 | 42.79–42.93 | 9.47 | 5.81 | 7.46 | 3.22 |
| q-c-Ph-06-5 | 6 | M19121776-M19811670 | 43.21–43.38 | 9.46 | 5.77 | 7.09 | 2.50 |
| q-c-Ph-06-6 | 6 | M19667942-M20735926 | 43.58–43.82 | 9.32 | 6.57 | 7.26 | 2.86 |
| q-c-Ph-06-7 | 6 | M19369196-M20709677 | 44.16–44.40 | 10.35 | 5.98 | 7.24 | 4.28 |
| q-c-Ph-06-8 | 6 | M21293030-M21651163 | 45.00–45.13 | 8.55 | 5.85 | 7.44 | 2.93 |
| q-c-Ph-07 | 7 | M37616796-M38050866 | 38.23–39.37 | 4.44 | 1.50 | −6.34 | 8.89 |
| q-c-Nms-06-1 | 6 | M17699008-M17967675 | 41.30–41.64 | 11.68 | 1.60 | 0.34 | 16.13 |
| q-c-Nms-06-2 | 6 | M19560132-M19720747 | 42.46–42.68 | 11.31 | 1.60 | 0.61 | 12.88 |
| q-c-Nms-06-3 | 6 | M19369235-M20733006 | 43.91–44.12 | 10.70 | 1.63 | 0.59 | 11.64 |
| q-c-Nms-06-4 | 6 | M20177466-M20805260 | 44.55–44.63 | 8.26 | 1.57 | 0.37 | 12.83 |
| q-c-Nms-06-5 | 6 | M33133144-M34575495 | 45.77–45.90 | 9.30 | 1.49 | 0.51 | 12.81 |
| q-c-Nms-06-6 | 6 | M39270735-M39375450 | 46.53–46.61 | 8.23 | 1.49 | 0.60 | 12.54 |
| q-c-Nms-10-1 | 10 | M45218626-M46706603 | 104.75–114.01 | 6.37 | −1.18 | 0.57 | 15.94 |
| q-c-Nms-10-2 | 10 | M46706431-M48656006 | 114.10–119.63 | 5.60 | −1.07 | 0.73 | 14.88 |
| q-c-Nms-12 | 12 | M16112509-M16124620 | 50.21–52.52 | 2.52 | −0.38 | 1.48 | 1.88 |
| q-c-Nms-13 | 13 | M27661716 -M27994124 | 58.09–59.06 | 1.44 | 0.94 | 0.79 | 2.48 |
| q-c-Nms-18 | 18 | M48705140-M49579192 | 65.25–66.40 | 3.31 | 0.59 | 0.88 | 0.10 |
| q-c-Nms-19-1 | 19 | M6070412-M27720002 | 35.41–36.43 | 2.39 | 0.85 | 0.24 | 5.09 |
| q-c-Nms-19-2 | 19 | M8911580-M23689027 | 38.91–39.00 | 2.45 | 0.84 | 0.07 | 3.46 |
| q-c-Nms-19-3 | 19 | M9387990-M27558601 | 39.61–39.72 | 3.27 | 0.84 | 0.20 | 4.73 |
| q-c-Nms-19-4 | 19 | M22949928-M25942777 | 39.89–39.95 | 2.38 | 0.89 | 0.15 | 3.40 |
| q-c-Nms-19-5 | 19 | M11105426-M28901311 | 41.82–41.90 | 2.00 | 0.82 | 0.28 | 3.76 |
| q-c-Nms-19-6 | 19 | M9388566-M30367852 | 42.27–43.17 | 3.17 | 0.82 | −0.15 | 6.19 |
| q-c-Nms-19-7 | 19 | M35378183-M36019184 | 49.66–50.41 | 2.73 | 0.78 | 0.46 | 1.97 |
| q-c-Bn-07 | 7 | M35251457-M37062168 | 30.93–36.01 | 5.05 | −0.73 | −0.40 | 5.90 |
| q-c-Pro-06 | 6 | M19369235-M20604864 | 43.85–44.04 | 1.43 | −0.05 | −1.00 | 2.16 |
| q-c-Oil-08 | 8 | M22523579-M22591404 | 13.38–14.34 | 2.91 | −0.15 | 0.56 | 6.00 |
| q-c-Oil-13 | 13 | M29950268-M30149493 | 68.90–70.00 | 2.67 | 0.30 | −0.42 | 0.80 |
| q-c-Oil-14 | 14 | M1320374-M47154263 | 15.27–77.19 | 2.65 | −1.19 | 1.26 | 5.49 |
| q-c-Sw-06-1 | 6 | M17559879-M19105624 | 39.66–40.66 | 15.97 | 2.13 | 0.63 | 19.42 |
| q-c-Sw-06-2 | 6 | M17905562-M17967675 | 41.30–41.57 | 18.55 | 2.19 | 0.48 | 24.63 |
| q-c-Sw-06-3 | 6 | M18905658-M19559998 | 52.11–42.33 | 18.70 | 2.17 | 0.74 | 21.39 |
| q-c-Sw-06-4 | 6 | M19667942-M20735926 | 43.58–43.82 | 17.66 | 2.17 | 0.85 | 21.50 |
| q-c-Sw-06-5 | 6 | M20805260-M20897356 | 44.63–44.68 | 14.27 | 2.15 | 0.81 | 21.14 |
| q-c-Sw-06-6 | 6 | M30266140-M34394104 | 45.71–45.79 | 17.88 | 2.09 | 0.78 | 20.95 |
| q-c-Sw-06-7 | 6 | M34575638-M38969070 | 46.24–46.30 | 19.38 | 2.17 | 0.75 | 21.98 |
| q-c-Sw-10-1 | 10 | M44554656-M45218626 | 99.56–104.75 | 8.62 | −1.44 | 0.46 | 17.26 |
| q-c-Sw-10-2 | 10 | M45218626-M46706603 | 104.75–114.01 | 8.80 | −1.53 | 0.27 | 17.54 |
| q-c-Sw-10-3 | 10 | M46706431-M48656006 | 114.10–119.63 | 6.65 | −1.40 | 0.02 | 11.42 |
| q-i-Ph-04-1 | 4 | M46343490-M46968746 | 37.50–40.50 | 3.60 | 4.40 | 0.64 | 6.39 |
| q-i-Ph-04-2 | 4 | M46968746-M48356273 | 38.50–42.50 | 3.65 | 4.41 | 0.56 | 6.42 |
| q-i-Ph-10-1 | 10 | M44554656-M45218626 | 103.50–105.50 | 3.71 | −3.90 | 2.67 | 6.50 |
| q-i-Ph-10-2 | 10 | M45218626-M46706603 | 104.50–107.50 | 3.76 | −3.97 | 2.51 | 6.56 |
| q-i-Nms-06 | 6 | M18032483-M18905642 | 41.50–42.50 | 11.20 | 1.57 | 0.47 | 20.31 |
| q-i-Nms-10-1 | 10 | M44554656-M45218626 | 101.50–104.50 | 5.46 | −1.05 | 0.56 | 9.87 |
| q-i-Nms-10-2 | 10 | M45218626-M46706603 | 108.50–110.50 | 5.59 | −1.10 | 0.59 | 11.24 |
| q-i-Bn-07 | 7 | M16407697-M17578608 | 29.50–32.50 | 4.81 | −0.64 | −0.49 | 9.69 |
| q-i-Bn-17 | 17 | M16966234-M17128072 | 31.50–32.50 | 3.16 | 0.19 | 0.67 | 6.27 |
| q-i-Bn-20 | 20 | M38397421-M38540760 | 89.50–91.50 | 2.92 | −0.47 | −0.15 | 5.78 |
| q-i-Snp-12-1 | 12 | M17630392-M17735810 | 63.50–64.50 | 31.50 | 0.10 | 117.49 | 7.40 |
| q-i-Snp-12-2 | 12 | M17735938-M17839525 | 64.50–66.50 | 22.52 | −0.26 | −94.98 | 7.18 |
| q-i-Swp-01 | 1 | M3606657-M27305021 | 81.50–86.50 | 2.57 | −7.89 | −9.14 | 27.90 |
| q-i-Pro-14 | 14 | M1853187-M10537655 | 122.50–128.50 | 5.74 | 2.45 | −2.71 | 8.60 |
| q-i-Oil-05 | 5 | M31258547-M35544983 | 8.50–13.50 | 3.41 | −0.48 | 0.09 | 9.67 |
| q-i-Oil-14 | 14 | M1853187-M10537655 | 120.50–127.50 | 8.92 | −1.60 | 1.77 | 11.14 |
| q-i-Oil-17 | 17 | M2326017- M13294959 | 18.50–23.50 | 2.82 | −0.24 | 0.65 | 9.33 |
| q-i-Sw-10-1 | 10 | M44554656-M45218626 | 102.50–105.50 | 9.90 | −1.54 | 0.26 | 15.85 |
| q-i-Sw-10-2 | 10 | M45218626-M46706603 | 108.50–110.50 | 10.40 | −1.61 | 0.23 | 17.94 |
| q-i-Sw-14 | 14 | M10499533-M10720836 | 162.50–163.50 | 2.69 | −0.80 | 0.24 | 3.82 |
| q-i-Sw-20 | 20 | M37170883-M38653687 | 75.50–81.50 | 2.57 | −0.42 | 0.82 | 3.43 |
| Final-QTL | QTL | Chr. | Genetic Distance (cM) | Physical Region (bp) | Raw QTL | Reported QTLs | ||
|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | |||||
| qPh04 | q-i-Ph1 | 04 | 37.50 | 42.50 | 46,343,490 | 48,356,273 | q-i-Ph-04-(1~2) (6.39~6.42) | Plant height 5-4,38-3 |
| qPh06.1 | q-c-Ph1 | 06 | 39.67 | 39.80 | 5,316,720 | 5,344,739 | q-c-Ph-06-1 (1.95) | |
| qPh06.2 | q-c-Ph2 | 06 | 40.94 | 44.40 | 17,853,562 | 20,735,926 | q-c-Ph-06-(2~7) (2.08~4.28) | Plant height 2-3,8-1,10-1,13-2, 17-6,17-9,18-4,19-3,21-2,30-2, 35-1;mqPlant height-004 |
| qPh06.3 | q-c-Ph3 | 06 | 45.00 | 45.13 | 21,293,030 | 21,651,163 | q-c-Ph-06-8 (2.93) | Plant height 19-3 |
| qPh07 | q-c-Ph4 | 07 | 38.23 | 39.37 | 37,616,796 | 38,050,866 | q-c-Ph-07 (8.89) | Plant height 37-5 |
| qPh10 | q-i-Ph2 | 10 | 103.50 | 107.50 | 44,554,656 | 46,706,603 | q-i-Ph-10-(1~2) (6.50~6.56) | Plant height 18-2,23-4,29-3,31-2 |
| qNms06.1 | q-c-Nms1 | 06 | 41.30 | 41.64 | 17,699,008 | 17,967,675 | q-c-Nms-06-1 (16.13) | |
| q-i-Nms1 | 06 | 41.50 | 42.50 | 18,032,483 | 18,905,642 | q-i-Nms-06 (20.31) | ||
| qNms06.2 | q-c-Nms2 | 06 | 42.46 | 44.63 | 19,369,235 | 20,805,260 | q-c-Nms-06-(2~4) (11.64~12.88) | Node number 2-2 |
| qNms06.3 | q-c-Nms3 | 06 | 45.77 | 46.61 | 33,133,144 | 34,575,495 | q-c-Nms-06-5 (12.81) | Node number 4-2 |
| qNms06.4 | q-c-Nms4 | 06 | 46.53 | 46.61 | 39,270,735 | 39,375,450 | q-c-Nms-06-6 (12.54) | Node number 4-2 |
| qNms10 | q-i-Nms2 | 10 | 101.50 | 110.50 | 44,554,656 | 46,706,603 | q-i-Nms-10-(1~2) (9.87~11.24) | |
| q-c-Nms5 | 10 | 104.75 | 119.63 | 45,218,626 | 48,656,006 | q-c-Nms-10-(1~2) (14.88~15.94) | ||
| qNms12 | q-c-Nms6 | 12 | 50.21 | 52.52 | 16,112,509 | 16,124,620 | q-c-Nms-12 (1.88) | |
| qNms13 | q-c-Nms7 | 13 | 58.09 | 59.06 | 27,661,716 | 27,994,124 | q-c-Nms-13 (2.48) | Node number 2-3 |
| qNms18 | q-c-Nms8 | 18 | 65.25 | 66.40 | 48,705,140 | 49,579,192 | q-c-Nms-18 (0.10) | |
| qNms19.1 | q-c-Nms9 | 19 | 35.41 | 43.17 | 6,070,412 | 30,367,852 | q-c-Nms-19-(1~6) (3.40~6.19) | |
| qNms19.2 | q-c-Nms10 | 19 | 49.66 | 50.41 | 35,378,183 | 36,019,184 | q-c-Nms-19-7 (1.97) | |
| qBn07.1 | q-i-Bn1 | 07 | 29.50 | 32.50 | 16,407,697 | 17,578,608 | q-i-Bn-07 (9.69) | |
| qBn07.2 | q-c-Bn1 | 07 | 30.93 | 36.01 | 35,251,457 | 37,062,168 | q-c-Bn-07 (5.90) | |
| qBn17 | q-i-Bn2 | 17 | 31.50 | 32.50 | 16,966,234 | 17,128,072 | q-i-Bn-17 (6.27) | |
| qBn20 | q-i-Bn3 | 20 | 89.50 | 91.50 | 38,397,421 | 38,540,760 | q-i-Bn-20 (5.78) | |
| qSnp12 | q-i-Snp1 | 12 | 63.50 | 66.50 | 17,630,392 | 17,839,525 | q-i-Snp-12-(1~2) (7.18~7.40) | |
| qSwp01 | q-i-Swp1 | 01 | 81.50 | 86.50 | 3,606,657 | 27,305,021 | q-i-Swp-01 (27.90) | |
| qPro06 | q-c-Pro1 | 06 | 43.85 | 44.04 | 19,369,235 | 20,604,864 | q-c-Pro-06 (2.16) | Seed protein 36-7 |
| qPro14 | q-i-Pro1 | 14 | 122.50 | 128.50 | 1,853,187 | 10,537,655 | q-i-Pro-14 (8.60) | Seed protein 1-6,4-10,21-8 |
| qOil05 | q-i-Oil1 | 05 | 8.50 | 13.50 | 31,258,547 | 35,544,983 | q-i-Oil-05 (9.67) | Seed oil 4-1 |
| qOil08 | q-c-Oil1 | 08 | 13.38 | 14.34 | 22,523,579 | 22,591,404 | q-c-Oil-08 (6.00) | |
| qOil13 | q-c-Oil2 | 13 | 68.90 | 70.00 | 29,950,268 | 30,149,493 | q-c-Oil-13 (0.80) | Seed oil 13-3,38-4 |
| qOil14.1 | q-c-Oil3 | 14 | 15.27 | 77.19 | 1,320,374 | 47,154,263 | q-c-Oil-14 (5.49) | Seed oil 30-4,34-2,37-4,42-11, 42-27,42-28,43-2;mqSeed Oil-005 |
| qOil14.2 | q-i-Oil2 | 14 | 120.50 | 127.50 | 1,853,187 | 10,537,655 | q-i-Oil-14 (11.14) | Seed oil 2-6,14-1,42-10,42-28 |
| qOil17 | q-i-Oil3 | 17 | 18.50 | 23.50 | 2,326,017 | 13,294,959 | q-i-Oil-17 (9.33) | Seed oil 5-5,23-3,24-22,37-1,39-7, 42-12,43-12;mqSeed Oil-011 |
| qSw06.1 | q-c-Sw1 | 06 | 39.66 | 44.68 | 17,559,879 | 20,897,356 | q-c-Sw-06-(1~5) (19.42~24.63) | Seed weight 6-5,15-1,16-1, 31-2,34-15,36-7,40-2,49-6 |
| qSw06.2 | q-c-Sw2 | 06 | 45.71 | 46.30 | 30,266,140 | 38,969,070 | q-c-Sw-06-(6~7) (20.95~21.98) | Seed weight 15-1,16-1,19-1,31-1, 34-16,34-2,35-2,40-3,49-6 |
| qSw10 | q-c-Sw3 | 10 | 99.56 | 119.63 | 44,554,656 | 48,656,006 | q-c-Sw-10-(1~3) (11.42~17.54) | Seed weight 34-8,35-8,36-8 |
| q-i-Sw1 | 10 | 102.50 | 110.50 | 44,554,656 | 46,706,603 | q-i-Sw-10-(1~2) (15.85~17.94) | Seed weight 34-8,35-8,36-8 | |
| qSw14 | q-i-Sw2 | 14 | 162.50 | 163.50 | 10,499,533 | 10,720,836 | q-i-Sw-14 (3.82) | Seed weight 3-8,4-10,13-2,23-1, 29-1,36-14 |
| qSw20 | q-i-Sw3 | 20 | 75.50 | 81.50 | 37,170,883 | 38,653,687 | q-i-Sw-20 (3.43) | Seed weight 36-5,37-11 |
| Total | 37 (3) | 14 | 63 | 71 (22) | ||||
| Final-QTL | Candidate Gene | No. of SNPs | Start (bp) | End (bp) | Gene Ontology Descriptions |
|---|---|---|---|---|---|
| h | Glyma.06G069500 | 9 (0;4) | 5,332,544 | 5,337,158 | Mitochondrial solute carrier protein |
| Glyma.06G069600 | 12 (0;3) | 5,338,316 | 5,344,365 | Cellulose synthase (UDP-forming) activity | |
| qPh06.2 | Glyma.06G207800 | 1 (0;1) | 20,207,077 | 20,207,940 | AP2/B3-like transcriptional factor family protein (E1) |
| qPh06.3 | Glyma.06G213200 | 1 (0;1) | 21,523,690 | 21,524,859 | |
| Glyma.06G213300 | 58 (0;6) | 21,548,690 | 21,565,666 | Translation initiation factor 2C and related proteins | |
| qPh07 | Glyma.07G207100 | 3 (1;1) | 37,628,251 | 37,629,081 | zinc ion binding nucleic acid binding |
| Glyma.07G209700 | 18 (1;7) | 38,049,934 | 38,051,000 | ||
| qPh10 | Glyma.10G221500 | 44 (0;2) | 45,294,735 | 45,316,121 | Regulation of photoperiodism, flowering (E2) |
| qNms06.1 | Glyma.06G196900 | 17 (0;4) | 17,770,435 | 17,777,991 | Protein kinase superfamily protein |
| Glyma.06G197100 | 2 (0;2) | 17,811,461 | 17,812,510 | F-box family protein | |
| Glyma.06G197200 | 5 (0;5) | 17,904,627 | 17,905,751 | F-box family protein | |
| Glyma.06G197500 | 17 (0;9) | 17,936,612 | 17,939,493 | ||
| Glyma.06G197600 | 10 (0;6) | 17,957,916 | 17,962,068 | Leucine-rich repeat protein kinase family protein | |
| Glyma.06G197700 | 5 (0;2) | 17,964,387 | 17,965,751 | Glycosyl hydrolase with C2H2-type zinc finger domain | |
| qNms06.2 | Glyma.06G204300 | 23 (0;5) | 19,210,586 | 19,213,448 | Transcription factor TCP (QNE1) |
| qNms06.3 | Glyma.06G227100 | 3 (0;1) | 34,252,103 | 34,254,142 | |
| Glyma.06G227300 | 7 (0;1) | 34,434,861 | 34,440,448 | Cytochrome P450 family 72 subfamily | |
| Glyma.06G227400 | 1 (0;1) | 34,446,404 | 34,449,305 | Cytochrome P450 family 72 subfamily | |
| Glyma.06G227800 | 1 (0;1) | 34,521,336 | 34,523,081 | ARM repeat superfamily protein | |
| qNms06.4 | Glyma.06G239300 | 3 (0;1) | 39,282,844 | 39,284,164 | Polynucleotidyl transferase protein |
| Glyma.06G239500 | 2 (0;1) | 39,371,341 | 39,373,375 | UDP-glucosyl transferase | |
| qNms10 | Glyma.10G221500 | 44 (0;2) | 45,294,735 | 45,316,121 | Regulation of photoperiodism, flowering (E2) |
| qNms12 | Glyma.12G136700 | 28 (0;1) | 16,135,929 | 16,145,471 | NB-ARC domain-containing disease resistance protein |
| qNms13 | Glyma.13G161300 | 7 (1;1) | 27,692,430 | 27,693,744 | Exostosin family protein |
| Glyma.13G164200 | 20 (1;5) | 27,922,878 | 27,924,801 | Transferring glycosyl groups | |
| qNms18 | Glyma.18G208200 | 15 (1;0) | 49,300,656 | 49,305,096 | Methyltransferases |
| Glyma.18G208600 | 4 (1;1) | 49,333,194 | 49,334,690 | UDP-glucosyl transferase 73B3 | |
| qNms19.2 | Glyma.19G104700 | 5 (0;2) | 35,430,695 | 35,431,194 | Nucleotidyltransferase activity |
| Glyma.19G104800 | 24 (0;1) | 35,437,052 | 35,439,310 | Beta carbonic anhydrase | |
| Glyma.19G105000 | 12 (0;12) | 35,452,898 | 35,454,069 | Hydroxyproline-rich glycoprotein family protein | |
| Glyma.19G105200 | 1 (0;1) | 35,492,974 | 35,493,519 | Polynucleotidyl transferase | |
| Glyma.19G105400 | 7 (0;1) | 35,541,994 | 35,545,213 | GRF zinc finger / Zinc knuckle protein | |
| Glyma.19G106000 | 5 (0;2) | 35,670,523 | 35,692,261 | ATP-dependent helicase activity | |
| Glyma.19G106100 | 11 (0;2) | 35,706,447 | 35,708,388 | Syntaxin/t-SNARE family protein | |
| Glyma.19G106300 | 56 (0;25) | 35,715,363 | 35,719,116 | DNA repair metallo-beta-lactamase family protein | |
| Glyma.19G106600 | 3 (0;3) | 35,767,221 | 35,768,987 | Xyloglucan endotransglucosylase | |
| Glyma.19G107000 | 4 (0;2) | 35,829,932 | 35,832,106 | Tetratricopeptide repeat (TPR)-like superfamily protein | |
| Glyma.19G107100 | 2 (0;2) | 35,840,827 | 35,841,117 | ||
| Glyma.19G107200 | 47 (0;8) | 35,854,636 | 35,862,179 | Alpha/beta-Hydrolases superfamily protein | |
| Glyma.19G107300 | 7 (0;1) | 35,879,955 | 35,888,760 | Acetyl-CoA synthetase | |
| Glyma.19G107400 | 1 (0;1) | 35,897,479 | 35,898,781 | Eukaryotic release factor | |
| Glyma.19G107500 | 73 (0;5) | 35,912,141 | 35,948,323 | ARM repeat superfamily protein | |
| Glyma.19G107700 | 14 (0;4) | 35,968,969 | 35,973,494 | Transferase family | |
| Glyma.19G107800 | 14 (0;9) | 35,979,733 | 35,982,244 | Replication factor-A C terminal domain | |
| Glyma.19G107900 | 2 (0;1) | 35,983,072 | 35,985,032 | DNA helicase PIF1/RRM3 | |
| qBn07.1 | Glyma.07G146700 | 30 (2;3) | 17,539,245 | 17,544,317 | PIF1 helicase |
| qBn17 | Glyma.17G172100 | 19 (0;2) | 16,767,027 | 16,775,605 | RING/U-box superfamily protein |
| qBn20 | Glyma.20G145700 | 7 (0;1) | 38,418,866 | 38,424,142 | PLP-dependent enzymes superfamily protein |
| Glyma.20G145900 | 14 (0;3) | 38,437,905 | 38,441,537 | Imidazoleglycerol-phosphate dehydratase | |
| Glyma.20G146100 | 3 (0;3) | 38,458,216 | 38,461,226 | FRIGIDA-like protein | |
| Glyma.20G146300 | 15 (0;2) | 38,475,899 | 38,478,861 | Cupin family protein | |
| Glyma.20G146400 | 2 (0;1) | 38,481,091 | 38,481,770 | ||
| Glyma.20G146800 | 4 (0;4) | 38,522,343 | 38,523,748 | Seed storage 2S albumin superfamily protein | |
| qSnp12 | Glyma.12G141000 | 2 (0;1) | 17,640,979 | 17,644,117 | Auxin-responsive GH3 family protein |
| Glyma.12g141100 | 10 (0;1) | 17,692,581 | 17,701,361 | Transducin/WD40 repeat-like superfamily protein | |
| Glyma.12G141300 | 8 (0;5) | 17,716,009 | 17,716,447 | DNAJ heat shock N-terminal domain-containing protein | |
| Glyma.12G141600 | 29 (0;3) | 17,823,834 | 17,827,414 | NAD(P)-linked oxidoreductase superfamily protein | |
| qPro06 | Glyma.06G207800 | 1 (0;1) | 20,207,077 | 20,207,940 | AP2/B3-like transcriptional factor family protein (E1) |
| qOil08 | Glyma.08G254800 | 1 (0;1) | 22,526,073 | 22,526,867 | Glutamine dumper 1 |
| Glyma.08G255000 | 7 (0;1) | 22,565,075 | 22,569,279 | Ribosomal RNA processing Brix domain protein | |
| qOil13 | Glyma.13G185600 | 3 (0;1) | 29,952,863 | 29,953,496 | |
| Glyma.13G186100 | 13 (0;5) | 29,985,014 | 29,988,798 | Root hair specific | |
| Glyma.13g186400 | 36 (0;5) | 30,001,282 | 30,008,506 | Zinc induced facilitator | |
| Glyma.13G186500 | 7 (0;1) | 30,016,526 | 30,025,554 | Zinc induced facilitator | |
| Glyma.13G186800 | 26 (0;3) | 30,049,481 | 30,055,924 | SU(VAR)3-9 homolog | |
| Glyma.13G186900 | 2 (0;2) | 30,050,935 | 30,051,293 | ||
| Glyma.13G187000 | 113 (0;3) | 30,060,197 | 30,072,759 | Subtilisin-like serine endopeptidase family protein | |
| Glyma.13G187300 | 6 (0;1) | 30,113,847 | 30,118,165 | Conserved developmentally regulated protein | |
| Glyma.13G187500 | 9 (0;2) | 30,128,663 | 30,133,972 | Myb-like DNA-binding domain | |
| Glyma.13G187600 | 9 (0;5) | 30,134,637 | 30,143,817 | Protein kinase superfamily protein | |
| Glyma.13G187700 | 19 (0;1) | 30,149,058 | 30,152,110 | ||
| qSw14 | Glyma.14G103900 | 2 (0;1) | 10,508,303 | 10,509,092 | EamA-like transporter family protein |
| Glyma.14G104000 | 11 (0;1) | 10,624,617 | 10,628,567 | EamA-like transporter family | |
| Glyma.14G104100 | 7 (0;1) | 10,634,347 | 10,639,492 | Monogalactosyl diacylglycerol synthase | |
| Glyma.14G104200 | 7 (0;3) | 10,674,108 | 10,675,950 | DnaJ/Hsp40 cysteine-rich domain superfamily protein | |
| Glyma.14G104500 | 1 (0;1) | 10,701,801 | 10,702,697 | ||
| Glyma.14G104700 | 4 (0;1) | 10,710,985 | 10,712,566 | CemA-like proton extrusion protein-related |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, M.; Qi, B.; Li, S.; Xu, H.; Wang, Y.; Zhao, Z.; Yu, X.; Pan, L.; Yang, J. Detection of Hub QTLs Underlying the Genetic Basis of Three Modules Covering Nine Agronomic Traits in an F2 Soybean Population. Agronomy 2022, 12, 3135. https://doi.org/10.3390/agronomy12123135
Fu M, Qi B, Li S, Xu H, Wang Y, Zhao Z, Yu X, Pan L, Yang J. Detection of Hub QTLs Underlying the Genetic Basis of Three Modules Covering Nine Agronomic Traits in an F2 Soybean Population. Agronomy. 2022; 12(12):3135. https://doi.org/10.3390/agronomy12123135
Chicago/Turabian StyleFu, Mengmeng, Bo Qi, Shuguang Li, Haifeng Xu, Yaqi Wang, Zhixin Zhao, Xiwen Yu, Liyuan Pan, and Jiayin Yang. 2022. "Detection of Hub QTLs Underlying the Genetic Basis of Three Modules Covering Nine Agronomic Traits in an F2 Soybean Population" Agronomy 12, no. 12: 3135. https://doi.org/10.3390/agronomy12123135