Genome-Wide Association Study for Plant Architecture and Bioenergy Traits in Diverse Sorghum and Sudangrass Germplasm

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growing Conditions
2.2. Phenotypic Traits and Repeatability
2.3. Experimental Design and Statistical Analysis
2.4. Genotyping
2.5. Population Structure, Relative Kinship, Principal Component Analysis (PCA), and Linkage Disequilibrium (LD)
2.6. Genome-Wide Association Study (GWAS) and Candidate Gene Identification
3. Results
3.1. Analysis of Variance, Trait Variation and Heritability
3.2. Trait Correlations
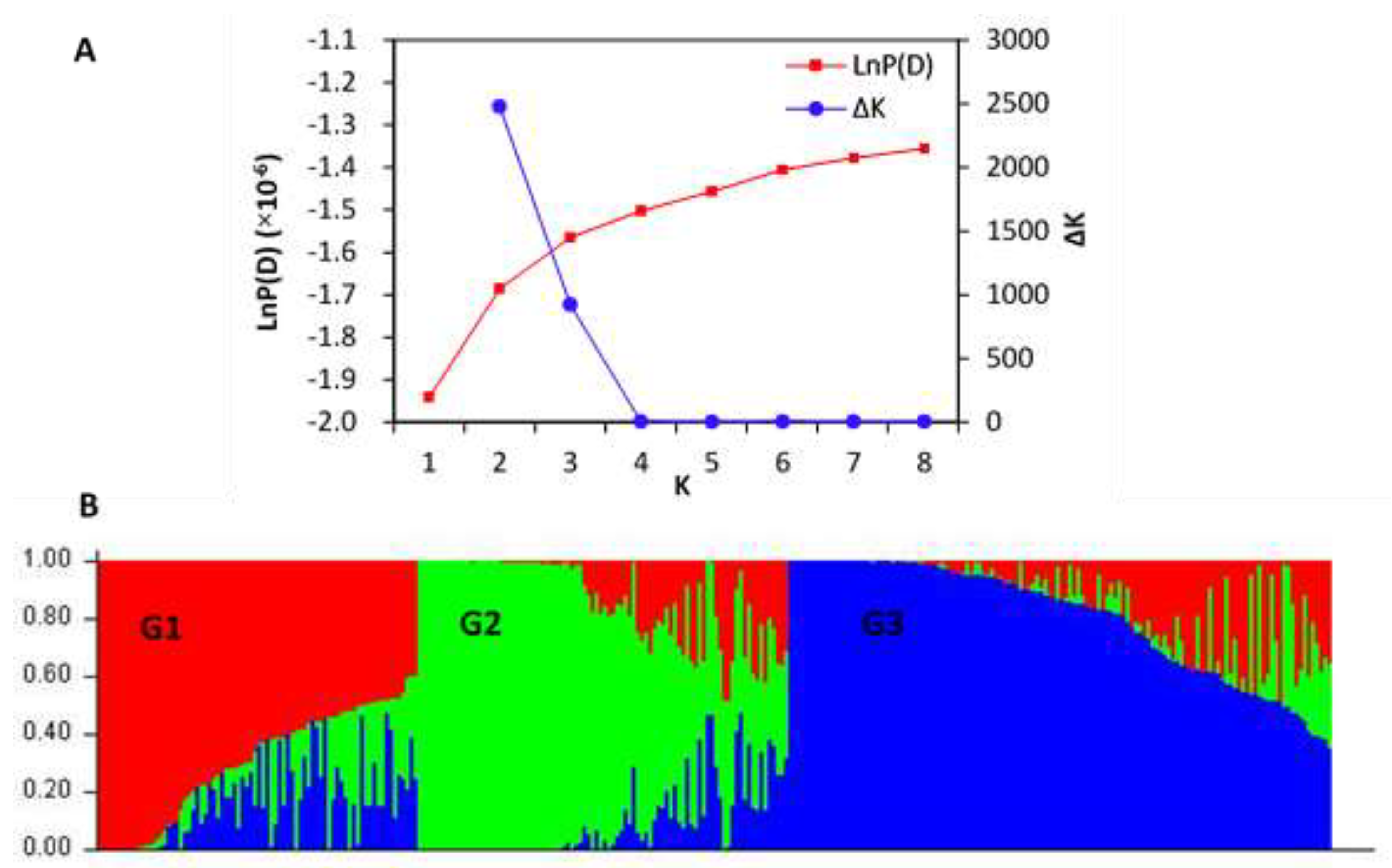
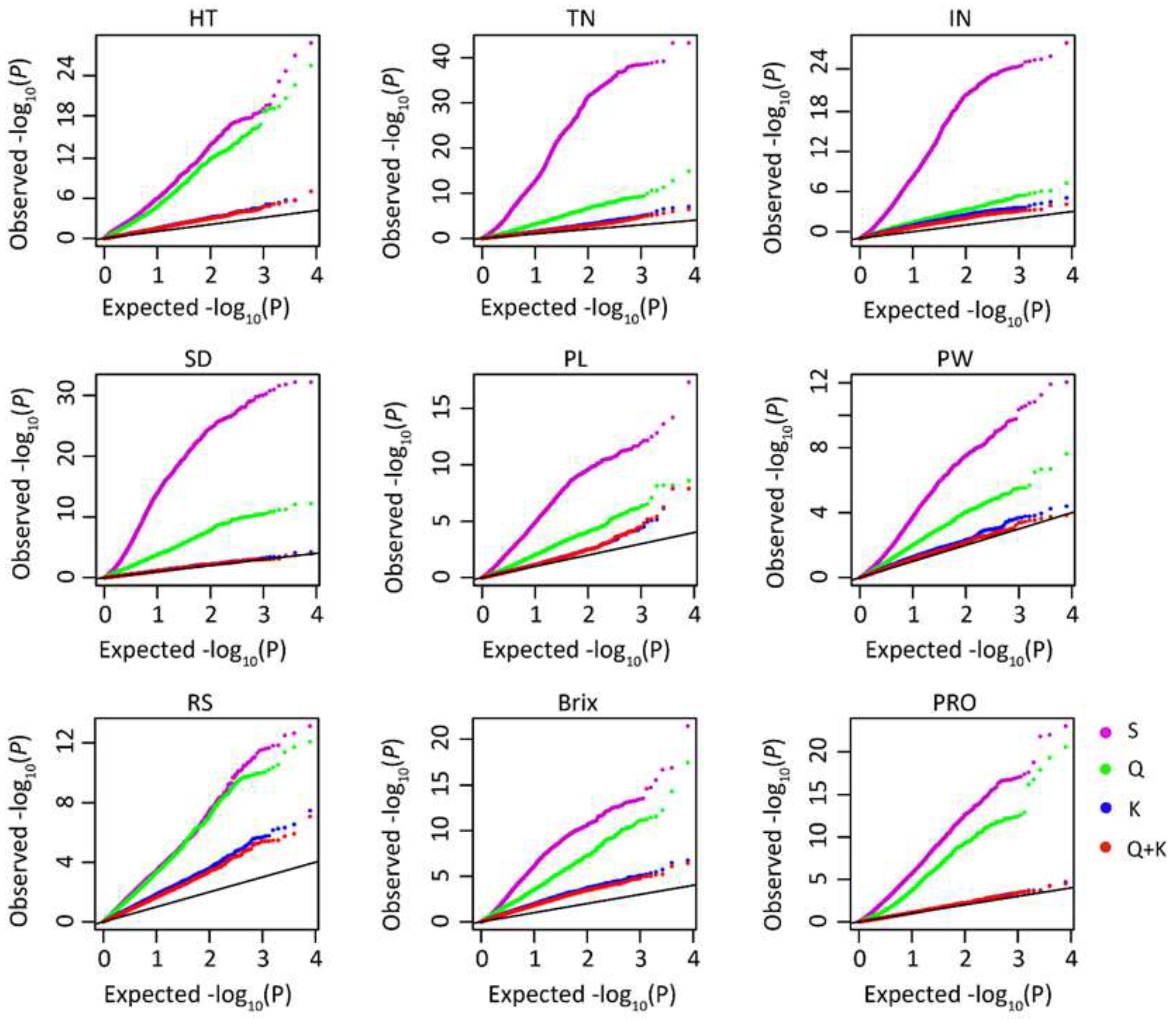
3.3. Population Structure and Its Effects on Traits
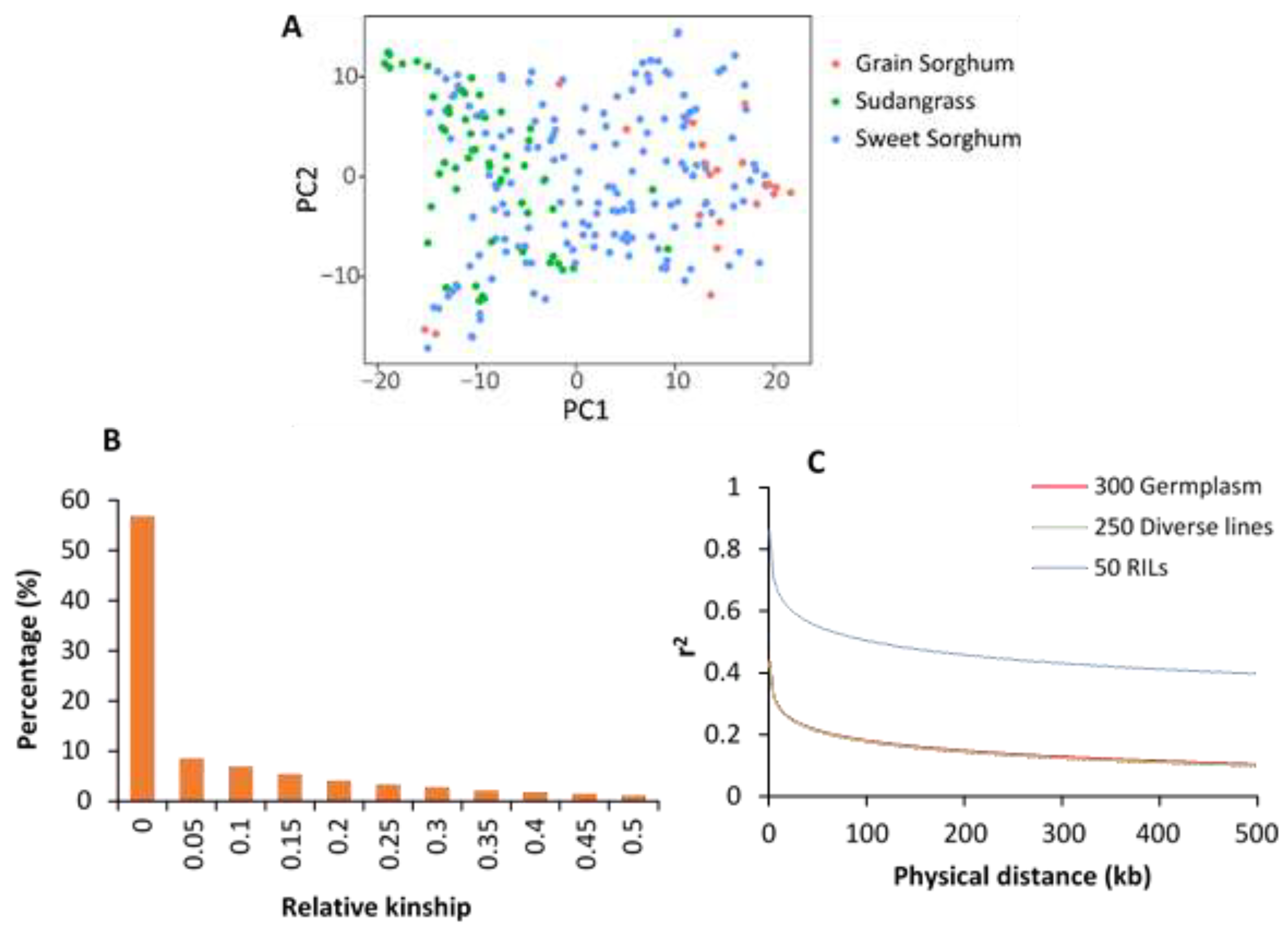
3.4. PCA, Relative Kinship and LD
3.5. GWAS
3.6. Candidate Genes
4. Discussion
4.1. Trait Variations and Correlations
4.2. LD Decay
4.3. Marker Effects
4.4. Significant SNPs across Environments
4.5. Significant SNPs for Individual Environment
4.6. Potential Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Research Council. Lost Crops of Africa: Volume I: Grains; National Academy Press: Washington, DC, USA, 1996. [Google Scholar]
- Wang, D.; Bean, S.; McLaren, J.; Seib, P.; Madl, R.; Tuinstra, M.; Shi, Y.; Lenz, M.; Wu, X.; Zhao, R. Grain sorghum is a viable feedstock for ethanol production. J. Ind. Microbiol. Biotech. 2008, 35, 313–320. [Google Scholar] [CrossRef]
- Nuessly, G.S.; Wang, Y.; Sandhu, H.; Larsen, N.; Cherry, R.H. Entomologic and agronomic evaluations of 18 sweet sorghum cultivars for biofuel in Florida. Fla. Entomol. 2013, 96, 512–528. [Google Scholar] [CrossRef]
- Ekefre, D.E.; Mahapatra, A.K.; Latimore, M., Jr.; Bellmer, D.D.; Jena, U.; Whitehead, G.J.; Williams, A.L. Evaluation of three cultivars of sweet sorghum as feedstocks for ethanol production in the Southeast United States. Heliyon 2017, 3, e00490. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Q.; Qian, Z. Heterosis utilization of hybrid between sorghum [Sorghum bicolor (L.) Moench] and sudangrass [Sorghum sudanense (Piper) Stapf]. Acta Agron. Sin. 2004, 30, 73–77. [Google Scholar]
- Lu, X.; Yun, J.; Gao, C.; Acharya, S. Quantitative trait loci analysis of economically important traits in Sorghum bicolor × S. sudanense hybrid. Can. J. Plant Sci. 2011, 9, 81–90. [Google Scholar]
- Venuto, B.; Kindiger, B. Forage and biomass feedstock production from hybrid forage sorghum and sorghum–sudangrass hybrids. Grassland Sci. 2008, 54, 189–196. [Google Scholar] [CrossRef]
- Mace, E.S.; Singh, V.; Van Oosterom, E.J.; Hammer, G.L.; Hunt, C.H.; Jordan, D.R. QTL for nodal root angle in sorghum (Sorghum bicolor L. Moench) co-locate with QTL for traits associated with drought adaptation. Theor. Appl. Genet. 2012, 124, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, H.D.; Wang, Y.; Sastry, D.V.; Dwivedi, S.L.; Prasad, P.V.; Burrell, A.M.; Klein, R.R.; Morris, G.P.; Klein, P.E. Association mapping of germinability and seedling vigor in sorghum under controlled low-temperature conditions. Genome 2016, 59, 137–145. [Google Scholar] [CrossRef]
- Ortiz, D.; Hu, J.; Salas Fernandez, M.G. Genetic architecture of photosynthesis in Sorghum bicolor under non-stress and cold stress conditions. J. Exp. Bot. 2017, 68, 4545–4557. [Google Scholar] [CrossRef]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5. [Google Scholar] [CrossRef]
- Morris, G.P.; Ramu, P.; Deshpande, S.P.; Hash, C.T.; Shah, T.; Upadhyaya, H.D.; Riera-Lizarazu, O.; Brown, P.J.; Acharya, C.B.; Mitchell, S.E.; et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. USA 2013, 110, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, X.; Fridman, E.; Tesso, T.T.; Yu, J. Dissecting repulsion linkage in the dwarfing gene Dw3 region for sorghum plant height provides insights into heterosis. Proc. Natl. Acad. Sci. USA 2015, 112, 11823–11828. [Google Scholar] [CrossRef] [Green Version]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E.; et al. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Mantilla Perez, M.B.; Hu, J.; Salas Fernandez, M.G. Genome-wide association study for nine plant architecture traits in sorghum. Plant Genome 2016, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Olatoye, M.O.; Marla, S.; Morris, G.P. An integrated genotyping-by-sequencing polymorphism map for over 10,000 sorghum Genotypes. Plant Genome 2019, 12, 180044. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Srinivasan, S.; Mirnezami, S.V.; Kusmec, A.; Fu, Q.; Attigala, L.; Fernandez, M.G.S.; Ganapathysubramanian, B.; Schnable, P.S. Semiautomated feature extraction from RGB images for sorghum panicle architecture GWAS. Plant Physiol. 2019, 179, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.H.; Hoffmann, L., Jr.; Rooney, W.L.; Ramu, P.; Morris, G.P.; Kresovich, S. Genome-wide association study of grain polyphenol concentrations in global sorghum [Sorghum bicolor (L.) Moench] germplasm. J. Agric. Food Chem. 2014, 62, 10916–10927. [Google Scholar] [CrossRef]
- Boyles, R.E.; Pfeiffer, B.K.; Cooper, E.A.; Rauh, B.L.; Zielinski, K.J.; Myers, M.T.; Brenton, Z.; Rooney, W.L.; Kresovich, S. Genetic dissection of sorghum grain quality traits using diverse and segregating populations. Theor. Appl. Genet. 2017, 130, 697–716. [Google Scholar] [CrossRef] [Green Version]
- Chopra, R.; Burow, G.; Burke, J.J.; Gladman, N.; Xin, Z. Genome-wide association analysis of seedling traits in diverse Sorghum germplasm under thermal stress. BMC Plant Biol. 2017, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tang, W.; Zhang, Y.-W.; Chen, K.-N.; Wang, C.; Liu, Y.; Zhan, Q.; Wang, C.; Wang, S.-B.; Xie, S.-Q.; et al. Genome-wide association studies for five forage quality-related traits in sorghum (Sorghum bicolor L.). Front. Plant Sci. 2018, 9, 1146. [Google Scholar] [CrossRef]
- Murray, S.C.; Sharma, A.; Rooney, W.L.; Klein, P.E.; Mullet, J.E.; Mitchell, S.E.; Kresovich, S. Genetic improvement of sorghum as a biofuel feedstock: I. QTL for stem sugar and grain nonstructural carbohydrates. Crop Sci. 2008, 48, 2165–2179. [Google Scholar] [CrossRef]
- Murray, S.C.; Rooney, W.L.; Hamblin, M.T.; Mitchell, S.E.; Kresovich, S. Sweet sorghum genetic diversity and association mapping for Brix and height. Plant Genome 2009, 2, 48. [Google Scholar] [CrossRef] [Green Version]
- Lv, P.; Ji, G.; Han, Y.; Hou, S.; Li, S.; Ma, X.; Du, R.; Liu, G. Association analysis of sugar yield-related traits in sorghum [Sorghum bicolor (L.)]. Euphytica 2013, 193, 419–431. [Google Scholar] [CrossRef]
- Burks, P.S.; Kaiser, C.M.; Hawkins, E.M.; Brown, P.K. Genome wide association for sugar yield in sweet sorghum. Crop Sci. 2015, 55, 2138–2148. [Google Scholar] [CrossRef] [Green Version]
- Disasa, T.; Feyissa, T.; Admassu, B.; Fetene, M.; Mendu, V. Mapping of QTLs associated with brix and biomass-related traits in sorghum using SSR markers. Sugar Tech. 2018, 20, 275–285. [Google Scholar] [CrossRef]
- Morris, G.P.; Rhodes, D.H.; Brenton, Z.; Ramu, P.; Thayil, V.M.; Deshpande, S.; Hash, C.T.; Acharya, C.; Mitchell, S.E.; Buckler, E.S.; et al. Dissecting genome-wide association signals for loss-of-function phenotypes in sorghum flavonoid pigmentation traits. G3 (Bethesda) 2013, 3, 2085–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenton, Z.W.; Cooper, E.A.; Myers, M.T.; Boyles, R.E.; Shakoor, N.; Zielinski, K.J.; Rauh, B.L.; Bridges, W.C.; Morris, G.F.; Kresovich, S. Genomic resource for the development, improvement, and exploitation of sorghum for bioenergy. Genetics 2016, 204, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.H.; Hoffmann, L., Jr.; Rooney, W.L.; Herald, T.J.; Bean, S.; Boyles, R.; Brenton, Z.W.; Kresovich, S. Genetic architecture of kernel composition in global sorghum germplasm. BMC Genom. 2017, 18, 15. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.J.; Rooney, W.L.; Franks, C.; Kresovich, S. Efficient mapping of plant height quantitative trait loci in a sorghum association population with introgressed dwarfing genes. Genetics 2008, 180, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja Reddy, R.; Madhusudhana, R.; Murali Mohan, S.; Chakravarthi, D.V.N.; Mehtre, S.P.; Seetharama, N.; Patil, J.V. Mapping QTL for grain yield and other agronomic traits in post-rainy sorghum [Sorghum bicolor (L.) Moench]. Theor. Appl. Genet. 2013, 126, 1921–1939. [Google Scholar] [CrossRef]
- Hirano, K.; Kawamura, M.; Araki-Nakamura, S.; Fujimoto, S.; Ohmae-Shinohara, K.; Yamaguchi, M.; Fujii, A.; Sasaki, H.; Kasuga, S.; Sazuka, T. Sorghum DW1 positively regulates brassinosteroid signaling by inhibiting the nuclear localization of BRASSINOSTEROID INSENSITIVE 2. Sci. Rep. 2017, 7, 126. [Google Scholar] [CrossRef]
- Hilley, J.L.; Weers, B.D.; Truong, S.K.; McCormick, R.F.; Mattison, A.J.; McKinley, B.A.; Morishige, D.T.; Mullet, J.E. Sorghum Dw2 encodes a protein kinase regulator of stem internode length. Sci. Rep. 2017, 7, 4616. [Google Scholar] [CrossRef] [Green Version]
- Bouchet, S.; Olatoye, M.O.; Marla, S.R.; Perumal, R.; Tesso, T.; Yu, J.; Tuinstra, M.; Morris, G.P. Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 2017, 206, 573–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.; Kim, C.; Zhang, D.; Guo, H.; Tan, X.; Jin, H.; Zhou, C.; Shuang, L.-S.; Goff, V.; Sezen, U.; et al. Genotyping by sequencing of 393 Sorghum bicolor BTx623 × IS3620C recombinant inbred lines improves sensitivity and resolution of QTL detection. G3 2018, 8, 2563–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Kong, W.; Robertson, J.; Goff, V.H.; Epps, E.; Kerr, A.; Mills, G.; Cromwell, J.; Lugin, Y.; Phillips, C.; et al. Genetic analysis of inflorescence and plant height components in sorghum (Panicoidae) and comparative genetics with rice (Oryzoidae). BMC Plant Biol. 2015, 15, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinby, J.R.; Karper, R.E. Inheritance of height in sorghum. Agron. J. 1954, 46, 211–216. [Google Scholar] [CrossRef]
- Multani, D.S.; Briggs, S.P.; Chamberlin, M.A.; Blakeslee, J.J.; Murphy, A.S.; Johal, G.S. Loss of an MDR transporter in compact stalks of maize br2 and sorghum Dw3 mutants. Science 2003, 302, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, C.; Lin, J.; Liu, J.; Liu, B.; Wang, J.; Huang, A.; Li, H.; Zhao, T. OsMPH1 regulates plant height and improves grain yield in rice. PLoS ONE 2017, 12, e0180825. [Google Scholar] [CrossRef] [Green Version]
- Anami, S.E.; Zhang, L.-M.; Xia, Y.; Zhang, Y.-M.; Liu, Z.-Q.; Jing, H.-C. Sweet sorghum ideotypes: Genetic improvement of the biofuel syndrome. Food Energy Secur. 2015, 4, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Boyles, R.E.; Brenton, Z.W.; Kresovich, S. Genetic and genomic resources of sorghum to connect genotype with phenotype in contrasting environments. Plant J. 2019, 97, 19–39. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Li, X.; Li, X.; Zhu, C.; Bai, G.; Perumal, R.; Tuinstra, M.R.; Prasad, P.V.V.; Mitchell, S.E.; Tesso, T.T.; et al. QTL mapping for grain yield, flowering time, and stay-green traits in sorghum with genotyping-by-sequencing markers. Crop Sci. 2016, 56, 1429–1442. [Google Scholar] [CrossRef]
- SAS Institute Inc. SAS Procedures Guide; Release 9.1 Edn.; SAS Institute: Cary, NC, USA, 2014. [Google Scholar]
- Wang, S.; Meyer, E.; McKay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Dou, J.; Mao, J.; Su, H.; Jiao, W.; Zhang, L.; Hu, X.; Huang, X.; Wang, S.; Bao, Z. RADtyping: An integrated package for accurate De Novo codominant and dominant rad genotyping in mapping populations. PLoS ONE 2013, 8, e79960. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Wang, M.; Zhu, C.; Barkley, N.A.; Chen, Z.; Erpelding, J.E.; Murray, S.C.; Tuinstra, M.R.; Tesso, T.; Pederson, G.A.; Yu, J. Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection. Theor. Appl. Genet. 2009, 120, 13–23. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Mace, E.S.; Innes, D.; Hunt, C.; Wang, X.; Tao, Y.; Baxter, J.; Hassall, M.; Hathorn, A.; Jordan, D.R. The Sorghum QTL Atlas: A powerful tool for trait dissection, comparative genomics and crop improvement. Theor. Appl. Genet. 2019, 132, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, T.; Okuno, K. QTL mapping for yield and yield-contributing traits in sorghum (Sorghum bicolor (L.) Moench) with genome-based SSR markers. Euphytica 2015, 203, 17–31. [Google Scholar] [CrossRef]
- Felderhoff, T.J.; Murray, S.C.; Klein, P.E.; Sharma, A.; Hamblin, M.T.; Kresovich, S.; Vermerris, W.; Rooney, W.L. QTLs for energy-related traits in a sweet × grain sorghum Sorghum bicolor (L.) Moench mapping population. Crop Sci. 2012, 52, 2040–2049. [Google Scholar] [CrossRef]
- Lin, Y.R.; Schertz, K.F.; Paterson, A.H. Comparative analysis of QTLs affecting plant height and maturity across the Poaceae, in reference to an interspecific sorghum population. Genetics 1995, 141, 391–411. [Google Scholar] [PubMed]
- Kajiya-Kanegae, H.; Takanashi, H.; Fujimoto, M.; Ishimori, M.; Ohnishi, N.; Fiona, W.W.; Omollo, E.A.; Kobayashi, M.; Yano, K.; Nakano, M.; et al. RAD-seq-based high-density linkage map construction and QTL mapping of biomass-related traits in sorghum using a Japanese landrace Takakibi NOG. Plant Cell Physiol. 2020, 61, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Feltus, F.A.; Hart, G.E.; Schertz, K.F.; Casa, A.M.; Kresovich, S.; Abraham, S.; Klein, P.E.; Brown, P.J.; Paterson, A.H. Alignment of genetic maps and QTLs between inter- and intra-specific sorghum populations. Theor. Appl. Genet. 2006, 112, 1295. [Google Scholar] [CrossRef]
- Takai, T.; Yonemaru, J.-I.; Kaidai, H.; Kasuga, S. Quantitative trait locus analysis for days-to-heading and morphological traits in an RIL population derived from an extremely late flowering F1 hybrid of sorghum. Euphytica 2012, 187, 411–420. [Google Scholar] [CrossRef]
- Wang, X.; Mace, E.; Hunt, C.; Cruickshank, A.; Henzell, R.F.; Parkes, H.; Jordan, D. Two distinct classes of QTL determine rust resistance in sorghum. BMC Plant Biol. 2014, 14, 366. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.M.; Mace, E.S.; Van Oosterom, E.J.; Cruickshank, A.; Hunt, C.H.; Hammer, G.L.; Jordan, D.R. QTL analysis in multiple sorghum populations facilitates the dissection of the genetic and physiological control of tillering. Theor. Appl. Genet. 2014, 127, 2253–2266. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.H.; Schertz, K.F.; Lin, Y.; Liu, S.; Chang, Y. The weediness of wild plants: Molecular analysis of genes influencing dispersal and persistence of johnsongrass, Sorghum halepense (L.) Pers. Proc. Natl. Acad. Sci. USA 1995, 92, 6127–6131. [Google Scholar] [CrossRef] [Green Version]
- Witt Hmon, K.P.; Shehzad, T.; Okuno, K. QTLs underlying inflorescence architecture in sorghum (Sorghum bicolor (L.) Moench) as detected by association analysis. Genet. Resour. Crop Evol. 2014, 61, 1545–1564. [Google Scholar] [CrossRef]
- Hart, G.; Schertz, K.; Peng, Y.; Syed, N.H. Genetic mapping of Sorghum bicolor (L.) Moench QTLs that control variation in tillering and other morphological characters. Theor. Appl. Genet. 2001, 103, 1232–1242. [Google Scholar] [CrossRef]
- Bai, C.; Wang, C.; Wang, P.; Zhu, Z.; Cong, L.; Li, D.; Liu, Y.; Zheng, W.; Lu, X. QTL mapping of agronomically important traits in sorghum (Sorghum bicolor L.). Euphytica 2017, 213, 285. [Google Scholar] [CrossRef]
- Shiringani, A.L.; Frisch, M.; Friedt, W. Genetic mapping of QTLs for sugar-related traits in a RIL population of Sorghum bicolor L. Moench. Theor. Appl. Genet. 2010, 121, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Ritter, K.B.; Jordan, D.R.; Chapman, S.C.; Godwin, I.D.; Mace, E.S.; McIntyre, C.L. Identification of QTL for sugar-related traits in a sweet × grain sorghum (Sorghum bicolor L. Moench) recombinant inbred population. Mol. Breed. 2008, 22, 367–384. [Google Scholar] [CrossRef]
- Wang, H.-L.; Zhang, H.-W.; Du, R.-H.; Chen, G.-L.; Liu, B.; Yang, Y.-B.; Qin, L.; Cheng, E.-Y.; Liu, Q.; Guanet, Y.-A. Identification and validation of QTLs controlling multiple traits in sorghum. Crop Pasture Sci. 2016, 67, 193–203. [Google Scholar] [CrossRef]
- Zou, G.; Zhai, G.; Feng, Q.; Yan, S.; Wang, A.; Zhao, Q.; Shao, J.; Zhang, Z.; Zou, J.; Han, B.; et al. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J. Exp. Bot. 2012, 63, 5451–5462. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Kumaravadivel, N. Understanding genetic diversity of sorghum using quantitative traits. Scientifica 2016, 2016, 3075023. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, L.; Li, J.; Zhan, Q.; Zhang, Q.; Li, J.; Fan, F. QTL mapping of forage yield and forage yield component traits in Sorghum bicolor × S. Sudanense. Genet. Mol. Res. 2015, 14, 3854–3861. [Google Scholar] [CrossRef]
- Guan, Y.-A.; Wang, H.-L.; Qin, L.; Zhang, H.-W.; Yang, Y.-B.; Gao, F.-J.; Li, R.-Y.; Wang, H.-G. QTL mapping of bio-energy related traits in Sorghum. Euphytica 2011, 182, 431–440. [Google Scholar] [CrossRef]
- Shukla, S.; Felderhoff, T.J.; Saballos, A.; Vermerris, W. The relationship between plant height and sugar accumulation in the stems of sweet sorghum (Sorghum bicolor (L.) Moench). Field Crop Res. 2017, 203, 181–191. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Liu, B.; Yang, Y.; Qin, L.; Chen, E.; Zhang, H.; Guan, Y. QTL analysis of salt tolerance in Sorghum bicolor during whole-plant growth stages. Plant Breed. 2020, 139, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, M.T.; Salas Fernandez, M.G.; Casa, A.M.; Mitchell, S.E.; Paterson, A.H.; Kresovich, S. Equilibrium processes cannot explain high levels of short- and medium-range linkage disequilibrium in the domesticated grass Sorghum bicolor. Genetics 2005, 171, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-H.; Upadhyaya, H.D.; Burrell, A.M.; Sahraeian, S.M.E.; Klein, R.R.; Klein, P.E. Genetic structure and linkage disequilibrium in a diverse, representative collection of the C4 model plant, Sorghum bicolor. G3 2013, 3, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchet, S.; Pot, D.; Deu, M.; Rami, J.-F.; Billot, C.; Perrier, X.; Rivallan, R.; Gardes, L.; Xia, L.; Wenzl, P.; et al. Genetic structure, linkage disequilibrium and signature of selection in sorghum: Lessons from physically anchored dart markers. PLoS ONE 2012, 7, e33470. [Google Scholar] [CrossRef] [Green Version]
- Marla, S.R.; Burow, G.; Chopra, R.; Hayes, C.; Olatoye, M.O.; Felderhoff, T.; Hu, Z.; Raymundo, R.; Perumal, R.; Morris, G.P. Genetic architecture of chilling tolerance in sorghum dissected with a nested association mapping population. G3 (Bethesda) 2019, 9, 4045–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Acharya, A.; Burrell, A.M.; Klein, R.R.; Klein, P.E.; Hasenstein, K.H. Mapping and candidate genes associated with saccharification yield in sorghum. Genome 2013, 56, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.C.; Rooney, R.L.; Mitchell, S.E.; Sharma, A.; Klein, P.E.; Mullet, J.E.; Kresovich, S. Genetic improvement of sorghum as a biofuel feedstock: II. QTL for stem and leaf structural carbohydrates. Crop Sci. 2008, 48, 2180–2193. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, R.; Liu, B.; Chen, E.; Yang, Y.; Qin, L.; Li, F.; Gao, F.; Cao, P.; Wang, H.; et al. Inclusive composite-interval mapping reveals quantitative trait loci for plant architectural traits in sorghum (Sorghum bicolor). Crop Pasture Sci. 2019, 70, 659–668. [Google Scholar] [CrossRef]
- Guo, Y.; Yuan, H.; Fang, D.; Song, L.; Liu, Y.; Liu, Y.; Wu, L.; Yu, J.; Li, Z.; Xu, X.; et al. An improved 2b-RAD approach (I2b-RAD) offering genotyping tested by a rice (Oryza sativa L.) F2 population. BMC Genom. 2014, 15, 956. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.-Y.; Wang, L.Y.; Wei, K.; Tan, L.-Q.; Su, J.-J.; Cheng, H. High-density SNP linkage map construction and QTL mapping for flavonoid-related traits in a tea plant (Camellia sinensis) using 2b-RAD sequencing. BMC Genom. 2018, 19, 955. [Google Scholar] [CrossRef] [Green Version]
- Hilley, J.; Truong, S.; Olson, S.; Morishige, D.; Mullet, J. Identification of Dw1, a regulator of sorghum stem internode length. PLoS ONE 2016, 11, e0151271. [Google Scholar] [CrossRef] [Green Version]
- Strader, L.C.; Culler, A.H.; Cohen, J.D.; Bartel, B. Conversion of endogenous indole-3-butyric acid to indole-3-acetic acid drives cell expansion in Arabidopsis seedlings. Plant Physiol. 2010, 153, 1577–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Wang, J.; Gao, Z.; Dong, J.; He, H.; Terzaghi, W.; Wei, N.; Deng, X.; Chen, H. Arabidopsis SAURs are critical for differential light regulation of the development of various organs. Proc. Natl. Acad. Sci. USA 2016, 113, 6071–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Nagpal, P.; Vitart, V.; McMorris, T.C.; Chory, J. A role for brassinosteroids in light-dependent development of Arabidopsis. Science 1996, 272, 398–401. [Google Scholar] [CrossRef]
- Schröder, F.; Lisso, J.; Lange, P.; Müssig, C. The extracellular EXO protein mediates cell expansion in Arabidopsis leaves. BMC Plant Biol. 2009, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Liang, G.; Yang, S.; Yu, D. Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid– and auxin-mediated signaling in jasmonic acid–induced leaf senescence. Plant Cell. 2014, 26, 230–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boorer, K.J.; Loo, D.D.; Wright, E.M. Steady-state and presteady-state kinetics of the H+/hexose cotransporter (STP1) from Arabidopsis thaliana expressed in Xenopus oocytes. J. Biol. Chem. 1994, 269, 20417–20424. [Google Scholar]
- Büttner, M.; Sauer, N. Monosaccharide transporters in plants: Structure, function and physiology. Biochim. Biophys. Acta 2000, 1465, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.A.; Thomas, M.A. The monosaccharide transporter gene family in Arabidopsis and rice: A history of duplications, adaptive evolution, and functional divergence. Mol. Biol. Evol. 2007, 24, 2412–2423. [Google Scholar] [CrossRef]
- Cordoba, E.; Aceves-Zamudio, D.L.; Hernández-Bernal, A.F.; Ramos-Vega, M.; León, P. Sugar regulation of SUGAR TRANSPORTER PROTEIN 1 (STP1) expression in Arabidopsis thaliana. J. Exp. Bot. 2015, 66, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, M.A.; Santos, E.; Flores, C.L.; Martínez-Zapater, J.M.; Salinas, J.; Gancedo, C. Isolation and molecular characterization of the Arabidopsis TPS1 gene, encoding trehalose-6-phosphate synthase. Plant J. 1998, 13, 685–689. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Min | Max | Mean | Heritability | Genotype (G) | Environment (E) | G × E |
|---|---|---|---|---|---|---|---|
| HT (cm) | 83.3 | 355.7 | 235.3 | 0.92 | *** | *** | *** |
| TN (number) | 0.67 | 6.44 | 2.71 | 0.89 | *** | ** | *** |
| IN (number) | 7.00 | 13.4 | 10.3 | 0.84 | *** | ** | *** |
| SD (cm) | 0.68 | 2.74 | 1.56 | 0.86 | *** | * | *** |
| PL (cm) | 14.0 | 42.4 | 26.2 | 0.80 | *** | *** | *** |
| PW (g) | 17.0 | 125.0 | 57.3 | 0.54 | *** | *** | *** |
| RS (g/L) | 0.14 | 1.21 | 0.41 | 0.76 | *** | ** | *** |
| Brix (%) | 6.70 | 22.4 | 14.1 | 0.73 | *** | *** | *** |
| PRO (%) | 2.79 | 7.99 | 4.94 | 0.52 | *** | *** | *** |
| HT | TN | IN | SD | PL | PW | RS | Brix | PRO | |
|---|---|---|---|---|---|---|---|---|---|
| HT | |||||||||
| TN | 0.43 *** | ||||||||
| IN | 0.21 *** | −0.53 *** | |||||||
| SD | −0.59 *** | −0.82 *** | 0.52 *** | ||||||
| PL | −0.08 | 0.33 *** | −0.38 *** | −0.14 * | |||||
| PW | −0.26 *** | −0.50 *** | 0.31 *** | 0.53 *** | 0.04 | ||||
| RS | 0.48 *** | −0.004 | 0.35 *** | −0.12 * | −0.35 *** | −0.09 | |||
| Brix | 0.52 *** | −0.15 * | 0.59 *** | 0.03 | −0.45 *** | −0.07 | 0.54 *** | ||
| PRO | −0.73 *** | −0.46 *** | 0.03 | 0.58 *** | −0.002 | 0.14 * | −0.38 *** | −0.27 *** |
| Group | HT (cm) | TN | IN | SD (cm) | PL (cm) | PW (g) | RS (g/L) | Brix (%) | PRO (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 238.8 ab | 2.4 b | 10.6 b | 1.6 b | 25.4 b | 66.2 a | 0.42 a | 14.3 b | 5.0 a |
| 2 | 250.4 a | 3.9 a | 9.1 c | 1.2 c | 28.7 a | 47.9 c | 0.35 b | 12.1 c | 4.5 b |
| 3 | 223.0 b | 2.1 c | 11.1 a | 1.8 a | 25.0 b | 58.5 b | 0.45 a | 15.2 a | 5.2 a |
| Traits | SNP Position | Chromosome | p-values | R2 (%) | Comments |
|---|---|---|---|---|---|
| HT | 53230521 | 3 | 5.11 × 10−6 | 9.5 | Novel region |
| 50424256 | 8 | 3.77 × 10−6 | 9.0 | Overlap [51] | |
| 56656748 | 9 | 1.78 × 10−7 | 11.3 | Overlap [19,36,52,53,54,55,56,57] | |
| TN | 63990205 | 2 | 2.68 × 10−6 | 9.1 | Overlap [58] |
| 46267204 | 6 | 3.61 × 10−7 | 10.1 | Overlap [36,55,59] | |
| 54924780 | 8 | 1.07 × 10−6 | 9.4 | Novel region | |
| 52354291 | 9 | 5.51 × 10−6 | 8.7 | Overlap [55] | |
| PL | 11646706 | 1 | 1.26 × 10−8 | 13.2 | Overlap [36] |
| 794419 | 2 | 6.11 × 10−6 | 8.5 | Overlap [51,60] | |
| 49724219 | 8 | 3.83 × 10−6 | 9.6 | Overlap [36] | |
| 1803717 | 10 | 5.63 × 10−7 | 10.3 | Overlap [61] | |
| Brix | 216438 | 5 | 8.12 × 10−7 | 11.2 | Novel region |
| 39878130 | 6 | 3.56 × 10−7 | 11.5 | Overlap [62] | |
| 40217639 | 8 | 6.02 × 10−6 | 8.9 | Overlap [63] | |
| RS | 50338536 | 1 | 3.35 × 10−6 | 10.7 | Overlap [52,64] |
| 50949572 | 1 | 3.86 × 10−6 | 8.9 | Overlap [52,64] | |
| 51456627 | 1 | 3.89 × 10−6 | 8.9 | Overlap [52,64] | |
| 51729456 | 1 | 5.08 × 10−6 | 9.9 | Overlap [52,64] | |
| 65963213 | 2 | 3.43 × 10−6 | 9.5 | Overlap [63] | |
| 70725655 | 2 | 1.24 × 10−6 | 9.8 | Novel region | |
| 20424005 | 3 | 5.49 × 10−6 | 8.7 | Overlap [64] | |
| 83605 | 9 | 5.60 × 10−6 | 8.9 | Novel region | |
| 43662454 | 9 | 1.14 × 10−7 | 12.5 | Novel region | |
| 57577048 | 10 | 1.82 × 10−6 | 10.3 | Novel region |
| Trait | SNP | Gene ID (V1.4) | Distance to peak SNP (kb) | Homolog to A. thaliana | Encoding Protein | Function/Biological Process | Gene ID (V3.1) |
|---|---|---|---|---|---|---|---|
| HT | S3-53230521 | Sb03g026400 | 40.1 | AT1G76190 | SAUR-like auxin-responsive protein | Responsive to auxin | Sobic.003G202000 |
| S8-50424256 | Sb08g019600 | 41.2 | AT2G25760 | Protein kinase | Protein binding | Sobic.008G146100 | |
| S9-56656748 | Sb09g026370 | 0.26 | AT4G14430 | Indole-3-butyric acid response 10 | Auxin metabolism | Sobic.009G207100 | |
| TN | S2-63990205 | Sb02g028870 | 3.7 | AT2G24430 | NAC domain containing protein 38 | Transcription factor | Sobic.002G253000 |
| S6-46267204 | Sb06g017100 | 0.47 | AT2G01170 | Bidirectional amino acid transporter 1 | Amino acid transport | Sobic.006G084600 | |
| S8-54924780 | Sb08g022790 | 33.4 | AT5G49660 | Receptor protein-tyrosine kinase | Growth and development | Sobic.008G186400 | |
| S9-52354291 | Sb09g022660 | 72.5 | AT2G28710 | C2H2 zinc finger protein | Transcription factor | Sobic.009G164600 | |
| PL | S1-11646700 | Sb01g012660 | 0.38 | AT1G67580 | Protein kinase family protein | Signal transduction | Sobic.001G145100 |
| S2-794419 | Sb02g000960 | 0.69 | AT1G69310 | WRKY57 | Hormone signaling | Sobic.002G008600 | |
| S8-49724219 | Sb08g019140 | 59.4 | AT5G19960 | RNA binding protein | Growth and development | Sobic.008G140700 | |
| S10-1803717 | Sb10g002190 | 52.3 | AT5G51550 | Exordium-like 3 | Brassinosteroid regulatory | Sobic.010G023400 | |
| Brix | S5-216438 | Sb05g000330 | 76.9 | AT2G35800 | Mitochondrial substrate carrier protein | Substrates transport | Sobic.005G001700 |
| S6-39878130 | Sb06g014370 | 94.5 | AT2G33490 | Hydroxyproline-rich glycoprotein protein | Cell wall composition | Sobic.006G055400 | |
| S8-40217639 | Sb08g015300 | 9.6 | AT4G18750 | Pentatricopeptide protein | Growth and development | Sobic.008G100400 | |
| RS | S1-50338536 | Sb01g028790 | 88.0 | AT2G44730 | MYB transcription factor | Alcohol dehydrogenase | Sobic.001G294700 |
| S1-50949572 | Sb01g029170 | 5.5 | AT3G60530 | Zinc finger family protein | GATA transcription factor | Sobic.001G299600 | |
| S1-51456627 | Sb01g029400 | 85.1 | AT2G35710 | Glucosyltransferase | Glucose transfer | Sobic.001G302200 | |
| S1-51729456 | Sb01g029590 | 79.7 | AT4G22590 | Trehalose-6-phosphate phosphatase | Trehalose biosynthesis | Sobic.001G303900 | |
| S2-65963213 | Sb02g030990 | 33.8 | AT1G75680 | Glycoside hydrolase 9B7 | Polysaccharide metabolism | Sobic.002G276600 | |
| S2-70725655 | Sb02g036310 | 5.1 | AT1G11260 | Sugar transporter 1 | Monosaccharide transport | Sobic.002G338500 | |
| S3-20424005 | Sb03g014780 | 23.7 | AT3G06400 | Chromatin-remodeling protein | Protein binding | Sobic.003G163200 | |
| S9-83605 | Sb09g000320 | 64.2 | AT1G57820 | C3HC4 zinc finger protein | Transcription factor | Sobic.009G001200 | |
| S9-43662454 | Sb09g017540 | 21.4 | AT3G06760 | Drought responsive family protein | Unknown function | Sobic.009G108600 | |
| S10-57577048 | Sb10g027790 | 46.8 | AT4G30080 | Auxin response factor 16 | Auxin-activated signaling | Sobic.010G236300 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, F.; Pei, Z.; Zhao, X.; Liu, H.; Jiang, Y.; Sun, S. Genome-Wide Association Study for Plant Architecture and Bioenergy Traits in Diverse Sorghum and Sudangrass Germplasm. Agronomy 2020, 10, 1602. https://doi.org/10.3390/agronomy10101602
Luo F, Pei Z, Zhao X, Liu H, Jiang Y, Sun S. Genome-Wide Association Study for Plant Architecture and Bioenergy Traits in Diverse Sorghum and Sudangrass Germplasm. Agronomy. 2020; 10(10):1602. https://doi.org/10.3390/agronomy10101602
Chicago/Turabian StyleLuo, Feng, Zhongyou Pei, Xiongwei Zhao, Huifen Liu, Yiwei Jiang, and Shoujun Sun. 2020. "Genome-Wide Association Study for Plant Architecture and Bioenergy Traits in Diverse Sorghum and Sudangrass Germplasm" Agronomy 10, no. 10: 1602. https://doi.org/10.3390/agronomy10101602