
Effect of the Acid Medium on the Synthesis of Polybenzimidazoles Using Eaton’s Reagent

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
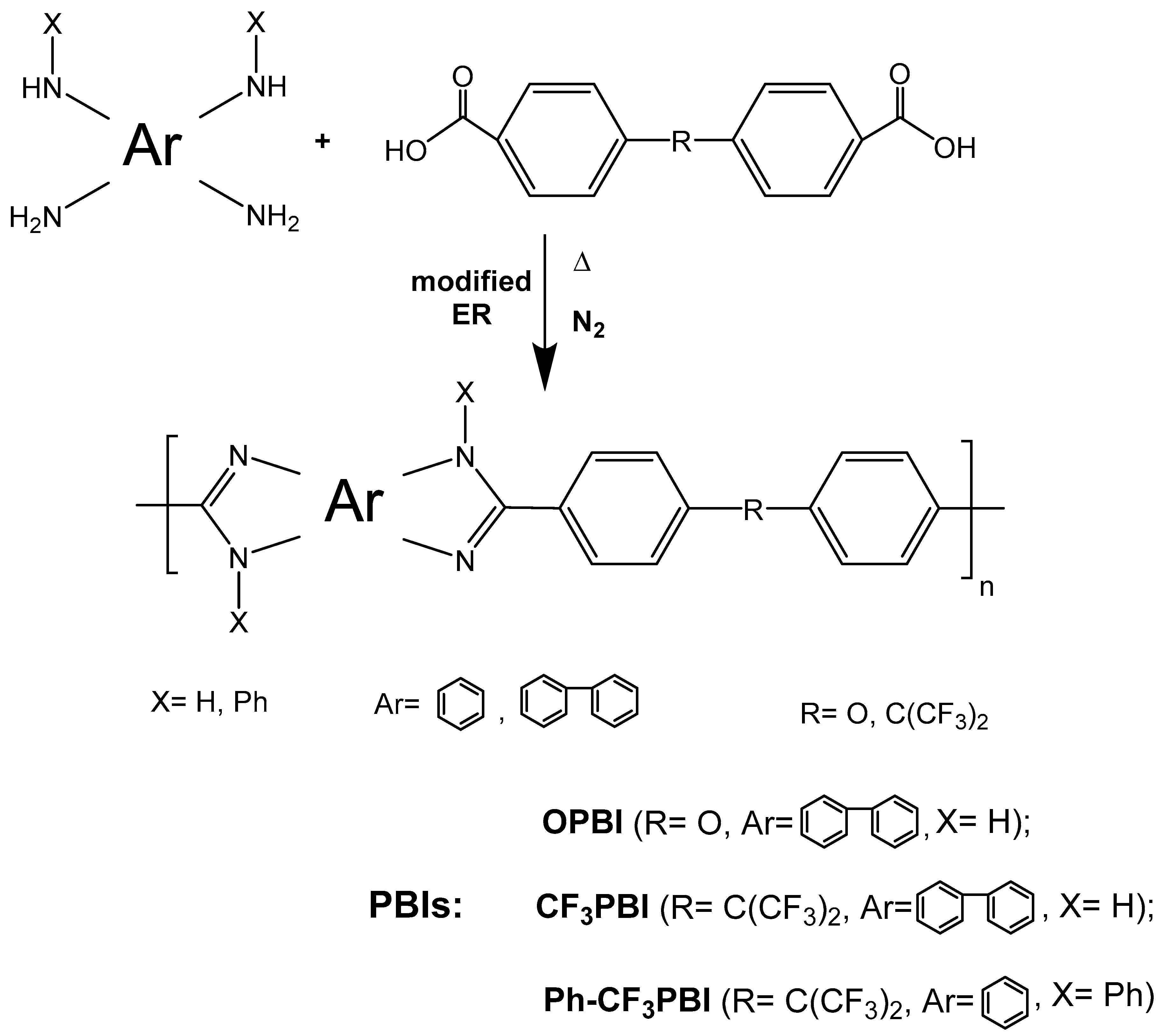
2.2. Syntheses of Polymers
2.3. Preparation of PBI Films
2.4. Characterization Methods
3. Results and Discussion
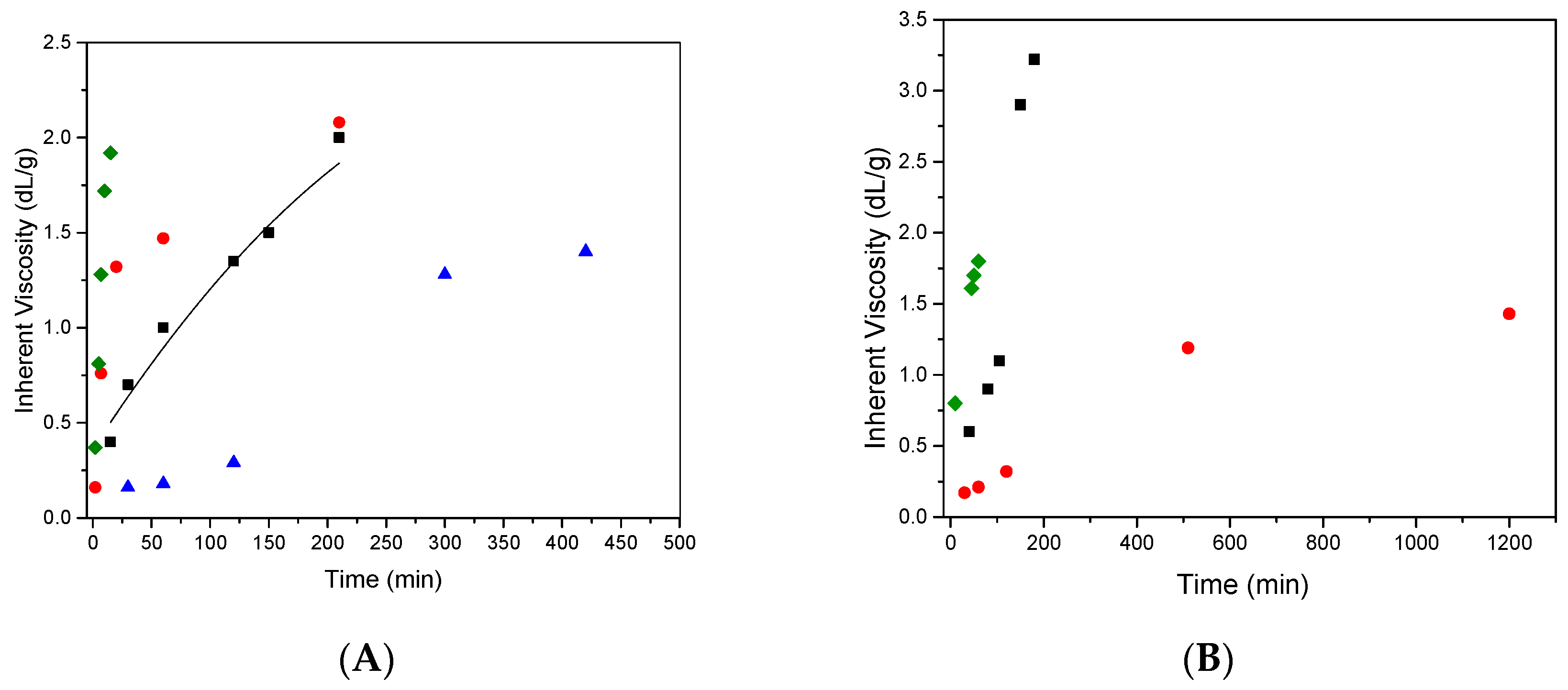
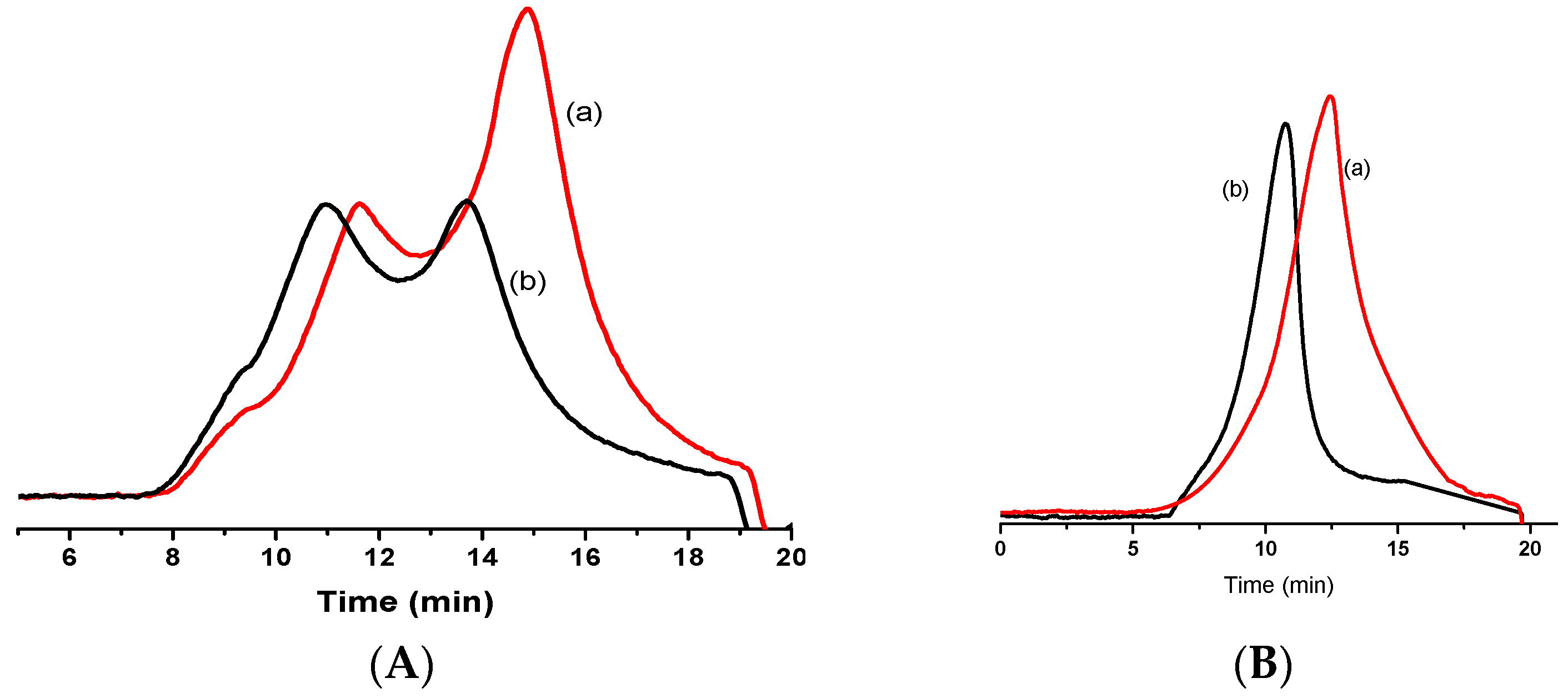
3.1. OPBI and CF3PBI Synthesis
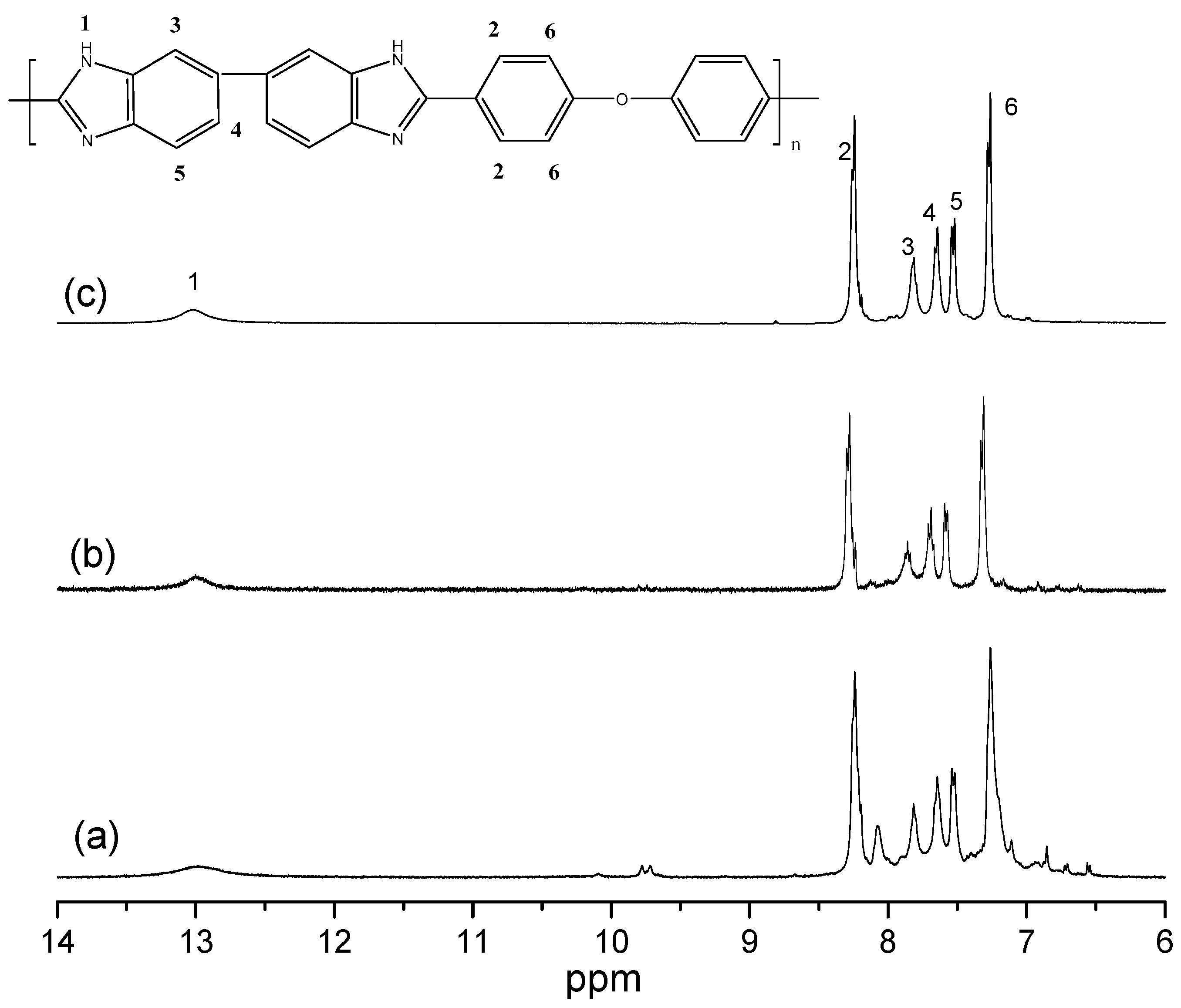
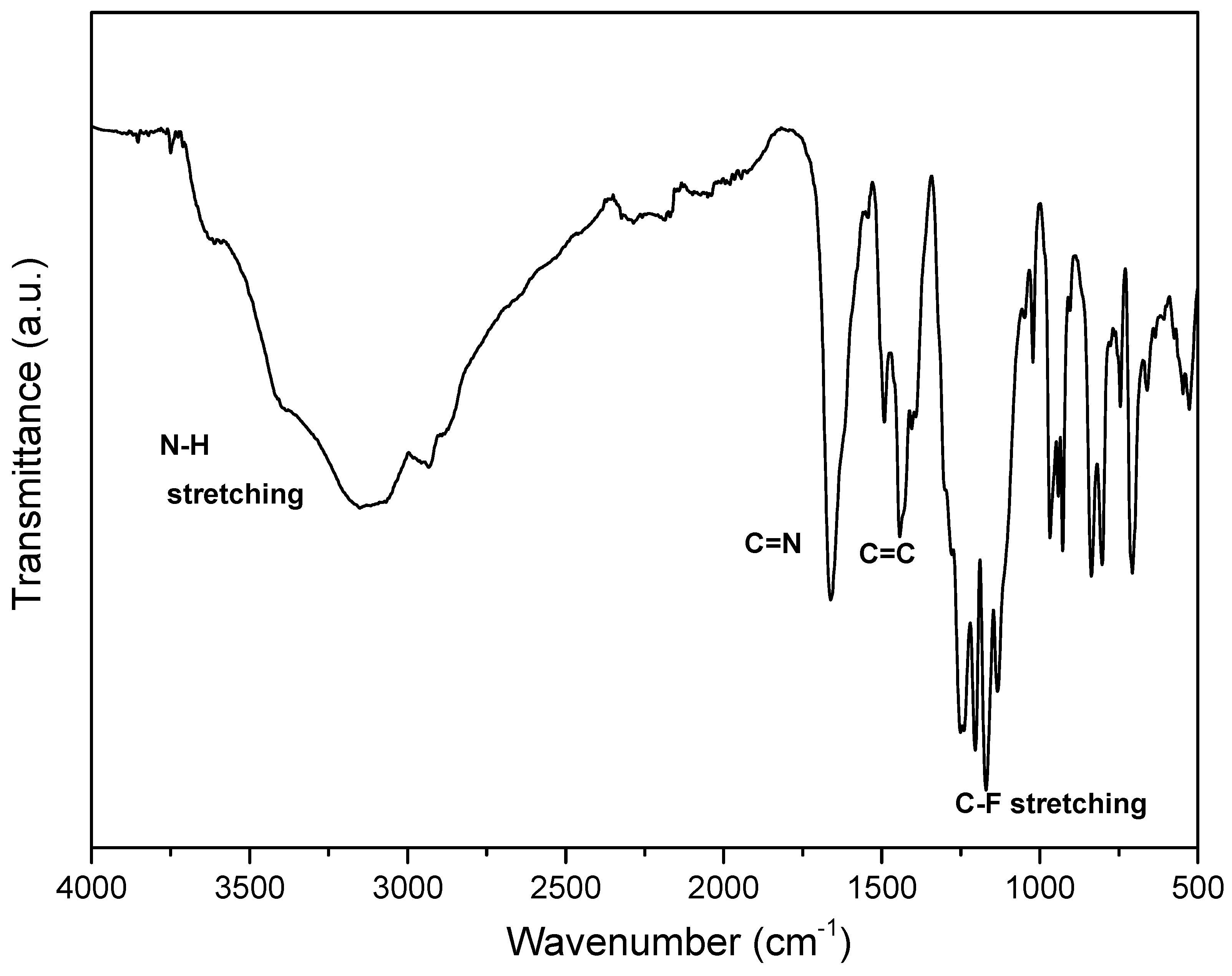
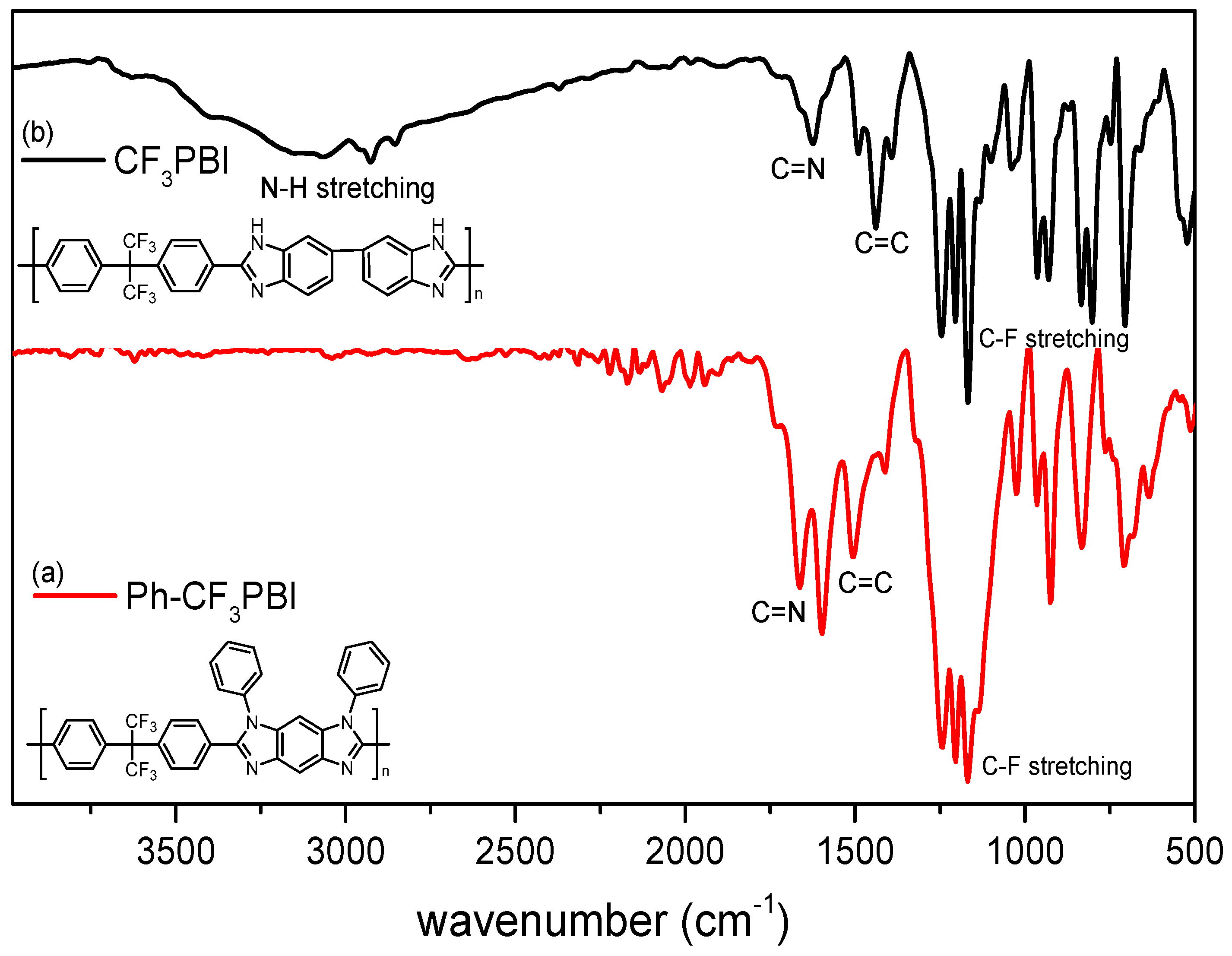
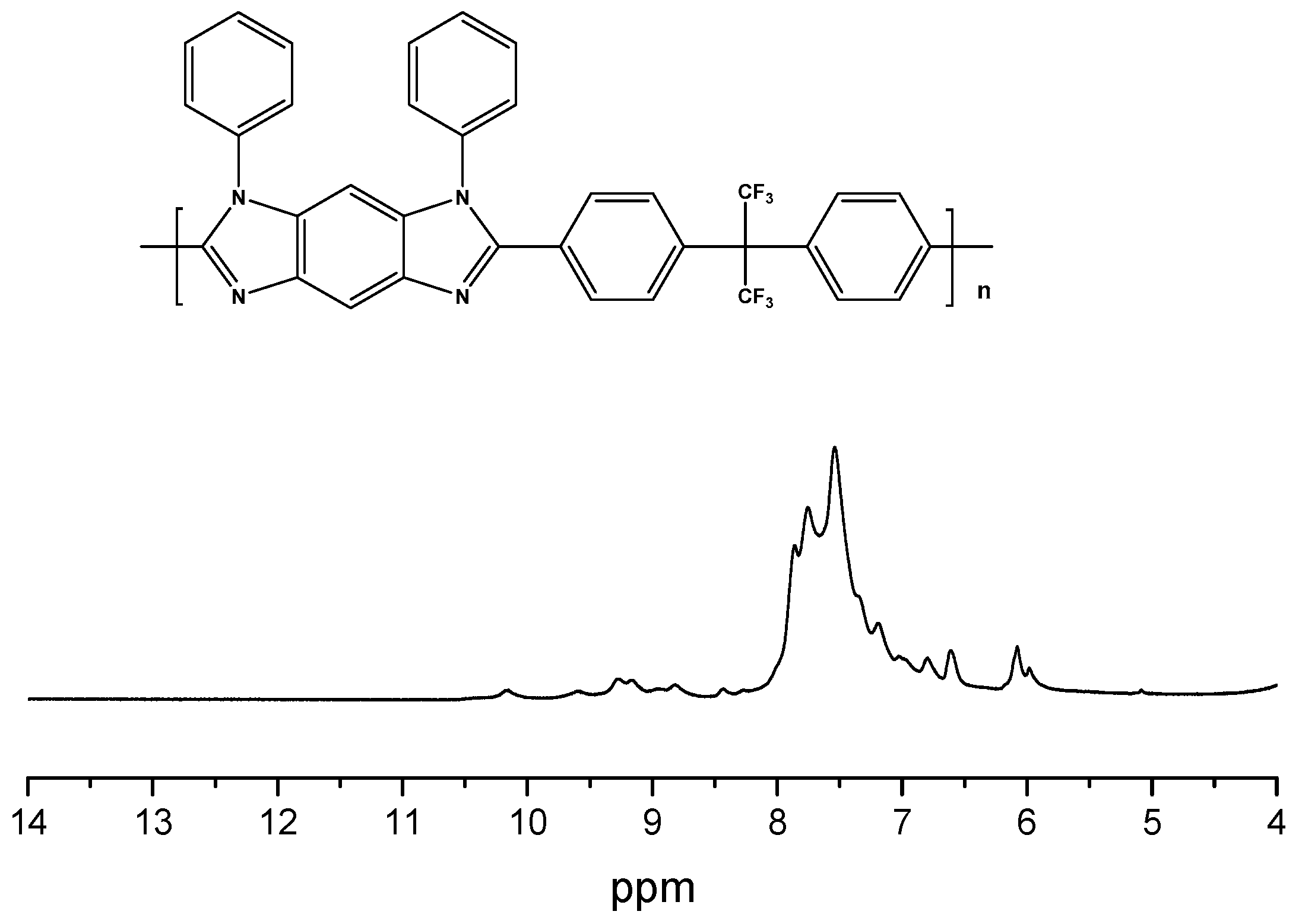
3.2. IR and NMR Studies
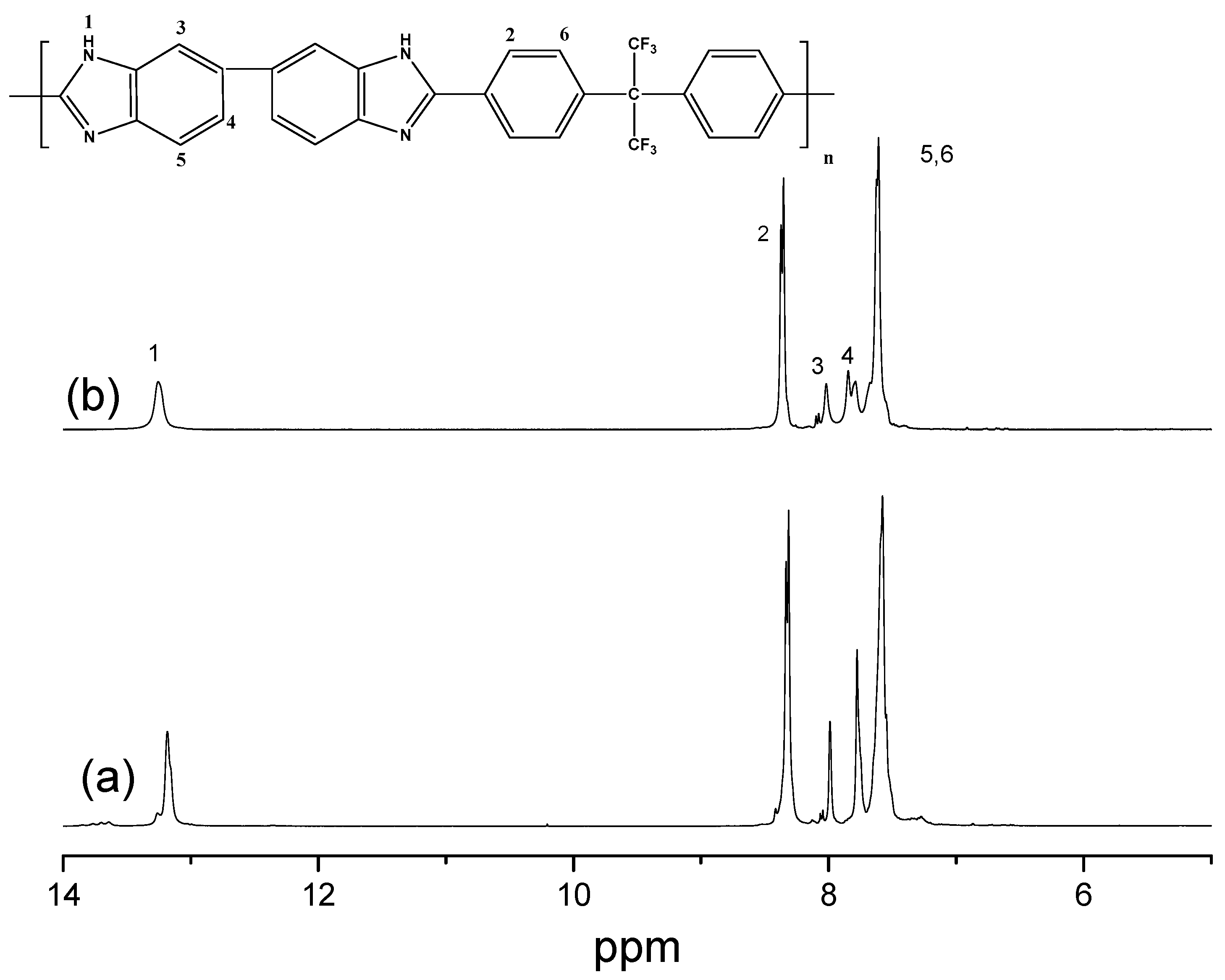
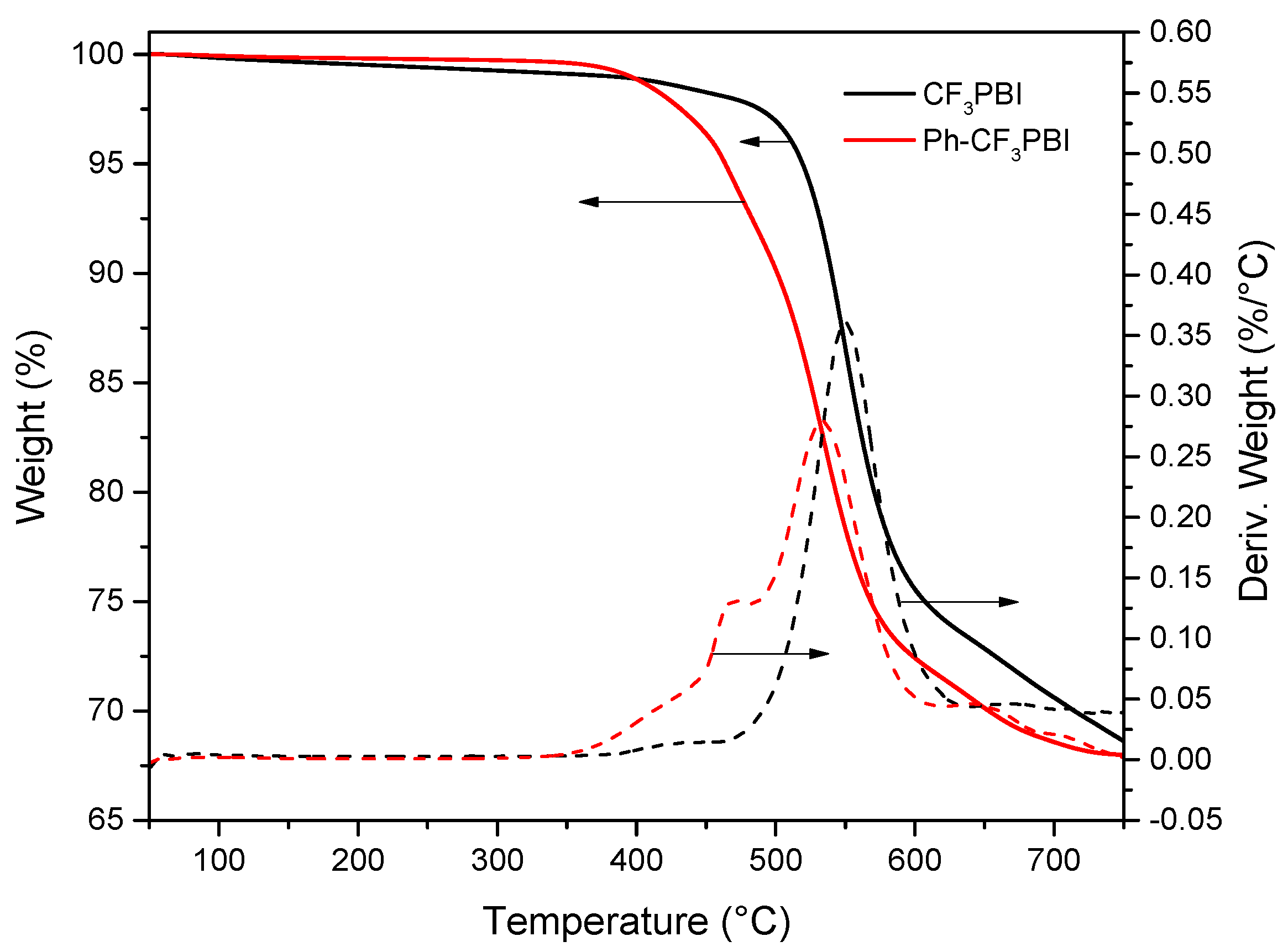
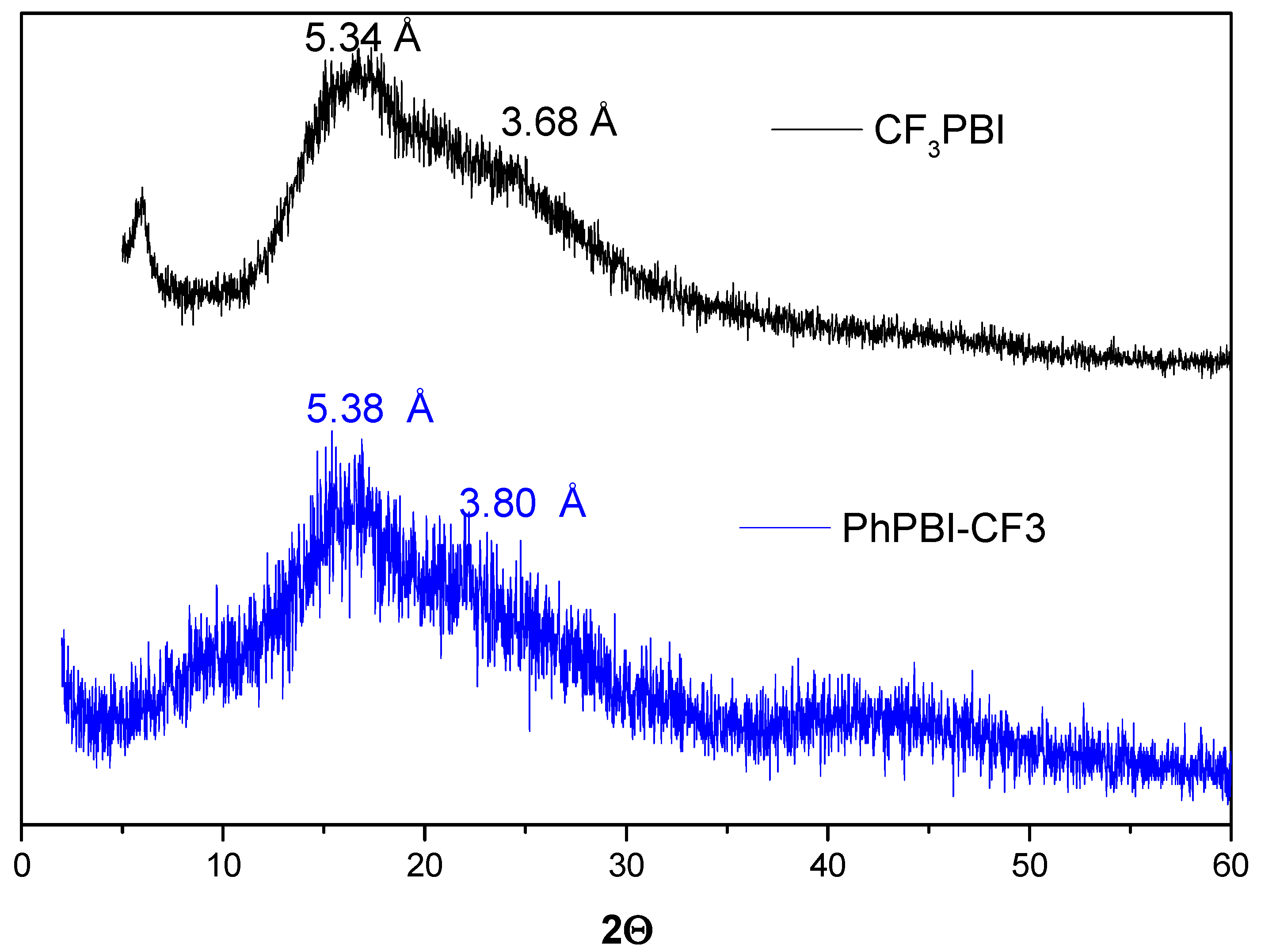
3.3. Synthesis of Ph-CF3PBI
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neuse, E.W. Aromatic polybenzimidazoles. Adv. Polym. Sci. 1982, 47, 1–42. [Google Scholar]
- Wang, Y.; Yang, T.; Fishel, K.; Benicewicz, B.C.; Chung, T.-S. Polybenzimidazoles. In Handbook of Thermoplastics, 2nd ed.; Olabisi, O., Adewale, K., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2016; pp. 617–667. ISBN 9780429101625. [Google Scholar] [CrossRef]
- Aili, D.; Yang, J.; Jankova, K.; Henkensmeier, D.; Li, O. From polybenzimidazoles to polybenzimidazoliums and polybenzimidazolides. J. Mater. Chem. A 2020, 8, 12854–12886. [Google Scholar] [CrossRef]
- Li, Q.; Jensen, J.O.; Savinell, R.F.; Bjerrum, N.J. High temperature proton exchange membranes based on polybenzimidazoles for fuel cells. Prog. Polym. Sci. 2009, 34, 449–477. [Google Scholar] [CrossRef] [Green Version]
- Ng, F.; Perón, J.; Jones, D.J.; Rozière, J. Synthesis of novel proton-conducting highly sulfonated polybenzimidazoles for PEMFC and the effect of the type of bisphenyl bridge on polymer and membrane properties. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 2107–2117. [Google Scholar] [CrossRef]
- Quartarone, E.; Mustarelli, P. Polymer fuel cells based on polybenzimidazole/H3PO4. Energy Environ. Sci. 2012, 5, 6436–6444. [Google Scholar] [CrossRef]
- Ponomarev, I.I.; Ponomarev, I.I.; Petrovskii, P.V.; Volkova, Y.A.; Razorenov, D.Y.; Goryunova, I.B.; Starikova, A.A.; Fomenkov, A.I.; Khokhlov, A.R. Synthesis of N-phosphonoethylated cardo polybenzimidazole and Testing of proton-conducting membranes made of it. Dokl. Akad. Nauk. 2010, 432, 168–174. [Google Scholar] [CrossRef]
- Escorihuela, J.; Olvera-Mancilla, J.; Alexandrova, L.; del Castillo, L.F.; Compañ, V. Recent Progress in the Development of Composite Membranes Based on Polybenzimidazole for High Temperature Proton Exchange Membrane (PEM) Fuel Cell Applications. Polymers 2020, 12, 1861. [Google Scholar] [CrossRef] [PubMed]
- Bye, K.P.; Loianno, V.; Pham, T.N.; Liu, R.; Riffle, J.S.; Galizia, M. Pure and Mixed fluid Sorption and Transport in Celazole Polybenzimidazole: Effect of Plasticization. J. Membr. Sci. 2019, 580, 235–247. [Google Scholar] [CrossRef]
- Borjigin, H.; Stevens, K.A.; Liu, R.; Moon, J.D.; Shaver, A.T.; Swinnea, S.; Freeman, B.; Riffle, J.; McGrath, J. Synthesis and Characterization of Polybenzimidazoles Derived from Tetraaminodiphenylsulfone for High Temperature Gas Separation Membranes. Polymers 2015, 71, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Singh, R.P.; Dudeck, K.W.; Berchtold, K.A.; Benicewicz, B.C. Influence of polybenzimidazole main chain structure on H2/CO2 separation at elevated temperatures. J. Membr. Sci. 2014, 461, 59–68. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, D.H.; Kim, D.Y.; Han, J.Y.; Yoon, C.W.; Ham, H.C.; Kim, J.-H.; Kim, H.-J.; Nam, S.W.; Lim, T.-H.; et al. A Highly Selective Polybenzimidazole-4,4′-(hexafluoroisopropylidene)bis(benzoic acid) Membrane for High Temperature Hydrogen Separation. J. Appl. Polym. Sci. 2015, 132, 42371. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, J.Y.; Kim, H.-J.; Kim, M.-H.; Han, S.G.; Jang, J.H.; Cho, E.A.; Yoo, S.J.; Henkensmeier, D. Synthesis and Charachterization of Fluorene-Based Polybenzimidazole Copolymer for Gas Separation. J. Appl. Polym. Sci. 2014, 131, 40521. [Google Scholar] [CrossRef]
- Kumbharkar, S.C.; Kharul, U.K. N-substitution of polybenzimidazoles: Synthesis and evaluation of physical properties. Eur. Polym. J. 2009, 45, 3363–3371. [Google Scholar] [CrossRef]
- Huang, C.-H.; Liu, Y.-L. Self-healing polymeric materials for membrane separation: An example of a polybenzimidazole-based membrane for pervaporation dehydration on isopropanol aqueous solution. RSC Adv. 2017, 7, 38360–38366. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.Y.; Xiao, Y.; Chung, T.-S. Chemically modified polybenzimidazole nanofiltration membrane for the separation of electrolytes and cephalexin. Chem. Eng. Sci. 2006, 61, 5807–5817. [Google Scholar] [CrossRef]
- Vogel, H.; Marvel, C.S. Polybenzimidazoles, new thermally stable polymers. J. Polym. Sci. 1961, 50, 511–539. [Google Scholar] [CrossRef]
- Iwakura, Y.; Uno, J.; Imai, Y. Polyphenylenebenzimidazoles. J. Polym. Sci. Part A Polym. Chem. 1964, 2, 2605. [Google Scholar] [CrossRef]
- Ueda, M.; Sato, M.; Mochizuki, A. Poly(Benzimidazole) Synthesis by Direct Reaction of Diacids and Tetramine. Macromolecules 1985, 18, 2723–2726. [Google Scholar] [CrossRef]
- Leykin, A.Y.; Fomenkov, A.I.; Galperin, E.G.; Stankevich, I.V.; Rusanov, A.L. Some aspects of polybenzimidazoles’ synthesis in P2O5 containing condensation media. Polymers 2010, 51, 4053–4057. [Google Scholar] [CrossRef]
- Qian, G.; Smith, D.W., Jr.; Benicewicz, B.C. Synthesis and characterization of high molecular weight perfluorocyclobutyl-containing polybenzimidazoles (PFCB-PBI) for high temperature polymer electrolyte membrane fuel cells. Polymers 2009, 50, 3911–3916. [Google Scholar] [CrossRef]
- Qing, S.; Huang, W.; Yan, D. Synthesis and characterization of thermally stable sulfonated polybenzimidazoles. Eur. Polym. J. 2005, 41, 1589–1595. [Google Scholar] [CrossRef]
- Kulkarni, M.; Potrekar, R.; Kulkarni, R.A.; Vernekar, P. Synthesis and characterization of novel polybenzimidazoles bearing pendant phenoxyamine groups. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 5776–5793. [Google Scholar] [CrossRef]
- Kim, E.-K.; Jung, J.; Cho, K.; Yun, G.-J.; Lee, J.-C. Synthesis of polybenzimidazoles having improved processability by introducing two and three ether groups in a repeating unit. Eur. Polym. J. 2022, 162, 110900. [Google Scholar] [CrossRef]
- Yang, J.; Xu, Y.; Zhou, L.; Che, Q.; He, R.-H.; Li, Q. Hydroxyl pyridine containing polybenzimidazole membrane for proton exchange membrane fuel cells. J. Membr. Sci. 2013, 446, 318–325. [Google Scholar] [CrossRef]
- Imai, Y.; Taoka, I.; Uno, K.; Iwakura, Y. Polybenzoxazoles and polybenzothiazoles. Die Makromol. Chem. Macromol. Chem. Phys. 1965, 83, 167–178. [Google Scholar] [CrossRef]
- Ghaffari, F.; Hodd, K.A. Studies in heterocyclic polymers. Part VI. A weight-loss study of the formation of a polybenzoxazole. Thermochim. Acta 1980, 41, 213–224. [Google Scholar] [CrossRef]
- So, Y.-H.; Heeschen, J.P. Mechanism of polyphosphoric acid and phosphorus pentoxide-methanesulfonic acid as synthetic reagents for benzoxazole formation. J. Org. Chem. 1997, 62, 3552–3561. [Google Scholar] [CrossRef]
- Korshak, V.V.; Rusanov, A.L.; Tugushi, D.S.; Cherkasova, G.M. Two-stage Synthesis of Poly(N-phenylbenzimidazoles). Macromolecules 1972, 5, 807–812. [Google Scholar] [CrossRef]
- Ponomarev, I.I.; Razorenov, D.Y.; Ponomarev, I.I.; Volkova, Y.A.; Skupov, K.M.; Lysova, A.A.; Yaroslavtsev, A.B.; Modestov, A.D.; Buzin, M.I.; Klemenkova, Z.S. Polybenzimidazoles via polyamidation: A more environmentally safe process to proton conducting membrane for hydrogen HT-PEM fuel cell. Eur. Polym. J. 2021, 156, 110613. [Google Scholar] [CrossRef]
- Neuse, E.; Loonat, M.S. Two-stage Polybenzimidazole Synthesis via Poly(azomethine) Intermediates. Macromolecules 1983, 16, 128–136. [Google Scholar] [CrossRef]
- Olvera-Mancilla, J.; Palacios-Alquisira, J.; Alexandrova, L. Eaton’s reagent in polybenzimidazole synthesis: The influence of temperature and microwave irradiation. High Perform. Polym. 2018, 30, 699–709. [Google Scholar] [CrossRef]
- Olvera-Mancilla, J.; Aguilar-Lugo, C.; Fernandez-Gijon, C.A.; Palacios-Alquisira, J.; Zolotukhin, M.; Alexandrova, L. Processable N-Substituted Polybenzimidazole; Direct Synthesis. Chem. Select. 2020, 5, 5082–5091. [Google Scholar] [CrossRef]
- Li, X.; Qian, G.; Chen, X.; Benicewicz, B.C. Synthesis and Characterization of a New Fluorine-Containing Polybenzimidazole (PBI) for Proton-Conducting Membranes in Fuel Cells. Fuel Cells 2013, 13, 832–842. [Google Scholar] [CrossRef]
- Lira, A.L.; Zolotukhin, M.G.; Fomina, L.; Fomine, S. Triflic-Acid-Mediated Polycondensation of Carbonyl Compounds with Aromatic Hydrocarbons–A Theoretical Study. Macromol. Theor. Simul. 2007, 16, 227–239. [Google Scholar] [CrossRef]
- Zolotukhin, M.G.; Fomina, L.; Salcedo, R.; Sansores, L.E.; Colquhoun, H.M.; Khalilov, L.M. Superelectrophiles in Polymer Chemistry. A Novel, One-Pot Synthesis of High-Tg, High-Temperature Polymers. Macromolecules 2004, 37, 5140–5141. [Google Scholar] [CrossRef]
- Olvera, L.I.; Guzmán-Gutiérrez, M.T.; Zolotukhin, M.G.; Fomine, S.; Cárdenas, F.; Ruiz-Trevino, F.A.; Villers, D.; Ezquerra, T.A.; Prokhorov, E. Novel high molecular weight aromatic fluorinated polymers from one-pot, metal-free step polymerizations. Macromolecules 2013, 46, 7245–7256. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; An, S.J.; Kim, J.-Y.; Moon, J.K.; Cho, S.Y.; Eun, Y.C.; Yoon, H.-K.; Park, Y.; Kweon, H.-J.; Shin, E.-M. Polybenzimidazoles for High Temperature Fuel Cell Applications. Macromol. Rapid Commun. 2004, 25, 1410–1413. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Shukla, A.; Ghosh, P.C.; Jana, T. Rational design of microporous polybenzimidazole framework for efficient proton exchange membrane fuel cells. J. Mater. Chem. A 2022, 10, 11074–11091. [Google Scholar] [CrossRef]
- Jouanneau, J.; Mercier, R.; Gonon, L.; Gebel, G. Synthesis of Sulfonated Polybenzimidazoles from Functionalized Monomers: Preparation of Ionic Conducting Membranes. Macromolecules 2007, 40, 983–990. [Google Scholar] [CrossRef]
- Rusanov, A.L.; Komarova, L.G. Synthesis of Heterochain and Heterocyclic Condensation Polymers in Novel Reaction Media. Polym. Sci. B 2005, 47, 284–303. [Google Scholar]
- Eaton, P.E.; Mueller, R.H. Peristylae System. J. Am. Chem. Soc. 1972, 94, 1014–1016. [Google Scholar] [CrossRef]
- Eaton, P.E.; Glenn, R.C.; Lee, J.T. Phosphorus pentoxide-methanesulfonic Acid. Convenient Alternative to Polyphosphoric Acid. J. Org. Chem. 1973, 38, 4071–4073. [Google Scholar] [CrossRef]
- Olvera-Mancilla, J.; Palacios-Alquisira, J.; Escobar-Barrios, V.A.; Alexandrova, L. Some aspects of polybenzimidazoles’ synthesis in Eaton reagent under different temperatures and microwave irradiation. J. Macromol. Sci. A 2019, 56, 609–617. [Google Scholar] [CrossRef]
- Ishida, H.; Wellinghoff, S.T.; Baer, E.; Koenig, J.L. Spectroscopic Studies of poly[N,N′-bis(phenoxyphenyl)pyromellitimide]. 1. Structures of the polyimide and three model compounds. Macromolecules 1980, 13, 826–834. [Google Scholar] [CrossRef]
- Preston, P.N. Benzimidazoles. In Benzimidazoles and Congeneric Tricyclic Compounds; Preston, P.N., Ed.; J. Wiley&Sons: New York, NY, USA, 1981; Volume 40, Part 1; pp. 63–65. [Google Scholar]
- Musto, P.; Karasz, F.E.; MacKnight, W.J. Fourier transform infrared spectroscopy on the thermo-oxidative degradation of polybenzimidazole and of a polybenzimidazole/polyetherimide blend. Polymers 1993, 34, 2934–2945. [Google Scholar] [CrossRef]
- Kojima, T. Proton NMR Studies of a Polybenzimidazole in Solution. J. Polym. Sci. Polym. Phys. Ed. 1980, 18, 1791–1800. [Google Scholar] [CrossRef]
- Sannigrahi, A.; Ghosh, S.; Lalnuntluanga, J.; Jana, T. How the Monomer Concentration of Polymerization Influences Various Properties of polybenzimidazole: A Case Study with Poly(4,4′-diphenylether-5,5′-bibenzimidazole). J. Appl. Polym. Sci. 2009, 111, 2194–2203. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Chiu, T.-H.; Lin, F.-J.; Chen, J.-C. Polybenzimidazole membranes derived from novel tetraamine containing 2,2′-disubstituted biphenyl structures for high temperature proton exchange membrane fuel cell application. J. Membr. Sci. 2022, 654, 120569. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Y.; Wang, L.; Zhao, Z.; He, M.; Song, Y. A simple and effective method of enhancing the proton conductivity of polybenzimidazole proton exchange membranes through protonated polymer during solvation. J. Power Sources 2020, 45, 227965. [Google Scholar] [CrossRef]
- Singh, R.P.; Li, X.; Dudeck, K.W.; Benicewicz, B.C.; Berchtold, K.A. Polybenzimidazole based random copolymers containing hexafluoroisopropylidene functional groups for gas separations at elevated temperatures. Polymers 2017, 119, 134–141. [Google Scholar] [CrossRef]
- Vogel, H.; Marvel, C.S. Polybenzimidazoles. II. J. Polym. Sci. Part A 1963, 1, 1531–1541. [Google Scholar] [CrossRef]
- Korshak, V.V.; Rode, V.V.; Kotsoyeva, N.M. Degradative Reactions of Polybenzimidazoles and of Corresponding Model Compounds. Vysokomol. Soedin. 1975, A 17, 1238–1242. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | MSA/TFSA (v/v) | Temperature (°C) | Time (min) | Inherent Viscosity (dL/g) | Gel Fraction (%) |
|---|---|---|---|---|---|
| OPBI-1 | 70/30 | 100 | 210 | 2.11 | ˂5 |
| OPBI-2 | 70/30 | 120 | 20 | - | 100 |
| OPBI-3 | 50/50 | 100 | 210 | 1.84 | ˂5 |
| OPBI-4 | 50/50 | 120 | 15 | - | 100 |
| OPBI-9 * | 100/0 | 100 | 600 | 1.2 | 22 |
| CF3PBI-1 | 70/30 | 140 | 360 | 0.75 | ˂5 |
| CF3PBI-2 | 50/50 | 120 | 900 | 0.36 | 30 |
| CF3PBI-3 | 50/50 | 140 | 180 | 3.22 | ˂5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Vargas, M.; Rojas-Rodríguez, M.; Palacios-Alquisira, J.; Fomina, L.; Aguilar-Lugo, C.; Alexandrova, L. Effect of the Acid Medium on the Synthesis of Polybenzimidazoles Using Eaton’s Reagent. Polymers 2023, 15, 2130. https://doi.org/10.3390/polym15092130
García-Vargas M, Rojas-Rodríguez M, Palacios-Alquisira J, Fomina L, Aguilar-Lugo C, Alexandrova L. Effect of the Acid Medium on the Synthesis of Polybenzimidazoles Using Eaton’s Reagent. Polymers. 2023; 15(9):2130. https://doi.org/10.3390/polym15092130
Chicago/Turabian StyleGarcía-Vargas, Miriam, Mario Rojas-Rodríguez, Joaquín Palacios-Alquisira, Lioudmila Fomina, Carla Aguilar-Lugo, and Larissa Alexandrova. 2023. "Effect of the Acid Medium on the Synthesis of Polybenzimidazoles Using Eaton’s Reagent" Polymers 15, no. 9: 2130. https://doi.org/10.3390/polym15092130