
A Thermo-Responsive Polymer Micelle with a Liquid Crystalline Core



Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Cholesteryl 6-(Methacryloyloxy)Hexanoate (ChM)
2.3. Preparation of PChM and PChM-PNIPAM
2.4. Preparation of PChM-PNIPAM Aqueous Solution
2.5. Measurements
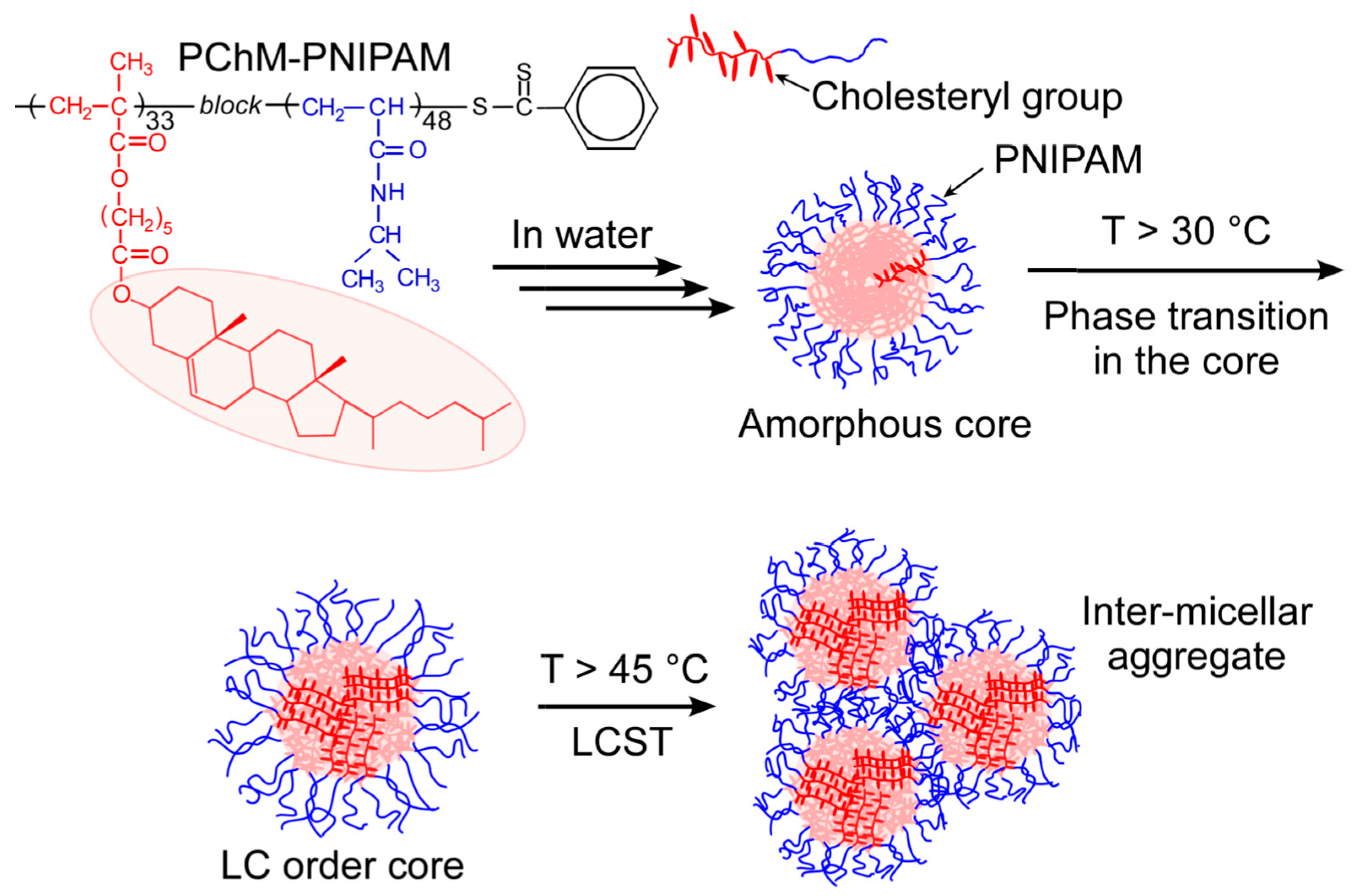
3. Results and Discussion
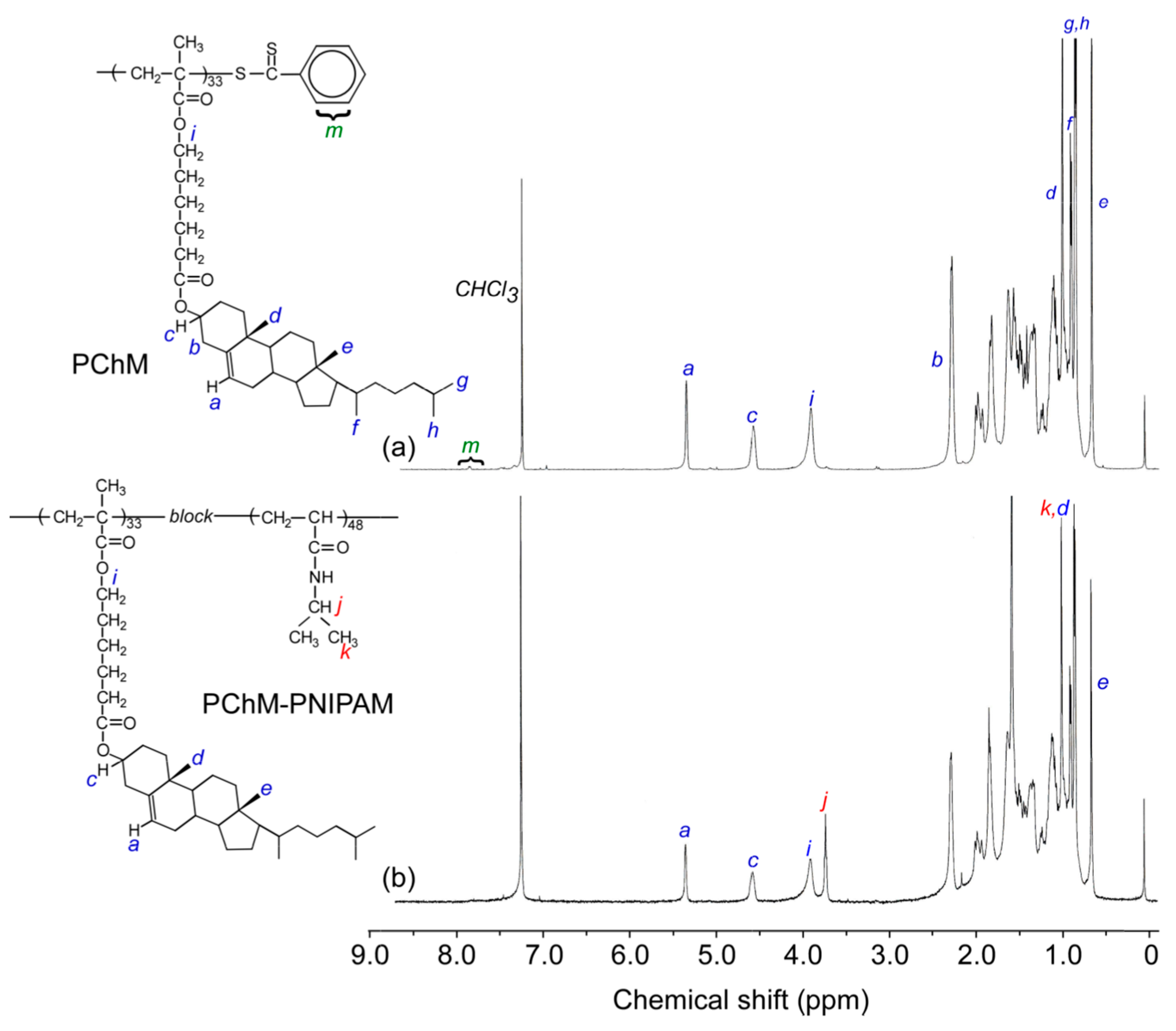
3.1. Polymer Characterization
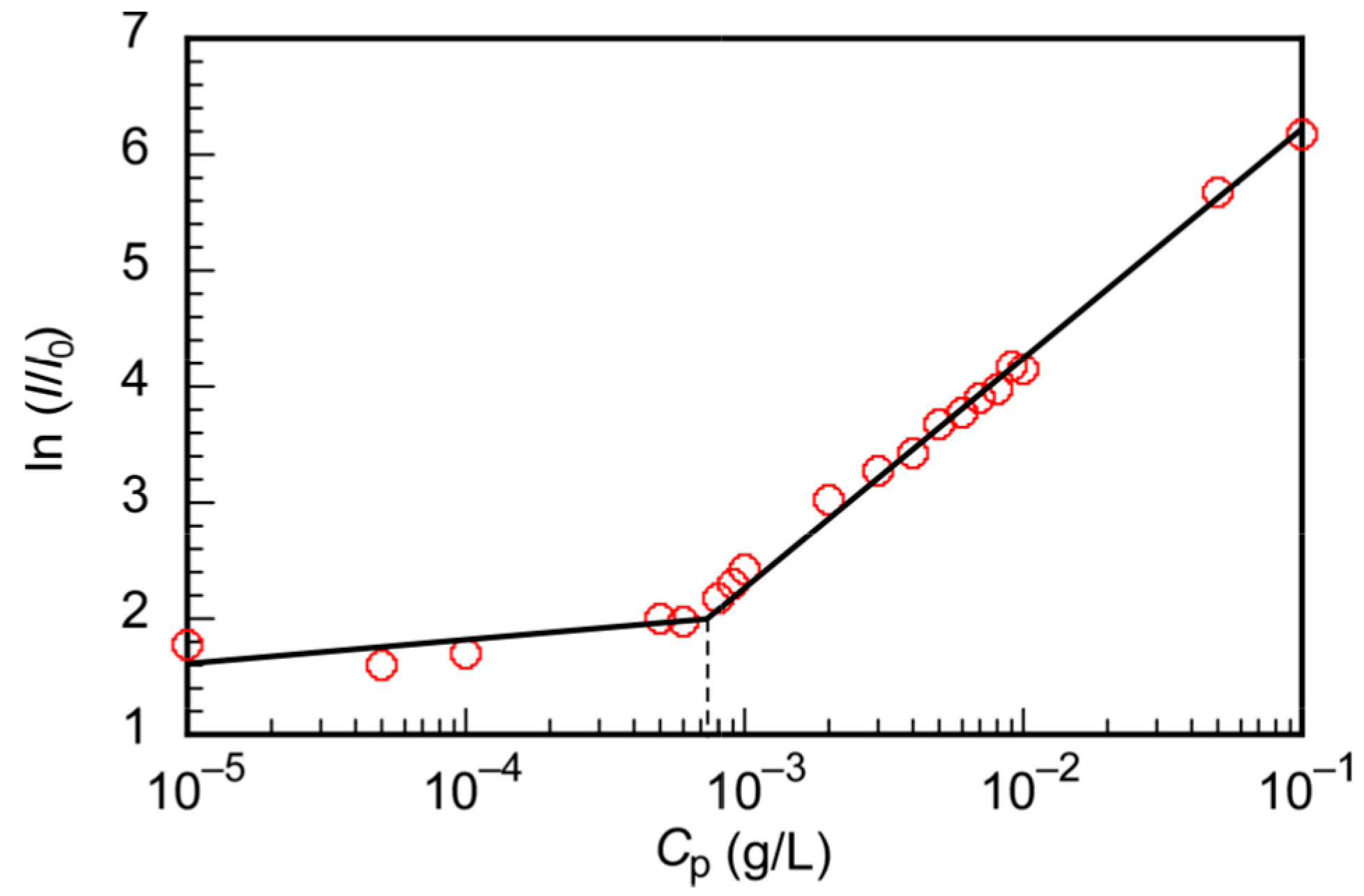
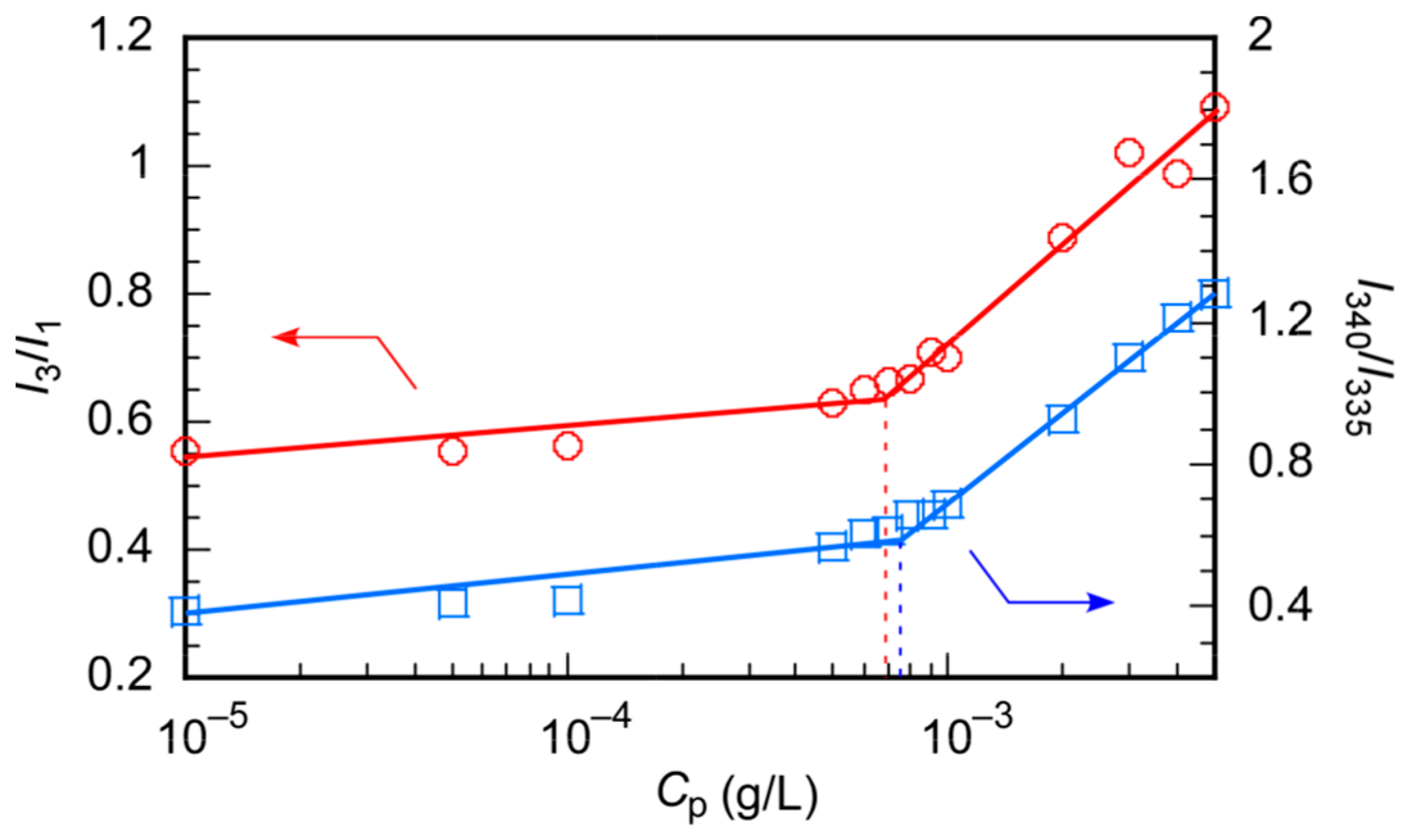
3.2. Self-Association Behavior of PChM-PNIPAM in Water
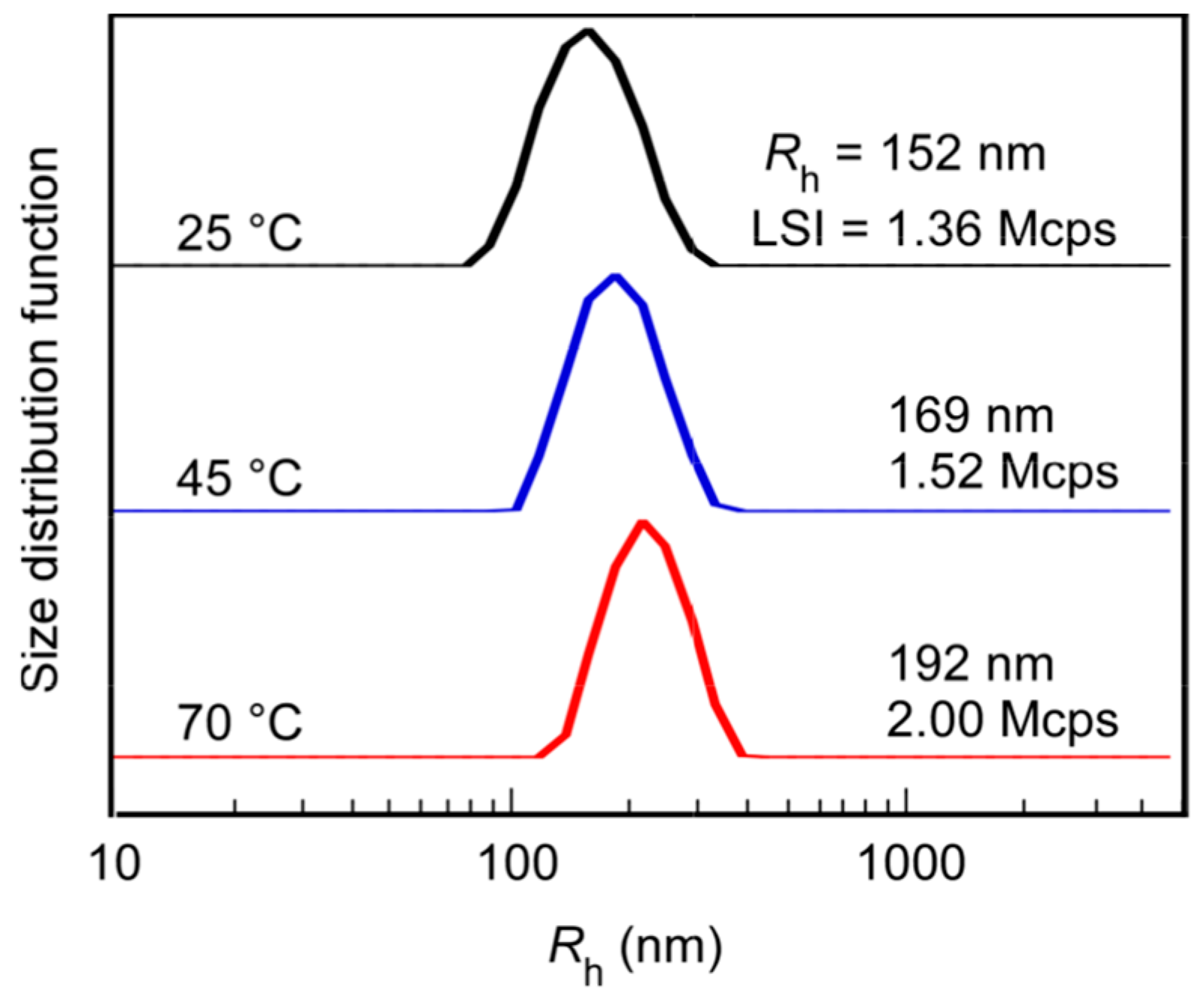
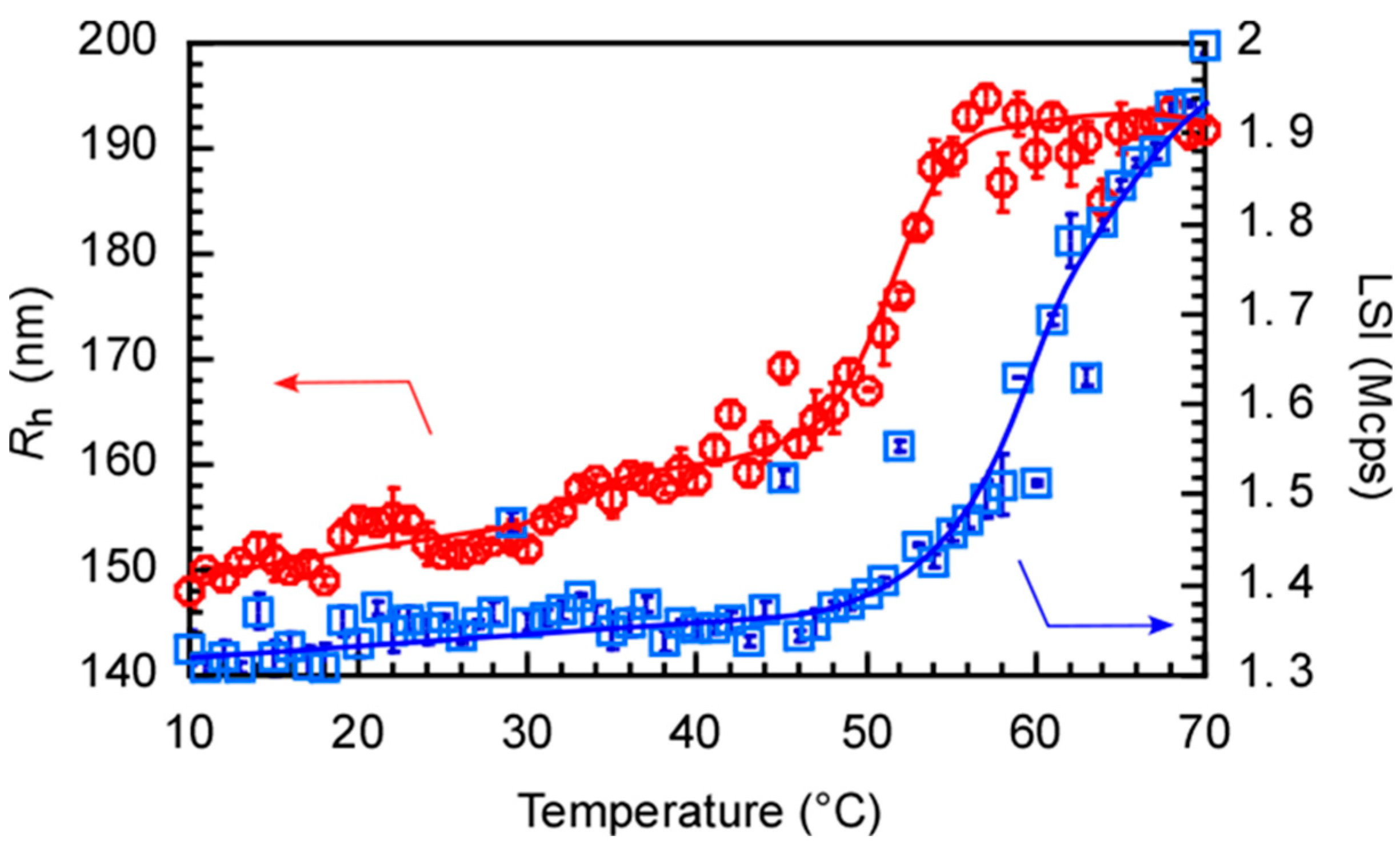
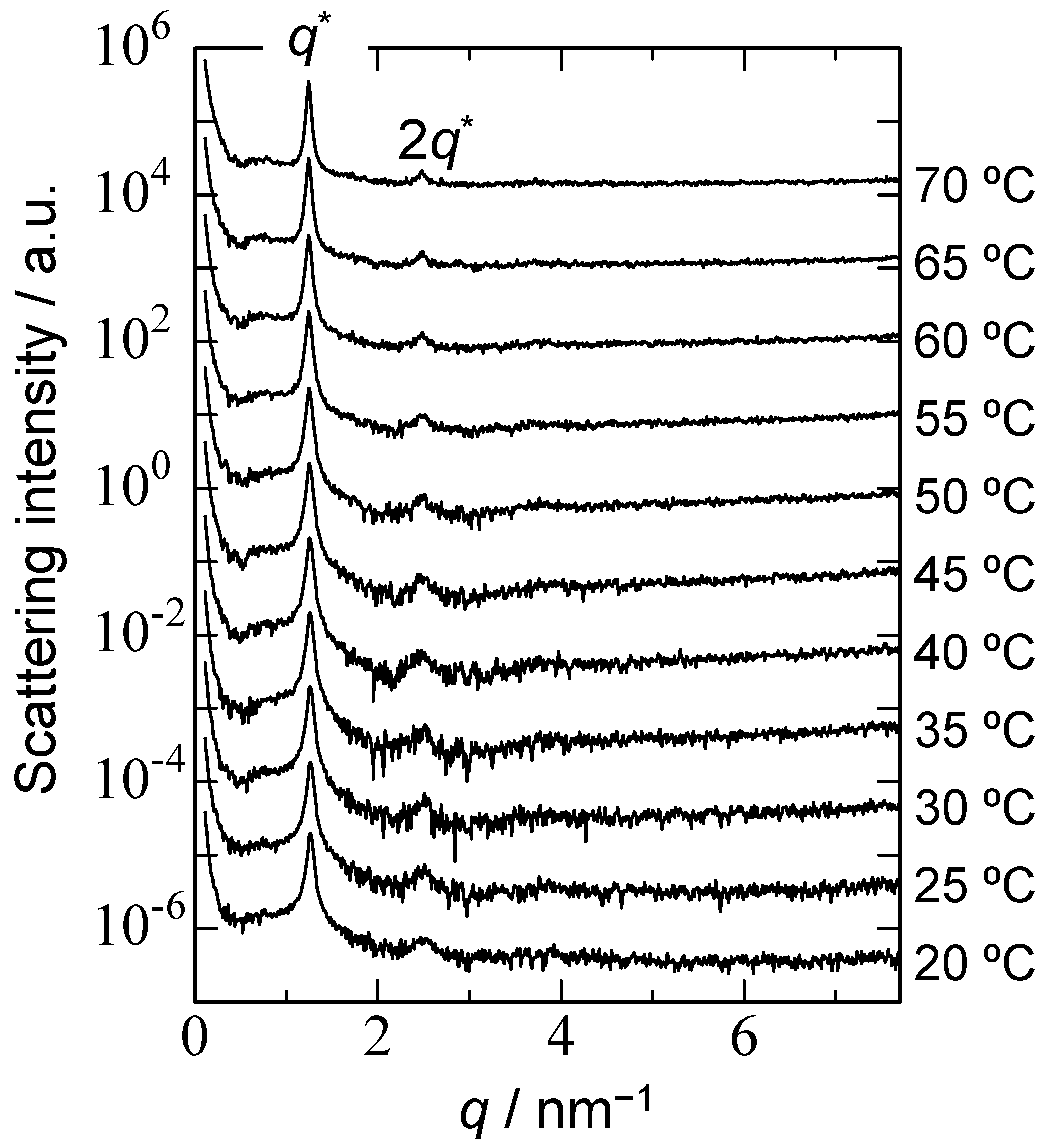
3.3. Thermo-Responsive Behavior of PChM-PNIPAM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Briand, V.A.; Sharma, N.; Ahn, S.; Kasi, R.M. Polymers comprising cholesterol: Synthesis, self-assembly, and applications. Materials 2009, 2, 636–660. [Google Scholar] [CrossRef]
- Ercole, F.; Whittaker, M.R.; Quinn, J.F.; Davis, T.P. Cholesterol modified self-assemblies and their application to nanomedicine. Biomacromolecules 2015, 16, 1886–1914. [Google Scholar] [CrossRef] [PubMed]
- Hosta-Rigau, L.; Zhang, Y.; Teo, B.M.; Postma, A.; Städler, B. Cholesterol–a biological compound as a building block in bionanotechnology. Nanoscale 2013, 5, 89–109. [Google Scholar] [CrossRef] [PubMed]
- Coates, D.; Gray, G.W. Optical studies of the amorphous liquid-cholesteric liquid crystal transition: The “blue phase”. Phys. Lett. A 1973, 45, 115–116. [Google Scholar] [CrossRef]
- Zapotocky, M.; Ramos, L.; Poulin, P.; Lubensky, T.C.; Weitz, D.A. Particle-stabilized defect gel in cholesteric liquid crystals. Science 1999, 283, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shang, Y.; Yu, L.; Zou, C.; Yao, W.; Zhao, D.; Song, P.; Yang, H.; Guo, L. Facet-dependent Cu2O nanocrystals in manipulating alignment of liquid crystals and photomechanical behaviors. Nano Res. 2016, 9, 2581–2589. [Google Scholar] [CrossRef]
- Fuller, S.; Shinde, N.N.; Tiddy, G.J.T.; Attard, G.S.; Howell, O. Thermotropic and lyotropic mesophase behavior of amphitropic diammonium surfactants. Langmuir 1996, 12, 1117–1123. [Google Scholar] [CrossRef]
- Bisoyi, H.K.; Kumar, S. Liquid-crystal nanoscience: An emerging avenue of soft self-assembly. Chem. Soc. Rev. 2011, 40, 306–319. [Google Scholar] [CrossRef]
- Nichifor, M.; Lopes, S.; Bastos, M.; Lopes, A. Self-aggregation of amphiphilic cationic polyelectrolytes based on polysaccharides. J. Phys. Chem. B 2004, 108, 16463–16472. [Google Scholar] [CrossRef]
- Nam, Y.S.; Kang, H.S.; Park, J.Y.; Park, T.G.; Han, S.H.; Chang, I.S. New micelle-like polymer aggregates made from PEI–PLGA diblock copolymers: Micellar characteristics and cellular uptake. Biomaterials 2003, 24, 2053–2059. [Google Scholar] [CrossRef]
- Jia, L.; Lévy, D.; Durand, D.; Impéror-Clerc, M.; Cao, A.; Li, M.H. Smectic polymer micellar aggregates with temperature-controlled morphologies. Soft Matter 2011, 7, 7395–7403. [Google Scholar] [CrossRef]
- Rahman, M.A.; Sha, Y.; Jui, M.S.; Lamm, M.E.; Ma, Y.; Tang, C. Facial amphiphilicity-induced self-assembly (FAISA) of amphiphilic copolymers. Macromolecules 2019, 52, 9526–9535. [Google Scholar] [CrossRef]
- Mizusaki, M.; Morishima, Y.; Winnik, F.M. Hydrophobically modified poly(sodium 2-acrylamido-2-methylpropanesulfonate) s bearing octadecyl groups: A fluorescence study of their solution properties in water. Macromolecules 1999, 32, 4317–4326. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A critical appraisal of RAFT-mediated polymerization-induced self-assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef]
- Alaboalirat, M.; Qi, L.; Arrington, K.J.; Qian, S.; Keum, J.K.; Mei, H.; Littrell, K.C.; Sumpter, B.G.; Carrillo, J.M.Y.; Verduzco, R.; et al. Amphiphilic bottlebrush block copolymers: Analysis of aqueous self-assembly by small-angle neutron scattering and surface tension measurements. Macromolecules 2019, 52, 465–476. [Google Scholar] [CrossRef]
- Williams, R.J.; Dove, A.P.; O’Reilly, R.K. Self-assembly of cyclic polymers. Polym. Chem. 2015, 6, 2998–3008. [Google Scholar] [CrossRef]
- Tan, J.; Sun, H.; Yu, M.; Sumerlin, B.S.; Zhang, L. Photo-PISA: Shedding light on polymerization-induced self-assembly. ACS Macro Lett. 2015, 4, 1249–1253. [Google Scholar] [CrossRef]
- Venkataraman, S.; Wei, G.; Mineart, K.P.; Hedrick, J.L.; Prabhu, V.M.; Yang, Y.Y. The effect of solvent quality on pathway-dependent solution-state self-assembly of an amphiphilic diblock copolymer. J. Appl. Phys. 2020, 127, 125104. [Google Scholar] [CrossRef]
- Ping, J.; Gu, K.; Zhou, S.; Pan, H.; Shen, Z.; Fan, X.H. Hierarchically self-assembled amphiphilic alternating copolymer brush containing side-chain cholesteryl units. Macromolecules 2016, 49, 5993–6000. [Google Scholar] [CrossRef]
- Raczkowska, J.; Stetsyshyn, Y.; Awsiuk, K.; Lekka, M.; Marzec, M.; Harhay, K.; Ohar, H.; Ostapiv, D.; Sharan, M.; Yaremchuk, I.; et al. Temperature-responsive grafted polymer brushes obtained from renewable sources with potential application as substrates for tissue engineering. Appl. Surf. Sci. 2017, 407, 546–554. [Google Scholar] [CrossRef]
- Stetsyshyn, Y.; Raczkowska, J.; Budkowski, A.; Awsiuk, K.; Kostruba, A.; Nastyshyn, S.; Harhay, K.; Lychkovskyy, E.; Ohar, H.; Nastishin, Y. Cholesterol-based grafted polymer brushes as alignment coating with temperature-tuned anchoring for nematic liquid crystals. Langmuir 2016, 32, 11029–11038. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Zhuang, D.; Yang, J.; Yang, J. Bionanoparticles of amphiphilic copolymers polyacrylate bearing cholesterol and ascorbate for drug delivery. J. Colloid Interface Sci. 2012, 377, 197–206. [Google Scholar] [CrossRef]
- Venkataraman, S.; Lee, A.L.; Maune, H.T.; Hedrick, J.L.; Prabhu, V.M.; Yang, Y.Y. Formation of disk-and stacked-disk-like self-assembled morphologies from cholesterol-functionalized amphiphilic polycarbonate diblock copolymers. Macromolecules 2013, 46, 4839–4846. [Google Scholar] [CrossRef]
- Li, L.; Zhou, F.; Li, Y.; Chen, X.; Zhang, Z.; Zhou, N.; Zhu, X. Cooperation of amphiphilicity and smectic order in regulating the self-assembly of cholesterol-functionalized brush-like block copolymers. Langmuir 2018, 34, 11034–11041. [Google Scholar] [CrossRef]
- Luo, G.F.; Chen, W.H.; Zhang, X.Z. 100th anniversary of macromolecular science viewpoint: Poly(N-isopropylacrylamide)-based thermally responsive micelles. ACS Macro Lett. 2020, 9, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Halperin, A.; Kröger, M.; Winnik, F.M. Poly(N-isopropylacrylamide) phase diagrams: Fifty years of research. Angew. Chem. Int. Ed. 2015, 54, 15342–15367. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Mäkinen, L.; Varadharajan, D.; Tenhu, H.; Hietala, S. Triple hydrophilic UCST–LCST block copolymers. Macromolecules 2016, 49, 986–993. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, Y. PNIPAM microgels for biomedical applications: From dispersed particles to 3D assemblies. Soft Matter 2011, 7, 6375–6384. [Google Scholar] [CrossRef]
- Boissé, S.; Rieger, J.; Di-Cicco, A.; Albouy, P.A.; Bui, C.; Li, M.H.; Charleux, B. Synthesis via RAFT of amphiphilic block copolymers with liquid-crystalline hydrophobic block and their self-assembly in water. Macromolecules 2009, 42, 8688–8696. [Google Scholar] [CrossRef]
- Mitsukami, Y.; Donovan, M.S.; Lowe, A.B.; McCormick, C.L. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules 2001, 34, 2248–2256. [Google Scholar] [CrossRef]
- Yusa, S.; Endo, T.; Ito, M. Synthesis of thermo-responsive 4-arm star-shaped porphyrin-centered poly(N,N-diethylacrylamide) via reversible addition-fragmentation chain transfer radical polymerization. J. Polym. Sci. A Polym. Chem. 2009, 47, 6827–6838. [Google Scholar] [CrossRef]
- Yusa, S.; Fukuda, K.; Yamamoto, T.; Iwasaki, Y.; Watanabe, A.; Akiyoshi, K.; Morishima, Y. Salt effect on the heat-induced association behavior of gold nanoparticles coated with poly(N-isopropylacrylamide) prepared via reversible addition−fragmentation chain transfer (RAFT) radical polymerization. Langmuir 2007, 23, 12842–12848. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.J. Synthesis of methacrylate and acrylate monomers of cholesteric esters via phase transfer catalysis. Macromolecules 1983, 16, 1677–1678. [Google Scholar] [CrossRef]
- Meier, M.A.R.; Lohmeijer, B.G.G.; Schubert, U.S. Characterization of defined metal-containing supramolecular block copolymers. Macromol. Rapid Commun. 2003, 24, 852–857. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Asada, T.; Hayashi, H.; Nakamura, N. Dependence of the packing structure of mesogenic groups on the flexible spacer length of liquid-crystalline side-chain polymers. Macromolecules 1989, 22, 1141–1144. [Google Scholar] [CrossRef]
- Sousa, R.G.; Magalhaes, W.F.; Freitas, R.F.S. Glass transition and thermal stability of poly(N-isopropylacrylamide) gels and some of their copolymers with acrylamide. Polym. Degrad. Stab. 1998, 61, 275–281. [Google Scholar] [CrossRef]
- Li, X.; Mya, K.Y.; Ni, X.; He, C.; Leong, K.W.; Li, J. Dynamic and static light scattering studies on self-aggregation behavior of biodegradable amphiphilic poly(ethylene oxide)−poly[(R)-3-hydroxybutyrate]−poly(ethylene oxide) triblock copolymers in aqueous solution. J. Phys. Chem. B 2006, 110, 5920–5926. [Google Scholar] [CrossRef] [PubMed]
- Akcasu, A.Z.; Han, C.C. Molecular weight and temperature dependence of polymer dimensions in solution. Macromolecules 1979, 12, 276–280. [Google Scholar] [CrossRef]
- Tahara, Y.; Sakiyama, M.; Takeda, S.; Nishimura, T.; Mukai, S.; Sawada, S.; Sasaki, Y.; Akiyoshi, K. Self-assembled nanogels of cholesterol-bearing hydroxypropyl cellulose: A thermoresponsive building block for nanogel tectonic materials. Langmuir 2016, 32, 12283–12289. [Google Scholar] [CrossRef]
- Wilhelm, M.; Zhao, C.L.; Wang, Y.; Xu, R.; Winnik, M.A.; Mura, J.L.; Riess, G.; Croucher, M.D. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: A fluorescence probe study. Macromolecules 1991, 24, 1033–1040. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Thomas, J.K. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar] [CrossRef]
- Xu, J.P.; Ji, J.; Chen, W.D.; Shen, J.C. Novel biomimetic surfactant: Synthesis and micellar characteristics. Macromol. Biosci. 2005, 5, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Y.; Meng, F.B.; Zang, B.L.; Hu, J.S. Liquid-crystalline elastomers containing sulfonic acid groups. Macromolecules 2003, 36, 3320–3326. [Google Scholar] [CrossRef]
- Zugenmaier, P. Structural models for some crystalline and liquid crystalline polymers. Macromol. Chem. Symp. 1986, 2, 33–46. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DP(theo) a | Mn(theo) b × 10−4 (g/mol) | DP(NMR) c | Mn(NMR) c × 10−4 (g/mol) | DP(GPC) d | Mn(GPC) d × 10−4 (g/mol) | Mw/Mnd |
|---|---|---|---|---|---|---|---|
| PChM | 36 | 2.05 | 33 | 1.89 | 41 | 2.34 | 1.13 |
| PChM-PNIPAM | 50 a | 2.45 | 48 a | 2.43 | 47 a | 2.42 | 1.12 |
| Mw(SLS) a (g/mol) | Naggb | Rhc (nm) | Rgd (nm) | Rg/Rh | ΦH e (g/cm3) |
|---|---|---|---|---|---|
| 7.73 × 109 | 2.84 × 105 | 152 | 114 | 0.75 | 0.873 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizoue, Y.; Takahashi, R.; Sakurai, K.; Yusa, S.-i. A Thermo-Responsive Polymer Micelle with a Liquid Crystalline Core. Polymers 2023, 15, 770. https://doi.org/10.3390/polym15030770
Mizoue Y, Takahashi R, Sakurai K, Yusa S-i. A Thermo-Responsive Polymer Micelle with a Liquid Crystalline Core. Polymers. 2023; 15(3):770. https://doi.org/10.3390/polym15030770
Chicago/Turabian StyleMizoue, Yoko, Rintaro Takahashi, Kazuo Sakurai, and Shin-ichi Yusa. 2023. "A Thermo-Responsive Polymer Micelle with a Liquid Crystalline Core" Polymers 15, no. 3: 770. https://doi.org/10.3390/polym15030770