
Additive Free Crosslinking of Poly-3-hydroxybutyrate via Electron Beam Irradiation at Elevated Temperatures

Abstract
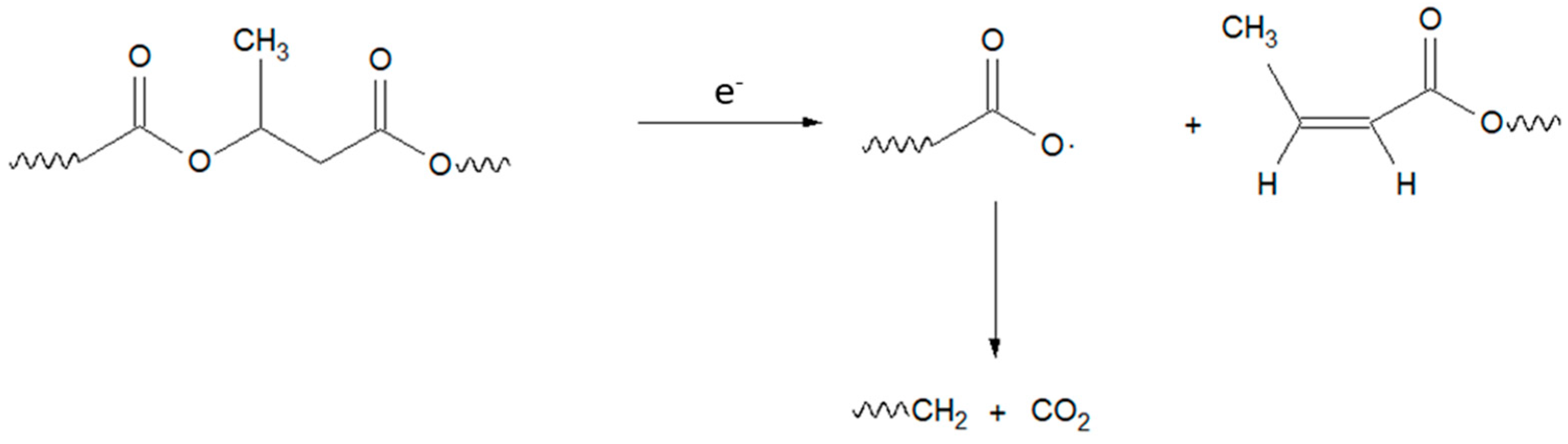
:1. Introduction
2. Materials and Methods
3. Results and Discussion
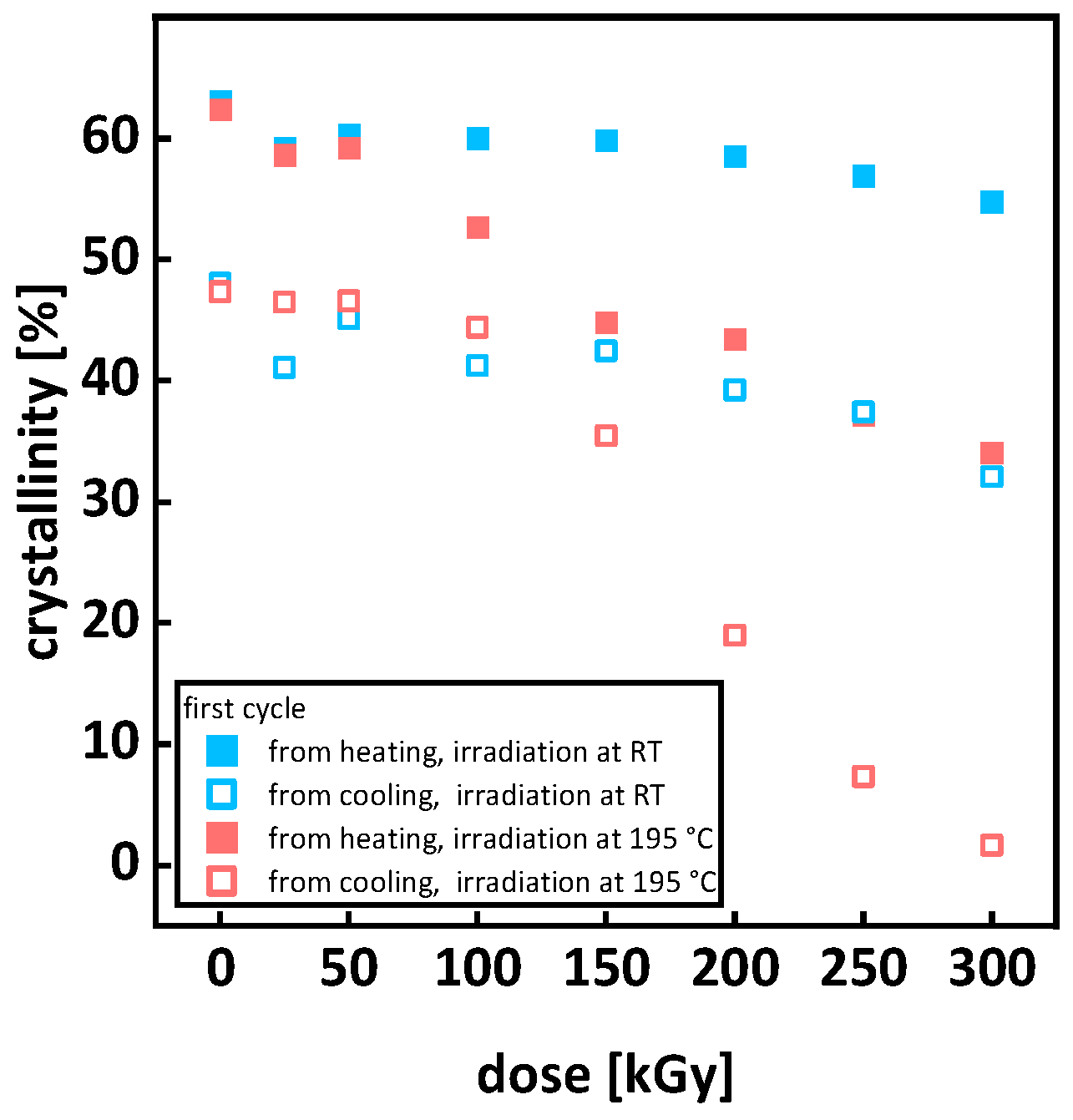
3.1. Optical Inspection of Samples after Irradiation
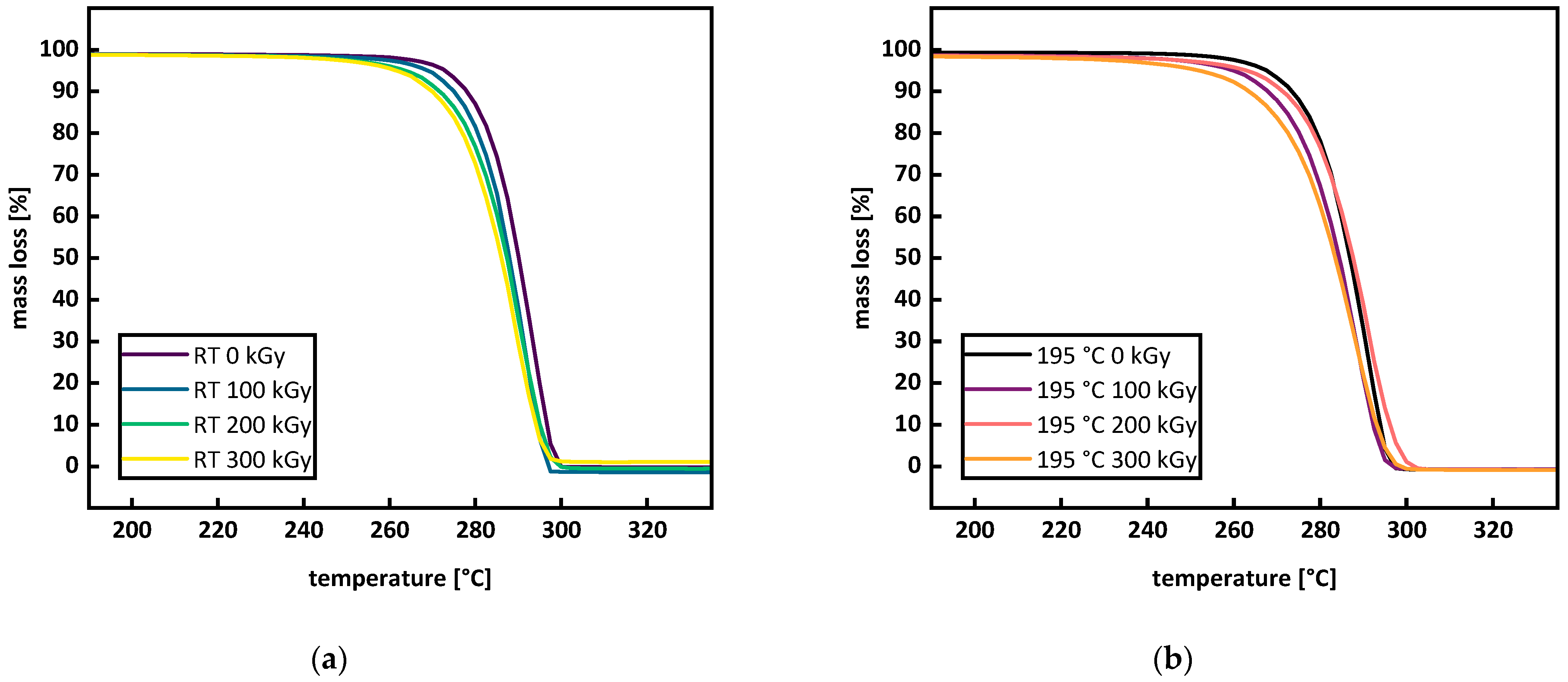
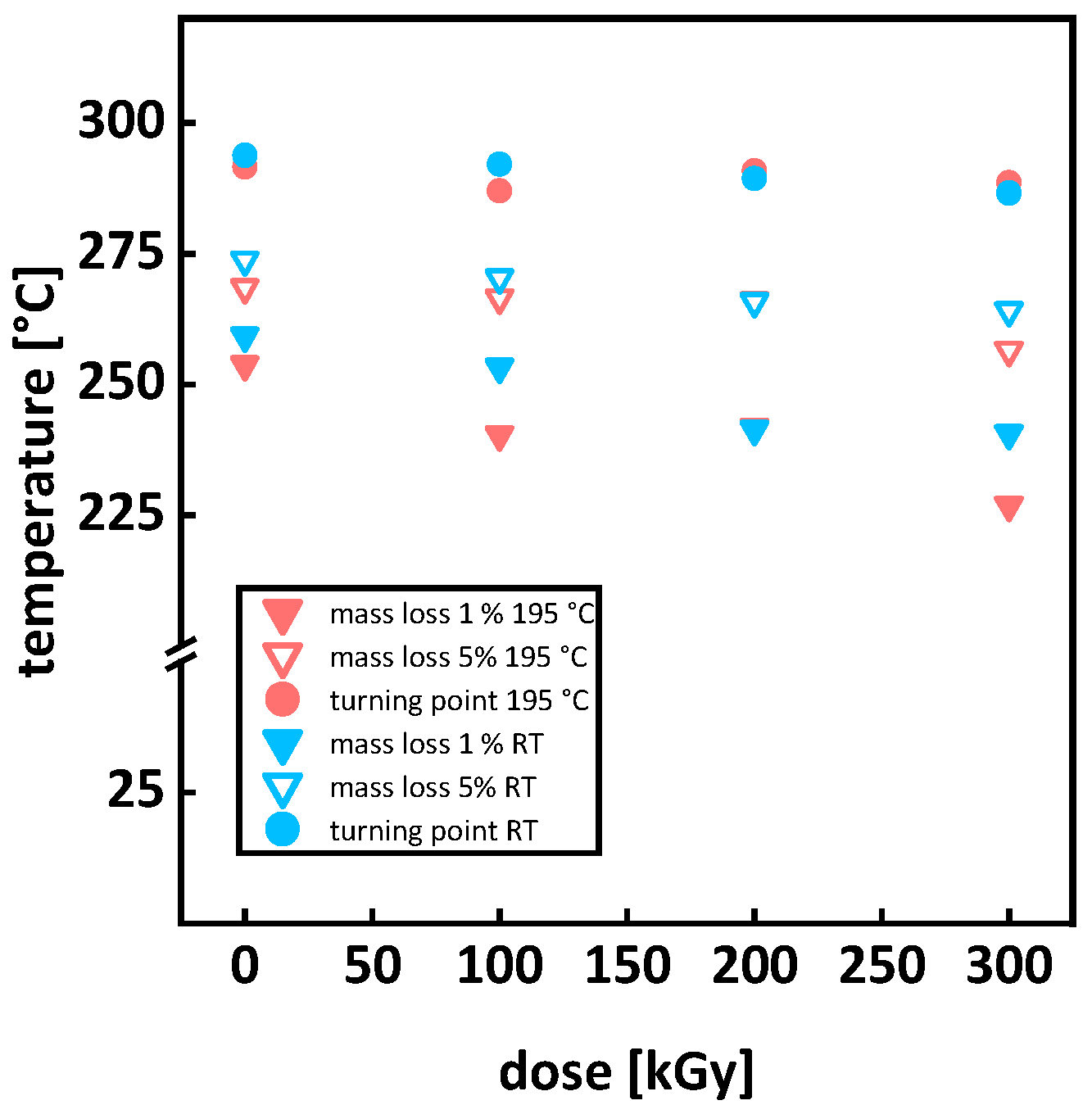
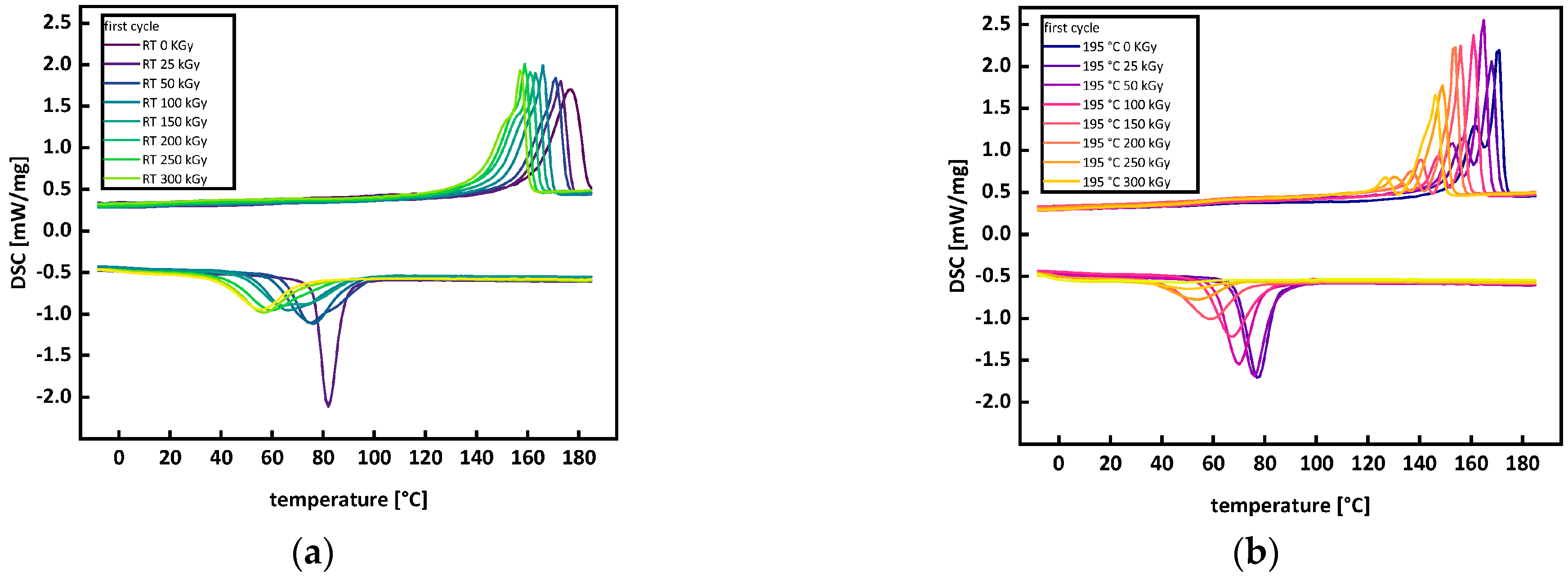
3.2. Thermal Analysis


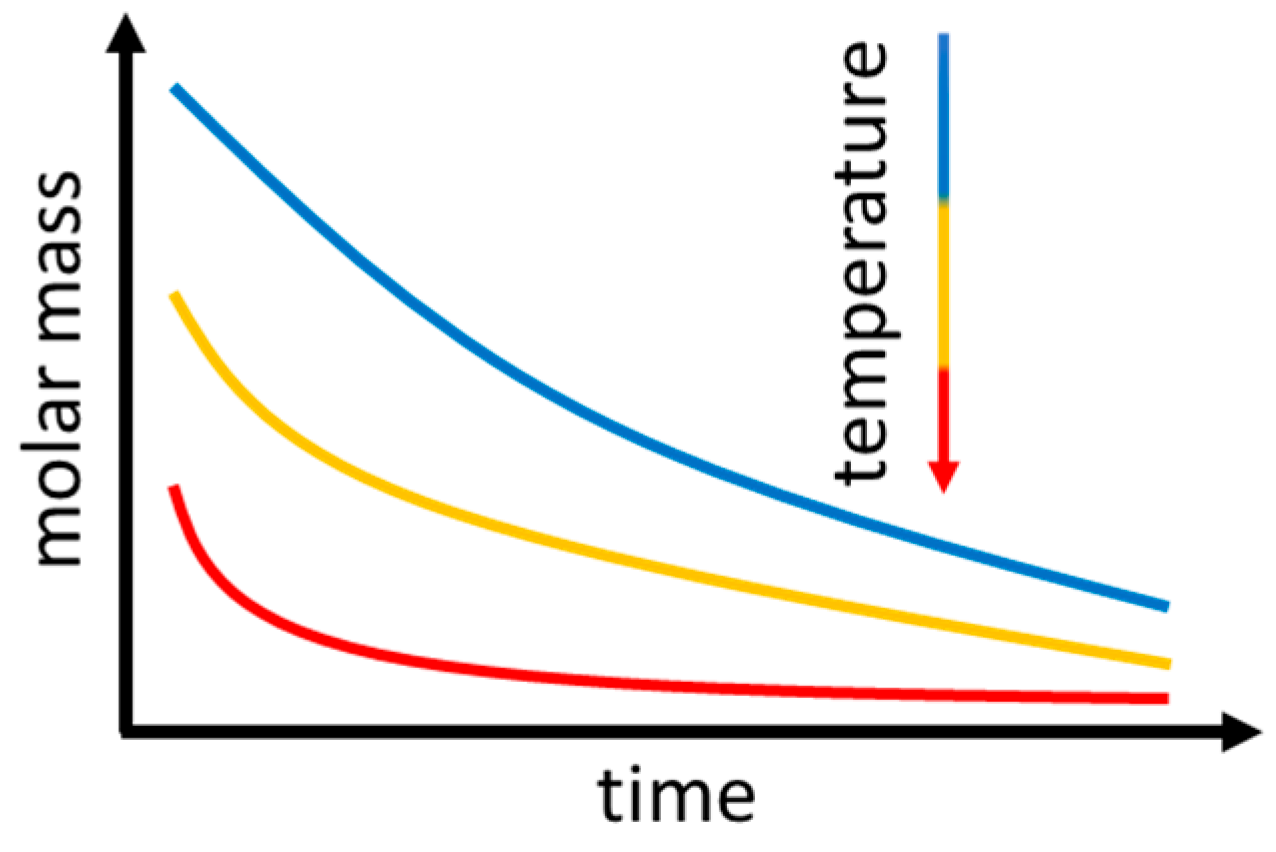
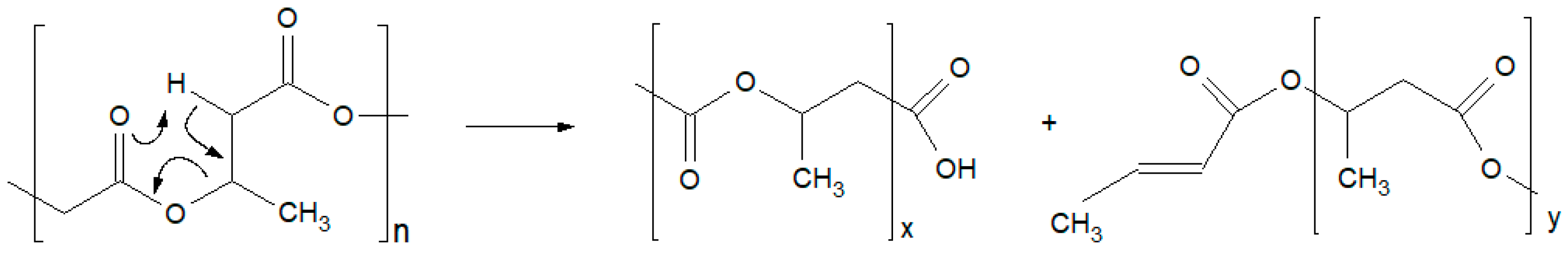
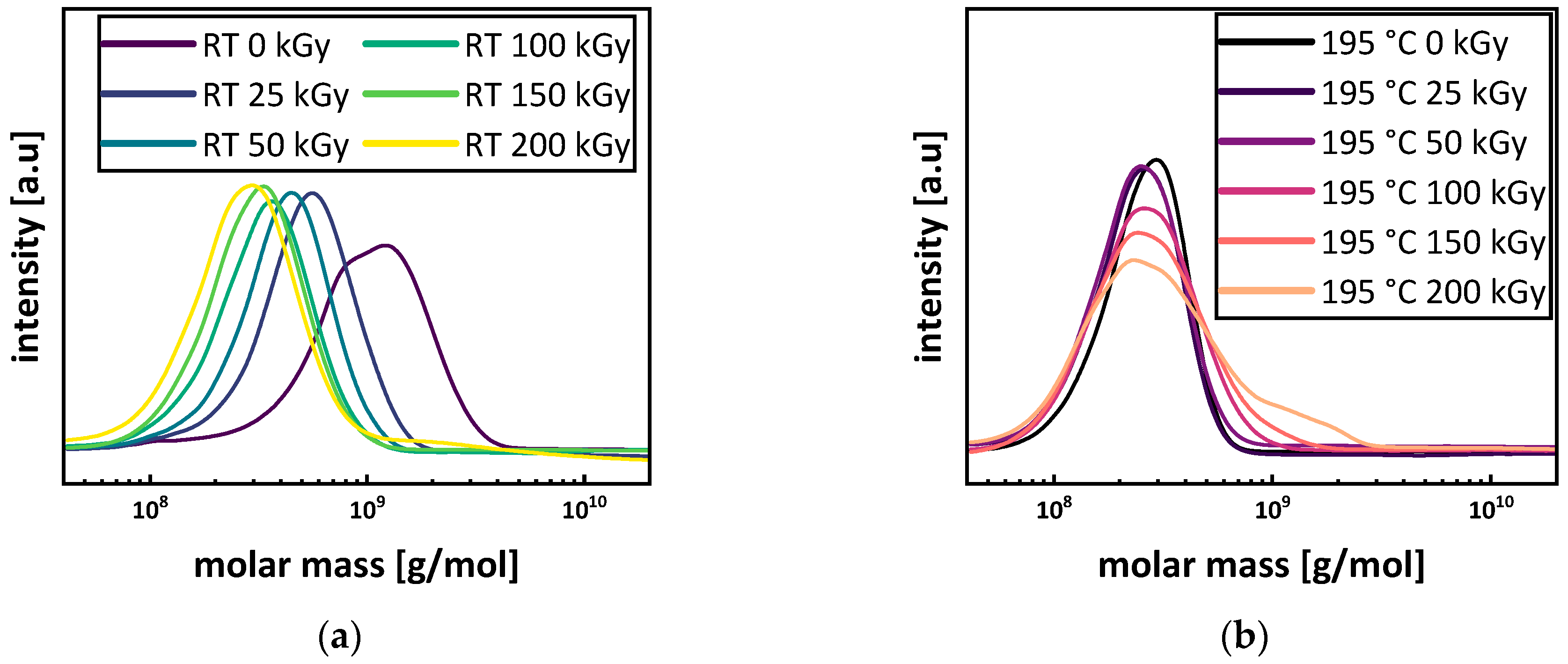
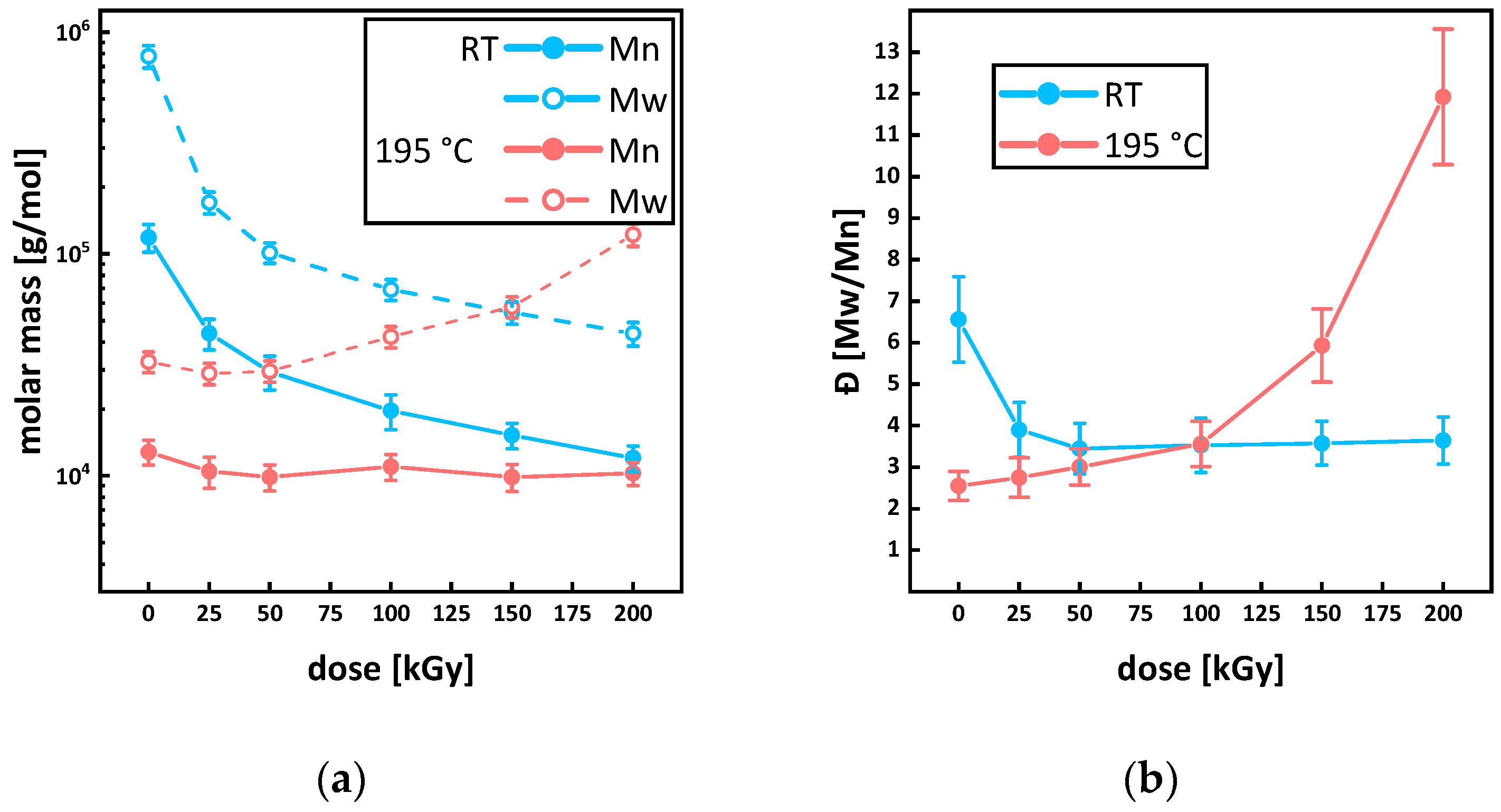
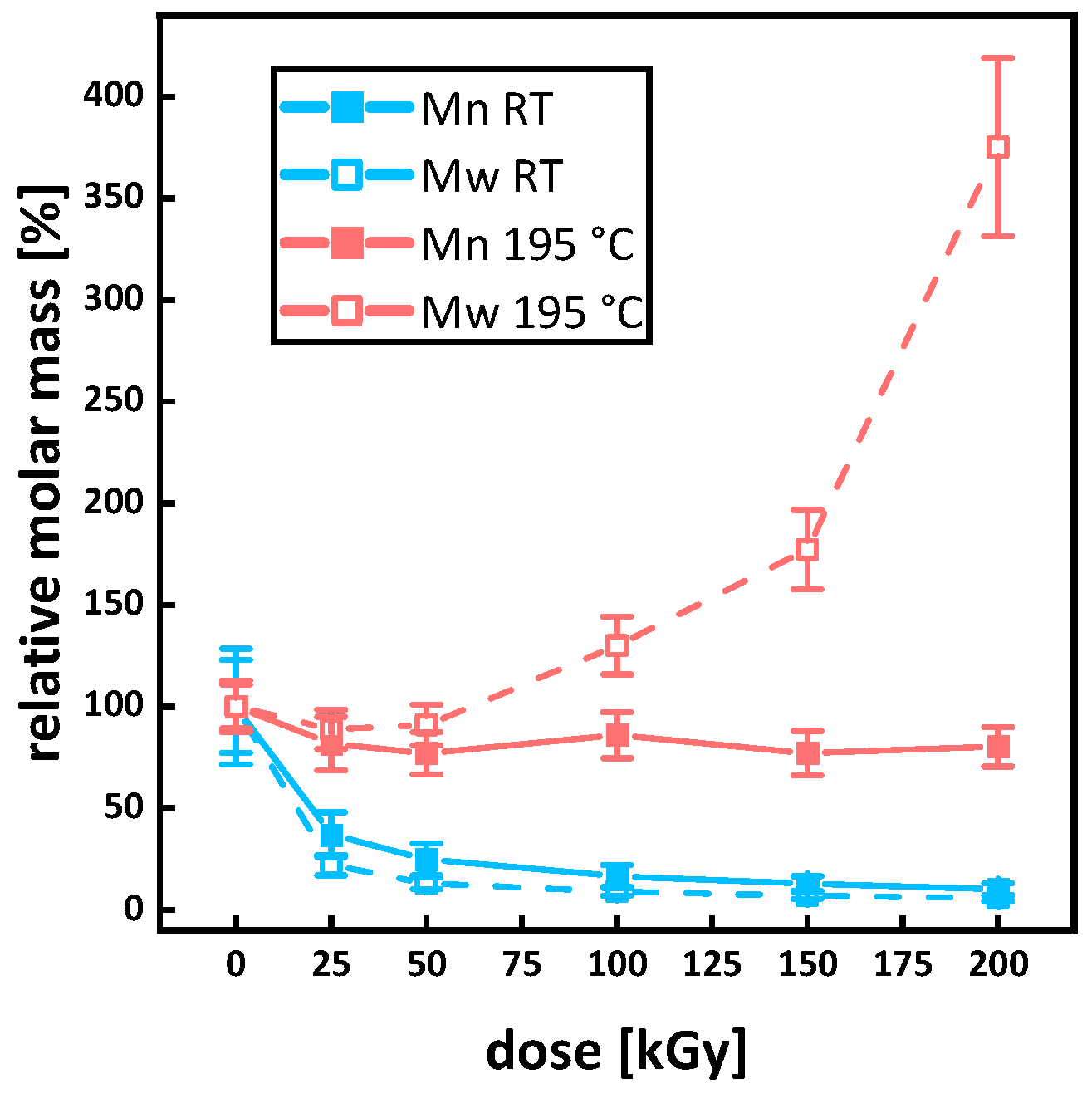
3.3. Molecular Mass and Branching



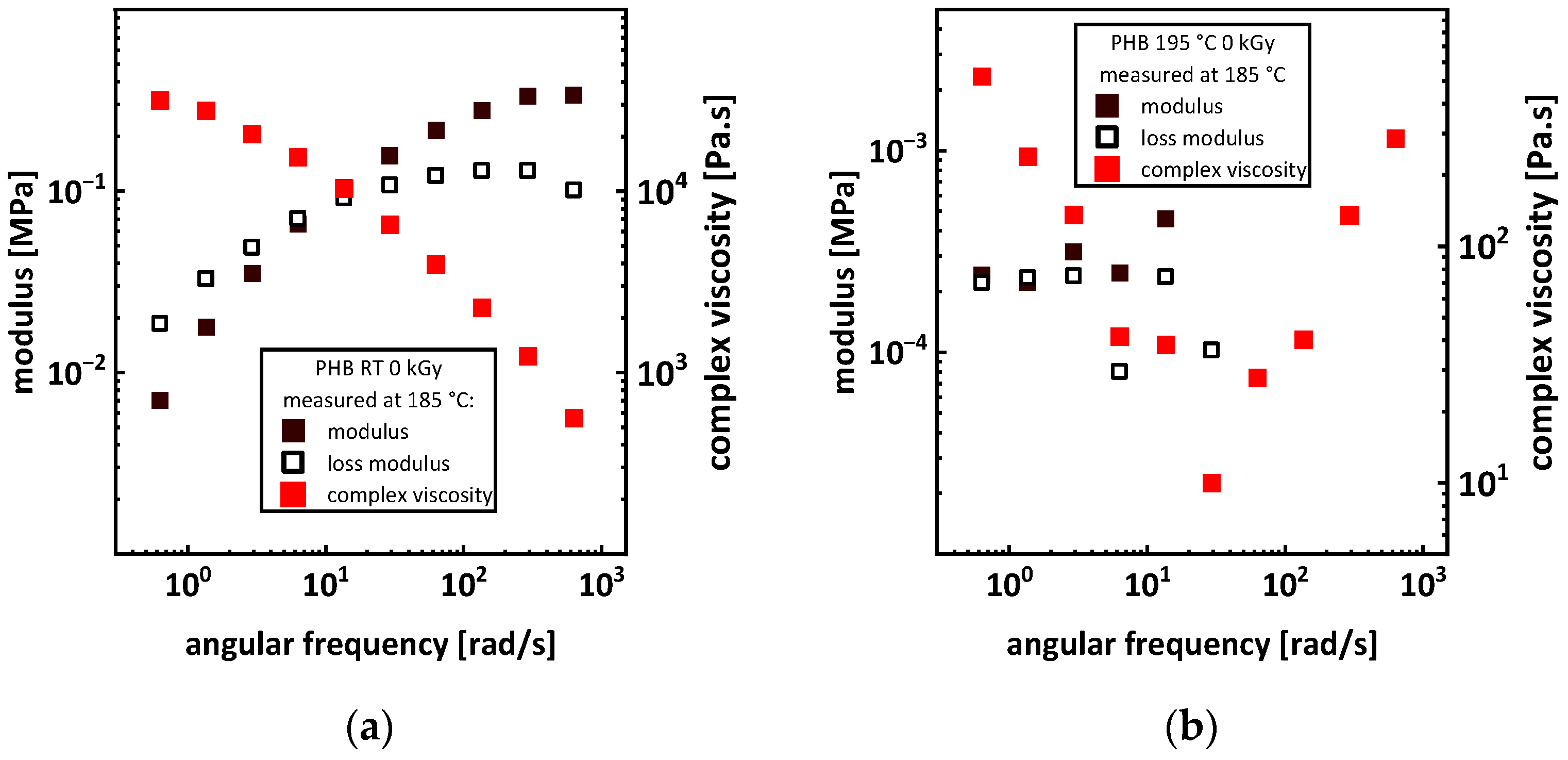
3.4. Rheology

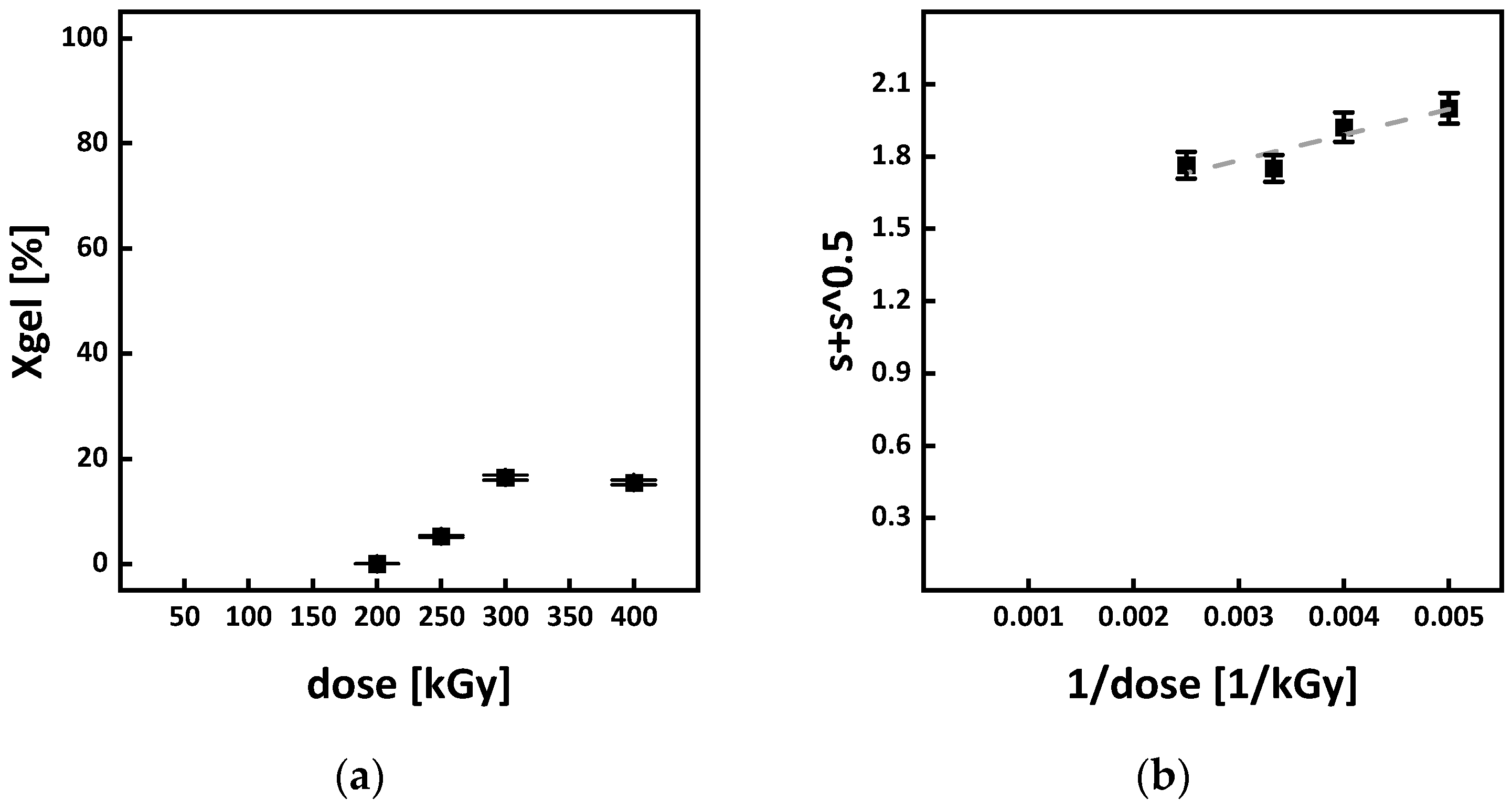
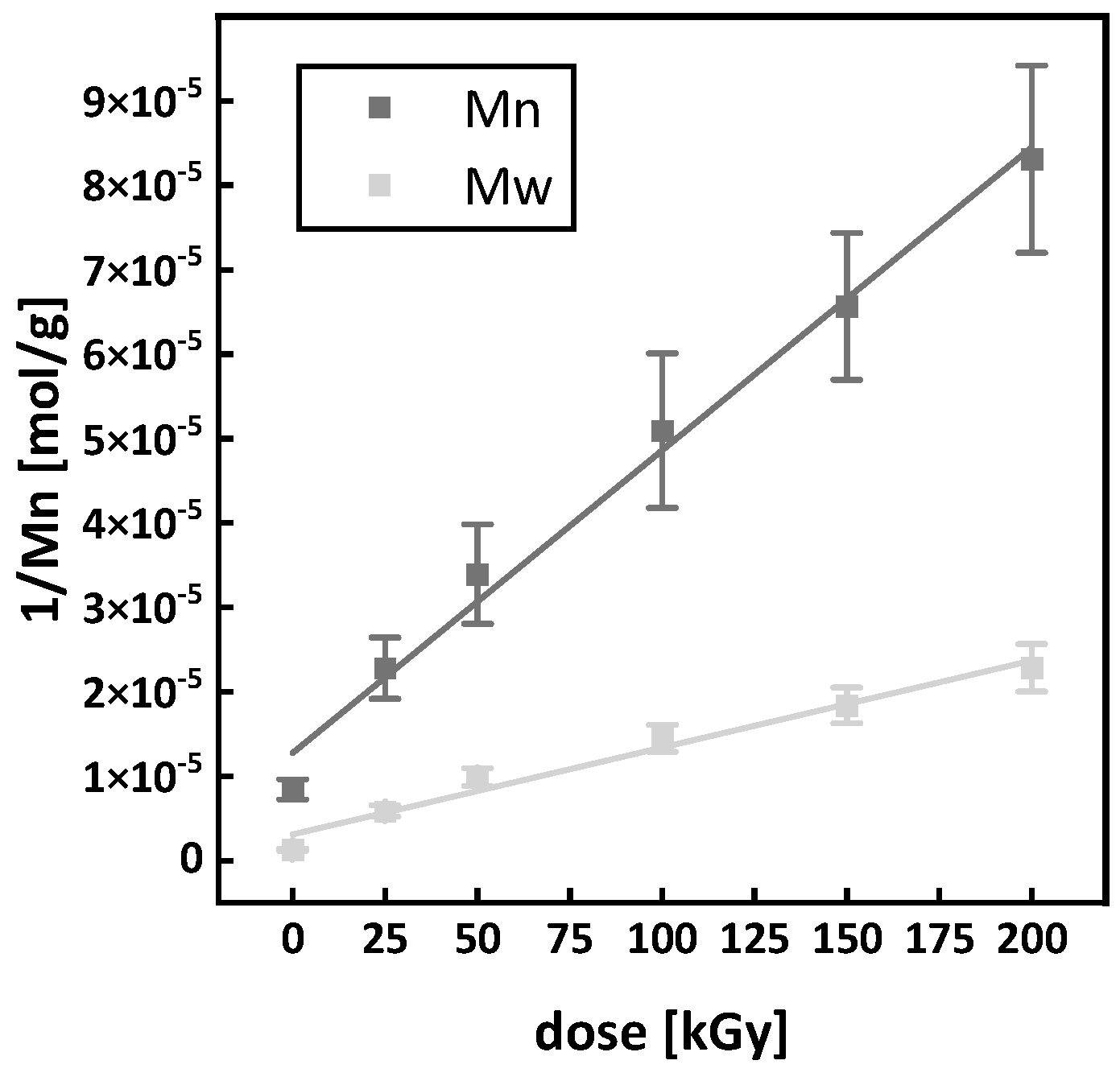
3.5. Gel Formation and G Values

4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
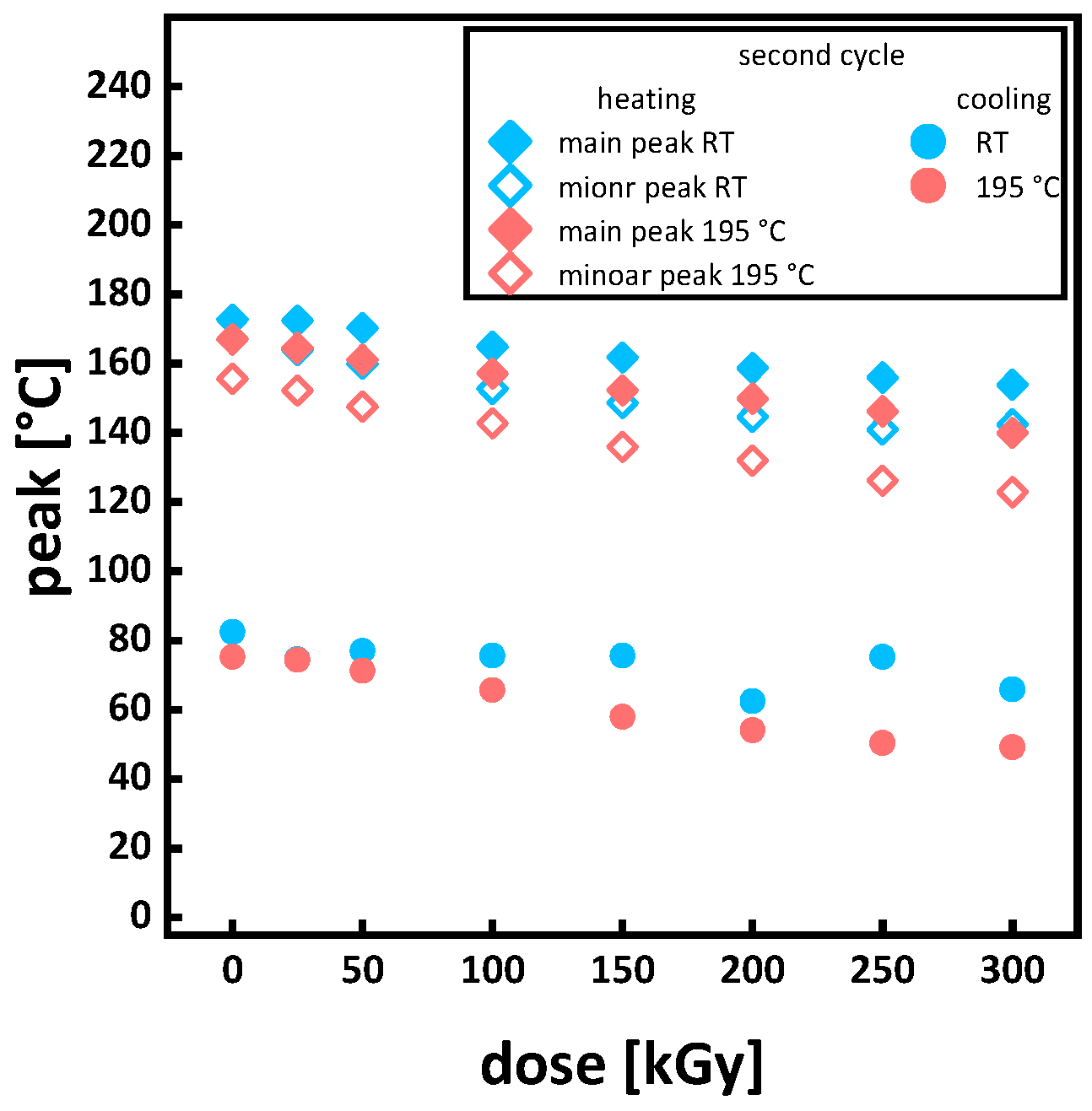
Appendix A.1. Thermal Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| heating rate | first cycle | ||||||||||||||||||||||||||||||||||||||
| 10 K/min | heating | cooling | |||||||||||||||||||||||||||||||||||||
| Tm | Tg | Tc | |||||||||||||||||||||||||||||||||||||
| peak area | main peak | minor peak | onset | end | onset | middle point | turning point | end | peak area | peak | onset | end | |||||||||||||||||||||||||||
| sample | [J/g] | [°C] | [°C] | [°C] | [°C] | [°C] | [°C] | [°C] | [°C] | [J/g] | [°C] | [°C] | [°C] | ||||||||||||||||||||||||||
| PHB RT 0 kGy | 103.4 | ± | 5.2 | 177 | ± | 9 | - | 163 | ± | 8 | 184 | ± | 9 | - | - | - | - | −78.7 | ± | 3.9 | 82 | ± | 4 | 76 | ± | 4 | 89 | ± | 4 | ||||||||||
| PHB RT 25 kGy | 97.1 | ± | 4.9 | 173 | ± | 9 | - | 161 | ± | 8 | 178 | ± | 9 | - | - | - | - | −67.4 | ± | 3.4 | 75 | ± | 4 | # | ± | 3 | 99 | ± | 5 | ||||||||||
| PHB RT 50 kGy | 98.9 | ± | 4.9 | 171 | ± | 9 | - | 162 | ± | 8 | 176 | ± | 9 | - | - | - | - | −74.0 | ± | 3.7 | 76 | ± | 4 | 59 | ± | 3 | 89 | ± | 4 | ||||||||||
| PHB RT 100 kGy | 98.4 | ± | 4.9 | 166 | ± | 8 | - | 158 | ± | 8 | 170 | ± | 8 | - | - | - | - | −67.7 | ± | 3.4 | 66 | ± | 3 | 51 | ± | 3 | 88 | ± | 4 | ||||||||||
| PHB RT 150 kGy | 98.1 | ± | 4.9 | 163 | ± | 8 | - | 156 | ± | 8 | 167 | ± | 8 | - | - | - | - | −69.6 | ± | 3.5 | 64 | ± | 3 | # | ± | 2 | 91 | ± | 5 | ||||||||||
| PHB RT 200 kGy | 96.0 | ± | 4.8 | 161 | ± | 8 | - | 154 | ± | 8 | 165 | ± | 8 | - | - | - | - | −64.4 | ± | 3.2 | 60 | ± | 3 | # | ± | 2 | 83 | ± | 4 | ||||||||||
| PHB RT 250 kGy | 93.3 | ± | 4.7 | 159 | ± | 8 | - | 153 | ± | 8 | 162 | ± | 8 | - | - | - | - | −61.4 | ± | 3.1 | 57 | ± | 3 | # | ± | 2 | 74 | ± | 4 | ||||||||||
| PHB RT 300 kGy | 89.8 | ± | 4.5 | 157 | ± | 8 | - | 152 | ± | 8 | 161 | ± | 8 | - | - | - | - | −52.6 | ± | 2.6 | 56 | ± | 3 | # | ± | 2 | 71 | ± | 4 | ||||||||||
| PHB 195 °C 0 kGy | 102.3 | ± | 5.1 | 171 | ± | 9 | 162 | ± | 8 | 167 | ± | 8 | 173 | ± | 9 | - | - | - | - | −77.6 | ± | 3.9 | 77 | ± | 4 | # | ± | 3 | 84 | ± | 4 | ||||||||
| PHB 195 °C 25 kGy | 96.1 | ± | 4.8 | 168 | ± | 8 | 157 | ± | 8 | 162 | ± | 8 | 171 | ± | 9 | - | - | - | - | −76.2 | ± | 3.8 | 76 | ± | 4 | # | ± | 3 | 84 | ± | 4 | ||||||||
| PHB 195 °C 50 kGy | 97.1 | ± | 4.9 | 165 | ± | 8 | 153 | ± | 8 | 160 | ± | 8 | 168 | ± | 8 | - | - | - | - | −76.4 | ± | 3.8 | 70 | ± | 3 | 61 | ± | 3 | 79 | ± | 4 | ||||||||
| PHB 195 °C 100 kGy | 86.3 | ± | 4.3 | 161 | ± | 8 | 147 | ± | 7 | 157 | ± | 8 | 164 | ± | 8 | - | - | - | - | −72.8 | ± | 3.6 | 67 | ± | 3 | 56 | ± | 3 | 80 | ± | 4 | ||||||||
| PHB 195 °C 150 kGy | 73.5 | ± | 3.7 | 156 | ± | 8 | 141 | ± | 7 | 152 | ± | 8 | 159 | ± | 8 | - | - | - | - | −58.1 | ± | 2.9 | 59 | ± | 3 | # | ± | 2 | 72 | ± | 4 | ||||||||
| PHB 195 °C 200 kGy | 71.2 | ± | 3.6 | 154 | ± | 8 | 137 | ± | 7 | 150 | ± | 8 | 156 | ± | 8 | −7 | ± | 1 | −2 | ± | 1 | −2 | ± | 1 | 2 | ± | 1 | −31.1 | ± | 1.6 | 54 | ± | 3 | 37 | ± | 2 | 69 | ± | 3 |
| PHB 195 °C 250 kGy | 61.0 | ± | 3.0 | 149 | ± | 7 | 130 | ± | 7 | 143 | ± | 7 | 152 | ± | 8 | −8 | ± | 1 | −2 | ± | 1 | −2 | ± | 1 | 2 | ± | 1 | −12.0 | ± | 1.0 | 51 | ± | 3 | 35 | ± | 2 | 66 | ± | 3 |
| PHB 195 °C 300 kGy | 55.8 | ± | 2.8 | 146 | ± | 7 | 127 | ± | 6 | 141 | ± | 7 | 150 | ± | 7 | −9 | ± | 1 | −3 | ± | 1 | −3 | ± | 1 | 1 | ± | 1 | −2.7 | ± | 1.0 | 49 | ± | 2 | # | ± | 2 | 62 | ± | 3 |
| second cycle | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| heating | cooling | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tg | Tcc | Tm | Tg | Tc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| onset | middle point | turning point | end | peak area | main peak | onset | end | peak area | main peak | minor peak | onset | end | onset | middle point | turning point | end | peak area | peak | onset | end | ||||||||||||||||||||||||||||||||||||||||||
| [°C] | [°C] | [°C] | [°C] | [J/g] | [°C] | [°C] | [°C] | [J/g] | [°C] | [°C] | [°C] | [°C] | [°C] | [°C] | [°C] | [°C] | [J/g] | [°C] | [°C] | [°C] | ||||||||||||||||||||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 104.1 | ± | 5.2 | 173 | ± | 9 | - | 165 | ± | 8 | 181 | ± | 9 | - | - | - | - | −82.9 | ± | 4.1 | 83 | ± | 4 | 77 | ± | 4 | 89 | ± | 4 | ||||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 95.8 | ± | 4.8 | 172 | ± | 9 | 164 | ± | 8 | 166 | ± | 8 | 178 | ± | 9 | - | - | - | - | −81.2 | ± | 4.1 | 75 | ± | 4 | 64 | ± | 3 | 97 | ± | 5 | ||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 93.6 | ± | 4.7 | 170 | ± | 9 | 160 | ± | 8 | 163 | ± | 8 | 176 | ± | 9 | - | - | - | - | −78.4 | ± | 3.9 | 77 | ± | 4 | 61 | ± | 3 | 94 | ± | 5 | ||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 87.6 | ± | 4.4 | 165 | ± | 8 | 153 | ± | 8 | 159 | ± | 8 | 168 | ± | 8 | - | - | - | - | −74.6 | ± | 3.7 | 76 | ± | 4 | 56 | ± | 3 | 98 | ± | 5 | ||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 82.7 | ± | 4.1 | 162 | ± | 8 | 149 | ± | 7 | 155 | ± | 8 | 165 | ± | 8 | - | - | - | - | −73.3 | ± | 3.7 | 76 | ± | 4 | 54 | ± | 3 | 97 | ± | 5 | ||||||||||||||||||||||||
| 0 | ± | 1 | 2 | ± | 1 | 3 | ± | 1 | 4 | ± | 1 | −2.6 | ± | 1.0 | 48 | ± | 2 | 39 | ± | 2 | 55 | ± | 3 | 78.5 | ± | 3.9 | 159 | ± | 8 | 145 | ± | 7 | 152 | ± | 8 | 162 | ± | 8 | - | - | - | - | −65.7 | ± | 3.3 | 63 | ± | 3 | 48 | ± | 2 | 90 | ± | 5 | ||||||||
| 1 | ± | 1 | 3 | ± | 1 | 2 | ± | 1 | 4 | ± | 1 | −4.8 | ± | 1.0 | 48 | ± | 2 | 40 | ± | 2 | 55 | ± | 3 | 77.1 | ± | 3.9 | 156 | ± | 8 | 141 | ± | 7 | 150 | ± | 7 | 160 | ± | 8 | - | - | - | - | −70.3 | ± | 3.5 | 75 | ± | 4 | 47 | ± | 2 | 98 | ± | 5 | ||||||||
| 0 | ± | 1 | 2 | ± | 1 | 2 | ± | 1 | 3 | ± | 1 | −7.1 | ± | 1.0 | 49 | ± | 2 | 39 | ± | 2 | 56 | ± | 3 | 69.9 | ± | 3.5 | 154 | ± | 8 | 143 | ± | 7 | 148 | ± | 7 | 158 | ± | 8 | - | - | - | - | −62.4 | ± | 3.1 | 66 | ± | 3 | 44 | ± | 2 | 94 | ± | 5 | ||||||||
| - | - | - | - | - | - | - | - | 100.7 | ± | 5.0 | 167 | ± | 8 | 156 | ± | 8 | 150 | ± | 8 | 171 | ± | 9 | - | - | - | - | −77.6 | ± | 3.9 | 75 | ± | 4 | 68 | ± | 3 | 83 | ± | 4 | ||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 96.0 | ± | 4.8 | 165 | ± | 8 | 152 | ± | 8 | 153 | ± | 8 | 168 | ± | 8 | - | - | - | - | −76.5 | ± | 3.8 | 74 | ± | 4 | 66 | ± | 3 | 88 | ± | 4 | ||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 98.0 | ± | 4.9 | 161 | ± | 8 | 148 | ± | 7 | 155 | ± | 8 | 165 | ± | 8 | - | - | - | - | −78.3 | ± | 3.9 | 71 | ± | 4 | 60 | ± | 3 | 97 | ± | 5 | ||||||||||||||||||||||||
| - | - | - | - | - | - | - | - | 88.9 | ± | 4.4 | 157 | ± | 8 | 143 | ± | 7 | 150 | ± | 8 | 161 | ± | 8 | - | - | - | - | −72.2 | ± | 3.6 | 657 | ± | 33 | 55 | ± | 3 | 78 | ± | 4 | ||||||||||||||||||||||||
| - | - | - | - | −2.9 | ± | 1.0 | 49 | ± | 2 | 40 | ± | 2 | 55 | ± | 3 | 77.4 | ± | 3.9 | 152 | ± | 8 | 136 | ± | 7 | 144 | ± | 7 | 156 | ± | 8 | - | - | - | - | −58.0 | ± | 2.9 | 58 | ± | 3 | 43 | ± | 2 | 71 | ± | 4 | ||||||||||||||||
| 0 | ± | 1 | 1 | ± | 2 | ± | 1 | 3 | ± | 1 | −24.8 | ± | 1.2 | 53 | ± | 3 | 43 | ± | 2 | 59 | ± | 3 | 77.0 | ± | 3.8 | 150 | ± | 7 | 132 | ± | 7 | 141 | ± | 7 | 154 | ± | 8 | −6.7 | ± | 1 | −2 | ± | 1 | −2.7 | ± | 1 | 1.7 | ± | 1 | −35.7 | ± | 1.8 | 54 | ± | 3 | 37 | ± | 2 | 71 | ± | 4 | |
| 0 | ± | 1 | 1 | ± | 1 | ± | 1 | 3 | ± | 1 | −39.9 | ± | 2.0 | 56 | ± | 3 | 47 | ± | 2 | 63 | ± | 3 | 66.7 | ± | 3.3 | 146 | ± | 7 | 126 | ± | 6 | 136 | ± | 7 | 151 | ± | 8 | −7.4 | ± | 1 | −2 | ± | 1 | −2.4 | ± | 1 | 1.5 | ± | 1 | −11.4 | ± | 0.6 | 50 | ± | 3 | 34 | ± | 2 | 66 | ± | 3 | |
| −1 | ± | 1 | 1 | ± | 1 | ± | 1 | 2 | ± | 1 | −49.8 | ± | 2.5 | 59 | ± | 3 | 49 | ± | 2 | 65 | ± | 3 | 63.3 | ± | 3.2 | 140 | ± | 7 | 123 | ± | 6 | 133 | ± | 7 | 148 | ± | 7 | −8.3 | ± | 1 | −3 | ± | 1 | −4.4 | ± | 1 | 0.5 | ± | 1 | −2.3 | ± | 0.1 | 49 | ± | 2 | 34 | ± | 2 | 65 | ± | 3 | |
| Temperature [°C] | ||||||||||||||||||
| Dose | 1% Mass Loss | 5% Mass Loss | Turning Point | 1% Mass Loss | 5% Mass Loss | Turning Point | ||||||||||||
| [kGy] | irradiation at 195 °C | irradiation at room temperature | ||||||||||||||||
| 0 | 254 | ± | 6 | 269 | ± | 7 | 292 | ± | 7 | 259 | ± | 6 | 274 | ± | 7 | 294 | ± | 7 |
| 100 | 240 | ± | 6 | 267 | ± | 7 | 287 | ± | 7 | 253 | ± | 6 | 270 | ± | 7 | 292 | ± | 7 |
| 200 | 242 | ± | 6 | 266 | ± | 7 | 291 | ± | 7 | 241 | ± | 6 | 266 | ± | 7 | 289 | ± | 7 |
| 300 | 227 | ± | 6 | 257 | ± | 6 | 289 | ± | 7 | 241 | ± | 6 | 264 | ± | 7 | 287 | ± | 7 |
Appendix A.2. Rheology

References
- Sirohi, R.; Prakash Pandey, J.; Kumar Gaur, V.; Gnansounou, E.; Sindhu, R. Critical overview of biomass feedstocks as sustainable substrates for the production of polyhydroxybutyrate (PHB). Bioresour. Technol. 2020, 311, 123536. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Mawatari, Y.; Onodera, R.; Nakamura, Y.; Yajima, Y.; Chang, Y.-C. Bacterial conversion of waste into polyhydroxybutyrate (PHB): A new approach of bio-circular economy for treating waste and energy generation. Bioresour. Technol. Rep. 2019, 7, 100246. [Google Scholar] [CrossRef]
- Chavan, S.; Yadav, B.; Tyagi, R.D.; Drogui, P. A review on production of polyhydroxyalkanoate (PHA) biopolyesters by thermophilic microbes using waste feedstocks. Bioresour. Technol. 2021, 341, 125900. [Google Scholar] [CrossRef]
- Ganesh Saratale, R.; Cho, S.-K.; Dattatraya Saratale, G.; Kadam, A.A.; Ghodake, G.S.; Kumar, M.; Naresh Bharagava, R.; Kumar, G.; Su Kim, D.; Mulla, S.I.; et al. A comprehensive overview and recent advances on polyhydroxyalkanoates (PHA) production using various organic waste streams. Bioresour. Technol. 2021, 325, 124685. [Google Scholar] [CrossRef]
- Kratzl, F.; Kremling, A.; Pflüger-Grau, K. Streamlining of a synthetic co-culture towards an individually controllable one-pot process for polyhydroxyalkanoate production from light and CO2. Eng. Life Sci. 2023, 23, e2100156. [Google Scholar] [CrossRef] [PubMed]
- McAdam, B.; Brennan Fournet, M.; McDonald, P.; Mojicevic, M. Production of Polyhydroxybutyrate (PHB) and Factors Impacting Its Chemical and Mechanical Characteristics. Polymers 2020, 12, 2908. [Google Scholar] [CrossRef]
- Boey, J.Y.; Lee, C.K.; Tay, G.S. Factors Affecting Mechanical Properties of Reinforced Bioplastics: A Review. Polymers 2022, 14, 3737. [Google Scholar] [CrossRef] [PubMed]
- López-Rubio, A.; Lagaron, J.M. Improvement of UV stability and mechanical properties of biopolyesters through the addition of β-carotene. Polym. Degrad. Stab. 2010, 95, 2162–2168. [Google Scholar] [CrossRef]
- van der Walle, G.A.; de Koning, G.J.; Weusthuis, R.A.; Eggink, G. Properties, modifications and applications of biopolyesters. Adv. Biochem. Eng. Biotechnol. 2001, 71, 263–291. [Google Scholar] [CrossRef] [PubMed]
- Majerczak, K.; Wadkin-Snaith, D.; Magueijo, V.; Mulheran, P.; Liggat, J.; Johnston, K. Polyhydroxybutyrate: A review of experimental and simulation studies of the effect of fillers on crystallinity and mechanical properties. Polym. Int. 2022, 71, 1398–1408. [Google Scholar] [CrossRef]
- Bergmann, A.; Teßmar, J.; Owen, A. Influence of electron irradiation on the crystallisation, molecular weight and mechanical properties of poly-(R)-3-hydroxybutyrate. J. Mater. Sci. 2007, 42, 3732–3738. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Gazzano, M.; Righetti, M.C. The Role of the Rigid Amorphous Fraction on Cold Crystallization of Poly(3-hydroxybutyrate). Macromolecules 2012, 45, 5684–5691. [Google Scholar] [CrossRef]
- Pachekoski, W.M.; Dalmolin, C.; Agnelli, J.A.M. The influence of the industrial processing on the degradation of poly(hidroxybutyrate)—PHB. Mater. Res. 2013, 16, 237–332. [Google Scholar] [CrossRef]
- Krieg, D.; Sergeieva, O.; Jungkind, S.; Rennert, M.; Nase, M. Influence of E-beam irradiation on compounds from linear low density polyethylene and thermoplastic vulcanized rubber consisting of a polypropylene and ethylene propylene diene monomer rubber phase. J. Appl. Polym. Sci. 2023, 140, e53765. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Araujo, P.L.B.; Araujo, E.S. The effect of gamma radiation on mechanical properties of biodegradable polymers poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Mat. Res. 2013, 16, 195–203. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Araújo, E.S.; Guedes, S.M. Gamma irradiation effects on poly(hydroxybutyrate). Polym. Degrad. Stab. 2006, 91, 2157–2162. [Google Scholar] [CrossRef]
- Parra, D.F.; Rosa, D.S.; Rezende, J.; Ponce, P.; Lugão, A.B. Biodegradation of γ Irradiated Poly 3-hydroxybutyrate (PHB) Films Blended with Poly(Ethyleneglycol). J. Polym. Env. 2011, 19, 918–925. [Google Scholar] [CrossRef]
- Radiation Effects on Biodegradable Polyesters; Malaysian Institute for Nuclear Technology Research: Kuala Lumpur, Malaysia, 1999.
- dos Santos, A.J.; Oliveira Dalla Valentina, L.V.; Hidalgo Schulz, A.A.; Tomaz Duarte, M.A. From obtaining to degradation of PHB: A literature review. Part II. Ing. Cienc. 2018, 14, 207–228. [Google Scholar] [CrossRef]
- Rivas, L.F.; Casarin, S.A.; Nepomuceno, N.C.; Alencar, M.I.; Agnelli, J.A.M.; de Medeiros, E.S.; Wanderley Neto, A.d.O.; de Oliveira, M.P.; de Medeiros, A.M.; Santos, A.S.F.e. Reprocessability of PHB in extrusion: ATR-FTIR, tensile tests and thermal studies. Polímeros 2017, 27, 122–128. [Google Scholar] [CrossRef]
- Weinmann, S.; Bonten, C. Thermal and rheological properties of modified polyhydroxybutyrate (PHB). Polym. Eng. Sci. 2019, 59, 1057–1064. [Google Scholar] [CrossRef]
- Seyed Khabbaz, H.; Garmabi, H. Modification of polylactide by reactive blending with polyhydroxybutyrate oligomers formed by thermal recycling through E1cB-elimination pathway. Eur. Polym. J. 2022, 166, 111043. [Google Scholar] [CrossRef]
- Zhang, Y.; Müller, M.T.; Boldt, R.; Stommel, M. Crystallinity effect on electron-induced molecular structure transformations in additive-free PLA. Polymer 2023, 265, 125609. [Google Scholar] [CrossRef]
- Huang, Y.; Brünig, H.; Müller, M.T.; Wießner, S. Melt spinning of PLA/PCL blends modified with electron induced reactive processing. J. Appl. Polym. Sci. 2022, 139, 51902. [Google Scholar] [CrossRef]
- Dorschner, H.; Jenschke, W.; Lunkwitz, K. Radiation field distributions of an industrial electron beam accelerator. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2000, 161–163, 1154–1158. [Google Scholar] [CrossRef]
- Geißler, U.; Dresden, D.E.; Hanke, R.; Dresden, D.E.; Körber, H.; Lappan, U.; Premnitz, D.E.; Lunkwitz, K. Physical and Chemical Modification Apparatus for Material Samples Comprises a Housing with a Radiation Beam Window, Sample Table on a Support and a Sample Pressure Applicator. DE19930742, 30 June 1999. [Google Scholar]
- Puente, J.A.S.; Esposito, A.; Chivrac, F.; Dargent, E. Effect of boron nitride as a nucleating agent on the crystallization of bacterial poly(3-hydroxybutyrate). J. Appl. Polym. Sci. 2013, 128, 2586–2594. [Google Scholar] [CrossRef]
- Charlesby, A.; Pinner, S.H. Analysis of the solubility behaviour of irradiated polyethylene and other polymers. Proc. R. Soc. Lond. A 1959, 249, 367–386. [Google Scholar] [CrossRef]
- Charlesby, A. Atomic Radiation and Polymers: International Series of Monographs on Radiation Effects in Materials, 1. Aufl.; Elsevier Reference Monographs: Amsterdam, The Netherlands, 2016; ISBN 9781483181301. [Google Scholar]
- Rosiak, J.M. Gel/sol analysis of irradiated polymers. Radiat. Phys. Chem. 1998, 51, 13–17. [Google Scholar] [CrossRef]
- Olejniczak, J.; Rosiak, J.; Charlesby, A. Gel/dose curves for polymers undergoing simultaneous crosslinking and scission. Int. J. Radiat. Appl. Instrumentation. Part. C Radiat. Phys. Chem. 1991, 37, 499–504. [Google Scholar] [CrossRef]
- Hill, D.J.; Preston, C.M.; Salisbury, D.J.; Whittaker, A.K. Molecular weight changes and scission and crosslinking in poly(dimethyl siloxane) on gamma radiolysis. Radiat. Phys. Chem. 2001, 62, 11–17. [Google Scholar] [CrossRef]
- O’Donnell, J.H. Chemistry of Radiation Degradation of Polymers; American Chemical Society: Washington, DC, USA, 1991; Volume 475. [Google Scholar]
- Da Silva, D.C.P.; de Menezes, L.R.; da Silva, P.S.R.C.; Tavares, M.I.B. Evaluation of thermal properties of zirconium–PHB composites. J. Therm. Anal. Calorim. 2021, 143, 165–172. [Google Scholar] [CrossRef]
- McKee, M.G.; Unal, S.; Wilkes, G.L.; Long, T.E. Branched polyesters: Recent advances in synthesis and performance. Prog. Polym. Sci. 2005, 30, 507–539. [Google Scholar] [CrossRef]
- Bibers, I.; Kalnins, M. Control of biopolymer poly-β-hydroxybutyrate characteristics by γ-irradiation. Mech. Compos. Mater. 1999, 35, 169–178. [Google Scholar] [CrossRef]
- You, J.; Yu, W.; Zhou, C. Accelerated Crystallization of Poly(lactic acid): Synergistic Effect of Poly(ethylene glycol), Dibenzylidene Sorbitol, and Long-Chain Branching. Ind. Eng. Chem. Res. 2014, 53, 1097–1107. [Google Scholar] [CrossRef]
- Vahabi, H.; Michely, L.; Moradkhani, G.; Akbari, V.; Cochez, M.; Vagner, C.; Renard, E.; Saeb, M.R.; Langlois, V. Thermal Stability and Flammability Behavior of Poly(3-hydroxybutyrate) (PHB) Based Composites. Materials 2019, 12, 2239. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Kao, W.A.; Loría-Bastarrachea, M.I.; Pérez-Padilla, Y.; Cauich-Rodríguez, J.V.; Vázquez-Torres, H.; Cervantes-Uc, J.M. Thermal degradation of poly(caprolactone), poly(lactic acid), and poly(hydroxybutyrate) studied by TGA/FTIR and other analytical techniques. Polym. Bull. 2018, 75, 4191–4205. [Google Scholar] [CrossRef]
- Aydemir, D.; Gardner, D.J. Biopolymer blends of polyhydroxybutyrate and polylactic acid reinforced with cellulose nanofibrils. Carbohydr. Polym. 2020, 250, 116867. [Google Scholar] [CrossRef] [PubMed]
- Fintzou, A.T.; Kontominas, M.G.; Badeka, A.V.; Stahl, M.R.; Riganakos, K.A. Effect of electron-beam and gamma-irradiation on physicochemical and mechanical properties of polypropylene syringes as a function of irradiation dose: Study under vacuum. Radiat. Phys. Chem. 2007, 76, 1147–1155. [Google Scholar] [CrossRef]
- Hassan, M.M.; El-kelesh, N.A.; Dessouki, A.M. The effect of gamma and electron beam irradiation on the thermal and mechanical properties of injection-moulded high crystallinity poly(propylene). Polym. Compos. 2008, 29, 883–889. [Google Scholar] [CrossRef]
- Kolahchi, A.R.; Kontopoulou, M. Chain extended poly(3-hydroxybutyrate) with improved rheological properties and thermal stability, through reactive modification in the melt state. Polym. Degrad. Stab. 2015, 121, 222–229. [Google Scholar] [CrossRef]
- Fei, B.; Chen, C.; Chen, S.; Peng, S.; Zhuang, Y.; An, Y.; Dong, L. Crosslinking of poly[(3-hydroxybutyrate)-co-(3-hydroxyvalerate)] using dicumyl peroxide as initiator. Polym. Int. 2004, 53, 937–943. [Google Scholar] [CrossRef]
- Schmack, G.; Kramer, S.; Dorschner, H.; Gliesche, K. Effect of electron irradiation on the properties of poly(3-hydroxybutyric acid) fibres and their in vitro degradation. Polym. Degrad. Stab. 2004, 83, 467–472. [Google Scholar] [CrossRef]
- Effects of gamma irradiation on the molecular structure and mechanical properties of biodegradable polymer poly(hydroxybutyrate). In Proceedings of the 2005 International Nuclear Atlantic Conference—INAC 2005, Santos, SP, Brazil, 28 August–2 September 2005.
- Rouxhet, L.; Legras, R. Modifications induced by swift heavy ions in poly(hydroxybutyrate–hydroxyvalerate) (PHB/HV) and poly(ε-caprolactone) (PCL) films. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2000, 171, 487–498. [Google Scholar] [CrossRef]
- Nugroho, P.; Mitomo, H.; Yoshii, F.; Kume, T. Degradation of poly(l-lactic acid) by γ-irradiation. Polym. Degrad. Stab. 2001, 72, 337–343. [Google Scholar] [CrossRef]







| Mn (g/mol) | G(S) (Scissions/100 eV) | G(X) (Crosslinks/100 eV) |
|---|---|---|
| 118,667 ± 5033 | 1.13 ± 0.18 | 0.38 ± 0.12 |
| 12,800 ± 346 | 10.45 ± 1.64 | 3.55 ± 1.08 |
| Source | This Work | [16] | [15] | [36] | ||||
|---|---|---|---|---|---|---|---|---|
| Material | PHB | PHB | PHBV | PHB | PHB | |||
| powder | film | film | powder | film | powder | film | ||
| irradiation temperature | room temperature | 195 °C | room temperature | room temperature | room temperature | |||
| G(S) (scissions/100 eV) | 4.1 | 1.13 to 10.45 | 15.7 | 12.9 | 15.7 | 5.4 | 5.9 | 6.0 |
| G(X) (crosslinks/100 eV) | 0.6 | 0.38 to 3.55 | - | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krieg, D.; Müller, M.T.; Boldt, R.; Rennert, M.; Stommel, M. Additive Free Crosslinking of Poly-3-hydroxybutyrate via Electron Beam Irradiation at Elevated Temperatures. Polymers 2023, 15, 4072. https://doi.org/10.3390/polym15204072
Krieg D, Müller MT, Boldt R, Rennert M, Stommel M. Additive Free Crosslinking of Poly-3-hydroxybutyrate via Electron Beam Irradiation at Elevated Temperatures. Polymers. 2023; 15(20):4072. https://doi.org/10.3390/polym15204072
Chicago/Turabian StyleKrieg, David, Michael Thomas Müller, Regine Boldt, Mirko Rennert, and Markus Stommel. 2023. "Additive Free Crosslinking of Poly-3-hydroxybutyrate via Electron Beam Irradiation at Elevated Temperatures" Polymers 15, no. 20: 4072. https://doi.org/10.3390/polym15204072