
Sustainable Polycaprolactone Polyol-Based Thermoplastic Poly(ester ester) Elastomers Showing Superior Mechanical Properties and Biodegradability

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthetic Procedures
2.2.1. Preparation of Polycaprolactone Diols
2.2.2. Synthesis of Thermoplastic Poly(ester ester) Elastomers Using PCL Diols
2.3. Biodegradable Test of TPEs
2.4. Characterization
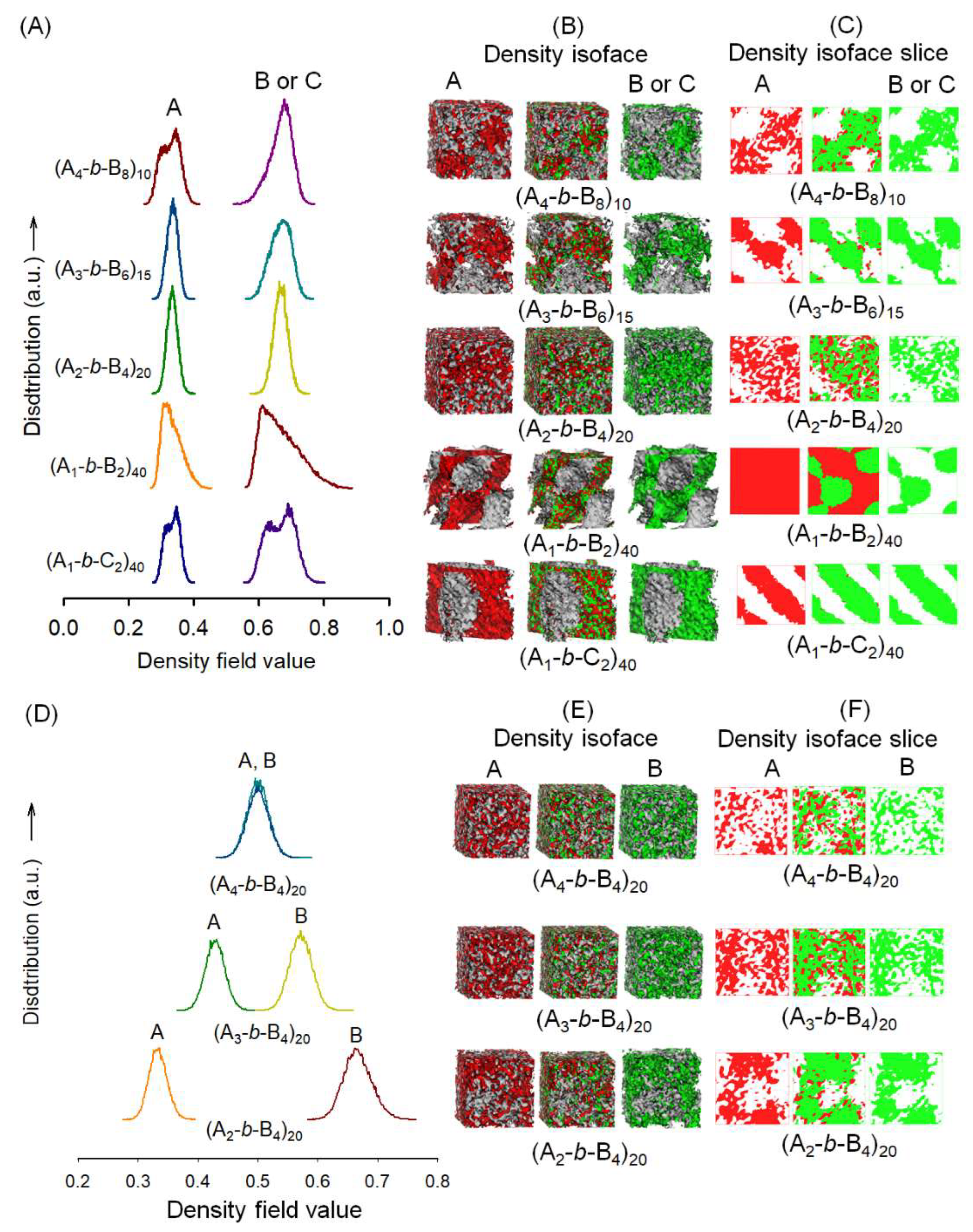
2.5. Mesoscale Simulation Method
3. Results and Discussion
3.1. Preparation of Polycaprolactone Diols with Different Molecular Weights
3.2. Synthesis of Thermoplastic Poly(ester ester) Elastomers
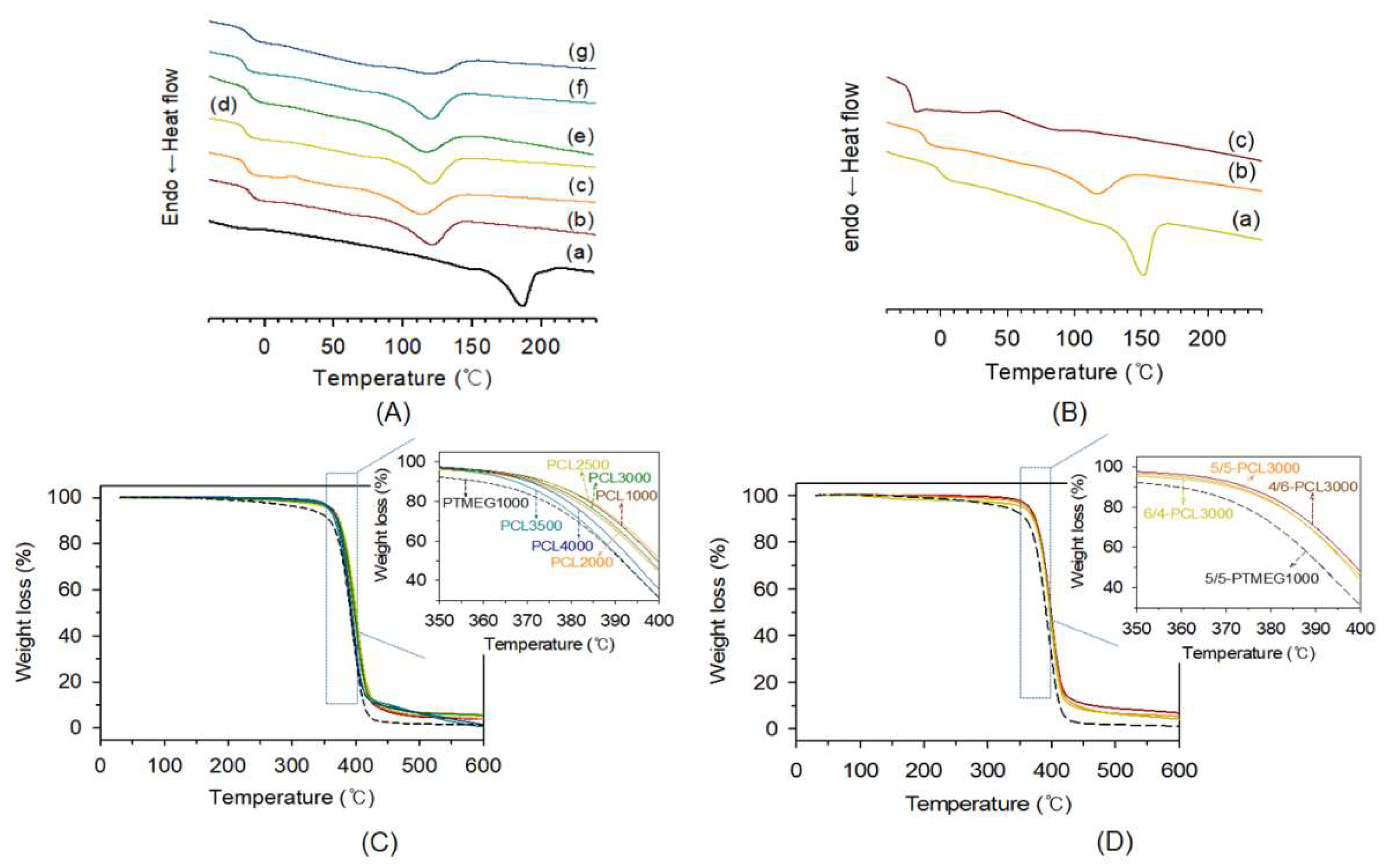
3.3. Thermal Properties of TPEs
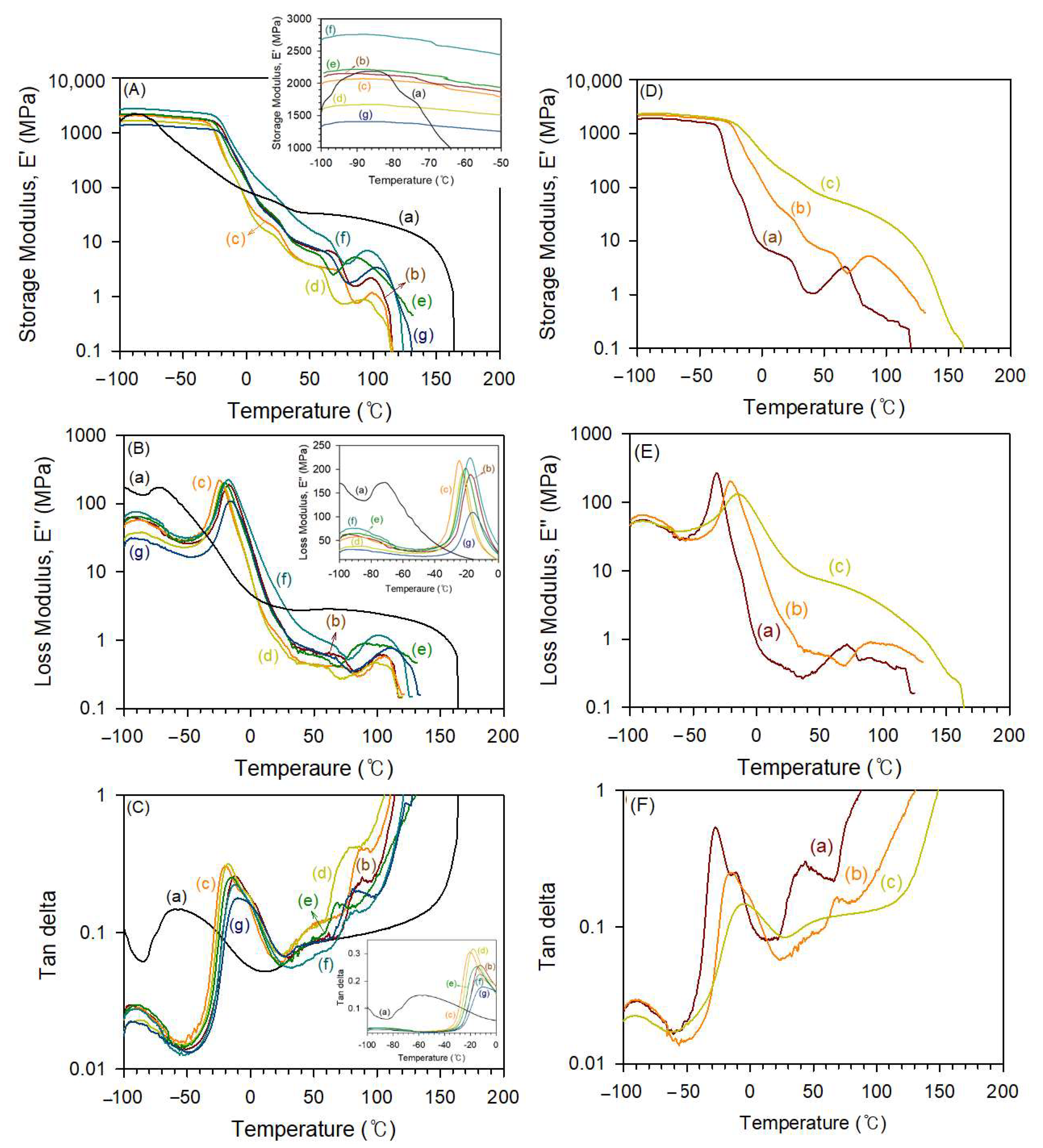
3.4. Tesile Properties of Thermoplastic Poly(Ester-Ester) Elastomers
3.5. WAXD Analysis of Thermoplastic Poly(Ester-Ester) Elastomers
3.6. Biodegradable Properties of Thermoplastic Poly(ester ester) Elastomers
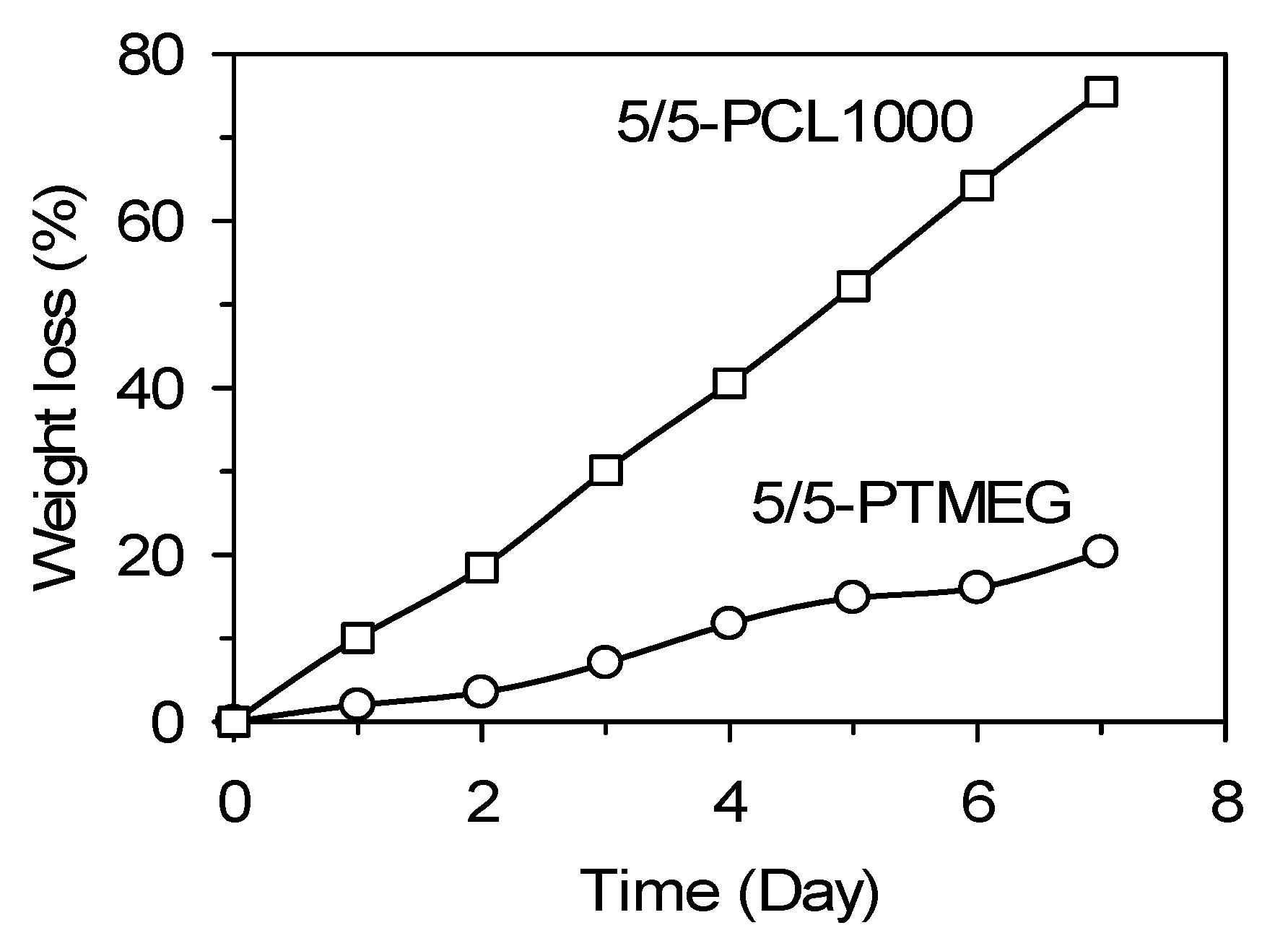
3.6.1. Hydrolytic Degradation Tests
3.6.2. Aerobic Degradation Tests
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Parameswaranpillai, J.; Midhun Dominic, C.D.; Rangappa, S.M.; Siengchin, S.; Ozbakkaloglu, T. Introduction to Elastombers; Elsevier Inc.: Amsterdam, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Alarif, I.M. A comprehensive review on advancements of elastomers for engineering applications. Adv. Ind. Eng. Polym. Res. 2023, in press. [CrossRef]
- Jagadeesh, P.; Rangappa, S.M.; Siengchin, S.; Puttegowda, M.; Thiagamani, S.M.K.; Rajeshkumar, G.; Kumar, M.H.; Oladijo, O.P.; Fiore, V.; Cuadrado, M.M.M. Sustainable recycling technologies for thermoplastic polymers and their composites: A review of the state of the art. Polym. Compos. 2022, 23, 5831–5862. [Google Scholar] [CrossRef]
- Wang, W.; Lu, W.; Goodwin, A.; Wang, H.; Yin, P.; Kang, N.G.; Hong, K.; Mays, J.W. Recent advances in thermoplastic elastomers from living polymerizations: Macromolecular architectures and supramolecular chemistry. Prog. Polym. Sci. 2019, 95, 1–31. [Google Scholar] [CrossRef]
- Drobny, J.G. Introduction and brief history of thermoplastic elastomers. In Handbook of Thermoplastic Elastomers, 2nd ed.; Drobny, J.G., Ed.; William Andrew Publishing: Oxford, UK, 2014; pp. 1–15. [Google Scholar] [CrossRef]
- Vinod, A.; Pulikkalparambil, H.; Jagadeesh, P.; Rangappa, S.M.; Siengchin, S. Recent advancements in lignocellulose biomass-based carbon fiber: Synthesis, properties, and applications. Heliyon 2023, 9, e13614. [Google Scholar] [CrossRef]
- Shi, L.; Zhu, T.; Gao, G.; Zhang, X.; Wei, W.; Liu, W.; Ding, S. Highly stretchable and transparent ionic conducting elastomers. Nat. Commun. 2018, 9, 2630. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.A. Polymer Science and Polymerization Methods toward Hybrid Materials. In Polymer Hybrid Materials and Nanocomposites; Saleh, T.A., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 59–103. [Google Scholar] [CrossRef]
- Jagadeesh, P.; Sanjay, M.R.; Siengchin, S. Advanced characterisation techniques for nanostructured materials in biomedical applications. Adv. Ind. Eng. Polym. Res. 2023, in press. [CrossRef]
- Pervez, T.; Al-Jahwari, F.S. Mechanical properties, sealability, and recyclability of elastomeric materials in petroleum industry. In Encyclopedia of Renewable and Sustainable Materials; Hashmi, S., Choudhury, I.A., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2020; Volume 5, pp. 131–147. [Google Scholar] [CrossRef]
- Legge, N.R. Thermoplastic elastomers—Three decades of progress. Rubber Chem. Technol. 1989, 62, 529–547. [Google Scholar] [CrossRef]
- Roslaniec, Z. Polyester thermoplastic elastomers: Synthesis, properties, and some applications. In Handbook of Condensation Thermoplastic Elastomers; Fakirov, D., Ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2005; pp. 75–116. [Google Scholar] [CrossRef]
- Ukielski, R. Terpoly(ester-b-ether-b-amide) thermoplastic elastomers: Synthesis, structure, and properties. In Handbook of Condensation Thermoplastic Elastomers; Fakirov, D., Ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2005; pp. 117–140. [Google Scholar] [CrossRef]
- Yamakawa, H.; Miyata, H. High performance thermoplastic aramid elastomers: Synthesis, properties, and applications. In Handbook of Condensation Thermoplastic Elastomers; Fakirov, D., Ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2005; pp. 141–166. [Google Scholar] [CrossRef]
- Sbrescia, S.; Ju, J.; Creton, C.; Engels, T.; Seitz, M. Effect of temperature, rate, and molecular weight on the failure behavior of soft block copoly(ether–ester) thermoplastic elastomers. Soft Matter 2023, in press. [CrossRef]
- Holland, B.J.; Hay, J.N. Analysis of comonomer content and cyclic oligomers of poly(ethylene terephthalate). Polymer 2002, 43, 1797–1804. [Google Scholar] [CrossRef]
- Lucherelli, M.A.; Duval, A.; Avérous, L. Biobased vitrimers: Towards sustainable and adaptable performing polymer materials. Prog. Polym. Sci. 2022, 127, 101515. [Google Scholar] [CrossRef]
- Thomas, J.; Bouscher, R.F.; Nwosu, J.; Soucek, M.D. Sustainable thermosets and composites based on the epoxides of norbornylized seed oils and biomass fillers. ACS Sustain. Chem. Eng. 2022, 10, 12342–12354. [Google Scholar] [CrossRef]
- Chamas, A.; Moon, H.; Zheng, J.; Qiu, Y.; Tabassum, T.; Jang, J.H.; Abu-Omar, M.; Scott, S.L.; Suh, S. Degradation rates of plastics in the environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. [Google Scholar] [CrossRef] [Green Version]
- Rosenboom, J.G.; Langer, R.; Traverso, G. Bioplastics for a circular economy. Nat. Rev. Mater. 2022, 7, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Albertsson, A.-C.; Varma, I.K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Dwivedi, R.; Kumar, S.; Pandey, R.; Mahajan, A.; Nandana, D.; Katti, D.S.; Mehrotra, D. Polycaprolactone as biomaterial for bone scaffolds: Review of literature. J. Oral. Biol. Craniofac. Res. 2020, 10, 381–388. [Google Scholar] [CrossRef]
- Bartnikowski, M.; Dargaville, T.R.; Ivanovski, S.; Hutmacher, D.W. Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog. Polym. Sci. 2019, 96, 1–20. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Guarino, V.; Gentile, G.; Sorrentino, L.; Ambrosio, L. Polycaprolactone: Synthesis, Properties, and Applications. In Encyclopedia of Polymer Science and Technology; Matyjaszewski, K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 1–36. [Google Scholar] [CrossRef]
- Guan, J.; Sacks, M.S.; Beckman, E.J.; Wagner, W.R. Synthesis, characterization, and cytocompatibility of elastomeric, biodegradable poly(ester-urethane)ureas based on poly(caprolactone) and putrescine. J. Biomed. Mater. Res. 2002, 61, 493–503. [Google Scholar] [CrossRef]
- Tang, D.; Macosko, C.W.; Hillmyer, M.A. Thermoplastic polyurethane elastomers from bio-based poly(δ-decalactone) diols. Polym. Chem. 2014, 5, 3231–3237. [Google Scholar] [CrossRef]
- Tran, C.H.; Pham, L.T.T.; Jang, H.B.; Kim, S.A.; Kim, I. Effect of α-, β-, γ-, and δ-dicarbonyl complexing agents on the double metal cyanide-catalyzed ring-opening polymerization of propylene oxide. Catal. Today 2021, 375, 429–440. [Google Scholar] [CrossRef]
- Tran, C.H.; Lee, M.W.; Park, S.W.; Jeong, J.E.; Lee, S.J.; Song, W.; Huh, P.; Kim, I. Heterogeneous Double Metal Cyanide Catalyzed Synthesis of Poly(ε-caprolactone) Polyols for the Preparation of Thermoplastic Elastomers. Catalysts 2021, 11, 1033. [Google Scholar] [CrossRef]
- Tran, C.H.; Lee, M.-W.; Lee, S.-J.; Choi, J.-H.; Lee, E.-G.; Choi, H.-K.; Kim, I. Highly Active Heterogeneous Double Metal Cyanide Catalysts for Ring-Opening Polymerization of Cyclic Monomers. Polymers 2022, 14, 2507. [Google Scholar] [CrossRef]
- Jeon, S.H.; Jeong, J.E.; Kim, S.; Jeon, S.; Choung, J.W.; Kim, I. Hardness Modulated Thermoplastic Poly(ether ester) Elastomers for the Automobile Weather-Strip Application. Polymers 2021, 13, 525. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, K.Y.; Han, M.H. Preparation and properties of highly functional copolyetheresters. J. Appl. Polym. Sci. 2003, 88, 139–145. [Google Scholar] [CrossRef]
- Mondal, S.; Martin, D. Hydrolytic degradation of segmented polyurethane copolymers for biomedical applications. Polym. Degrad. Stab. 2012, 97, 1553–1561. [Google Scholar] [CrossRef]
- Chuah, H.H.; Lin-Vien, D.; Soni, U. Poly(trimethylene terephthalate) molecular weight and Mark–Houwink equation. Polymer 2001, 42, 7137–7139. [Google Scholar] [CrossRef]
- Accelrys, Inc. Materials Studio Release Notes, Release 4.2; Accelrys, Inc.: San Diego, CA, USA, 2007. [Google Scholar]
- Mu, D.; Li, Q.; Feng, Y. MesoDyn modeling study of the phase morphologies of miktoarm poly(ethylene oxide)-b-poly(methyl methacrylate) copolymers doped with nanoparticles. Polym. Int. 2014, 63, 568–575. [Google Scholar] [CrossRef]
- Kang, H.; Cheon, M.; Lee, C.H.; Kim, T.-H.; Hong, Y.T.; Nam, S.Y.; Park, C.H. Mesoscale simulation based on the dynamic mean-field density functional method on block-copolymeric ionomers for polymer electrolyte membranes. Membranes 2023, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, S.; Wu, J. Mesoscopic simulation on phase behavior of ternary copolymeric solution in the absence and presence of shear. Macromolecules 2006, 39, 6631–6642. [Google Scholar] [CrossRef]
- Báez, J.E.; Marcos-Fernández, Á.; Lebrón-Aguilar, R.; Martínez-Richa, A. A novel route to α,ω-telechelic poly(ε-caprolactone) diols, precursors of biodegradable polyurethanes, using catalysis by decamolybdate anion. Polymer 2006, 47, 8420–8429. [Google Scholar] [CrossRef]
- Cebe, P.; Hong, S.D. Crystallization behaviour of poly(ether-ether-ketone). Polymer 1986, 27, 1183–1192. [Google Scholar] [CrossRef]
- Bassett, D.C.; Olley, R.H.; Raheil, I.A.M. On crystallization phenomena in PEEK. Polymer 1988, 29, 1745–1754. [Google Scholar] [CrossRef]
- Lee, Y.; Porter, R.S.; Lin, J.S. On the double-melting behavior of poly(ether ether ketone). Macromolecules 1989, 22, 1756–1760. [Google Scholar] [CrossRef]
- Jonas, A.M.; Russell, T.P.; Yoon, D.Y. Synchrotron x-ray scattering studies of crystallization of poly(ether-ether-ketone) from the glass and structural changes during subsequent heating-cooling processes. Macromolecules 1995, 28, 8491–8503. [Google Scholar] [CrossRef]
- Jeong, Y.G.; Jo, W.H.; Lee, S.C. Cocrystallization Behavior of Poly(butylene terephthalate-co-butylene 2,6-naphthalate) Random Copolymers. Macromolecules 2000, 33, 9705–9711. [Google Scholar] [CrossRef]
- Lilaonitkul, A.; Cooper, S.L. Properties of Polyether-Polyester Thermoplastic Elastomers. Rubber Chem. Technol. 1977, 50, 1–23. [Google Scholar] [CrossRef]
- John, J.V.; Kim, K.R.; Baek, S.T.; Yoon, J.H.; Suh, H.; Kim, I. Effect of chain-extender modification on the structure and properties of thermoplastic poly(ether ester) elastomers. J. Appl. Polym. Sci. 2016, 133, 42888. [Google Scholar] [CrossRef]
- Boulares, A.; Tessier, M.; Maréchal, E. Synthesis and characterization of poly(copolyethers-block-polyamides) II. Characterization and properties of the multiblock copolymers. Polymer 2000, 41, 3561–3580. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, I. Polycaprolactone Polyol-Based Thermoplastic Poly (ester ester) Elastomers showing High-End Properties. Annu. Meet. Program Book Polym. Soc. Korea 2022, 47, 74. [Google Scholar]
- Djonlagic, J.; Nikolic, M.S. Thermoplastic copolyester elastomers. In Handbook of Engineering and Specialty Thermoplastics; Thomas, S., Visakh, P.M., Eds.; Scrivrener Publishing LLC: Salem, MA, USA, 2011; pp. 377–428. [Google Scholar] [CrossRef]
- Harrison, J.P.; Boardman, C.; O’Callaghan, K.; Delort, A.-M.; Song, J. Biodegradability standards for carrier bags and plastic films in aquatic environments: A critical review. R. Soc. Open Sci. 2018, 5, 171792. [Google Scholar] [CrossRef] [Green Version]
- Kunioka, M.; Ninomiya, F.; Funabashi, M. Biodegradation of poly(butylene succinate) powder in a controlled compost at 58 °C evaluated by naturally-occurring carbon 14 amounts in evolved CO2 based on the ISO 14855-2 method. Int. J. Mol. Sci. 2009, 10, 4267–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnik, E. Biodegradability testing of compostable Polymer Materials. In Compostable Polymer Materials; Rudnik, E., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2008; pp. 112–166. [Google Scholar] [CrossRef]
- Pepic, D.; Nikolic, M.S.; Djonlagic, J. Synthesis and characterization of biodegradable aliphatic copolyesters with poly(tetramethylene oxide) soft segments. J. Appl. Polym. Sci. 2007, 106, 1777–1786. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Mm a (g/mol) | Vm b (cm3/mol) | δ c (J/cm3)0.5 | Cn d | NMeso e |
|---|---|---|---|---|---|
| PCL | 114.14 | 103.61 | 17.78 | 5.78 | ~2 |
| PTMEG | 72.11 | 71.24 | 18.11 | 5.99 | ~2 |
| DMT/BDO | 220.23 | 178.07 | 19.27 | 4.15 | ~1 |
| Sample | [CL]0/ [EG]0 | Mnb (g mol−1) | Ð b | OH value c | F c | Tgd (°C) | Tmd (°C) | |
|---|---|---|---|---|---|---|---|---|
| Theor. | Measured b | |||||||
| PCL1000 | 9 | 1000 | 1000 | 1.3 | 107.04 | 1.91 | –61.0 | 31.3/36.3 |
| PCL2000 | 17 | 2000 | 2200 | 1.4 | 50.11 | 1.97 | –60.0 | 44.6/45.9 |
| PCL2500 | 22 | 2500 | 2400 | 1.5 | 36.37 | 1.99 | –61.2 | 46.8/48.7 |
| PCL3000 | 26 | 3000 | 3000 | 1.5 | 37.77 | 2.02 | –57.8 | 45.5/49.5 |
| PCL3500 | 30 | 3500 | 3500 | 1.6 | 32.70 | 2.04 | –65.4 | 49.9/51.1 |
| PCL4000 | 35 | 4000 | 4200 | 1.8 | 23.28 | 1.74 | –60.1 | 50.8/51.9 |
| Sample Code | HS/SS | Tg1 a (°C) | Tg1 b (°C) | Tg2 b (°C) | Tm a (°C) | η (dL g−1) | Mv c (g mol−1) | Mn d (g mol−1) | |
|---|---|---|---|---|---|---|---|---|---|
| Feed | NMR | ||||||||
| 5/5-PTMEG | 50/50 | 45/55 | −22.9 | −54.9 | 42.6 | 168.9 | 1.70 | 113,300 | - |
| 5/5-PCL1000 | 50/50 | 40/60 | −11.1 | −11.8 | 88.3 | 120.5 | 1.90 | 139,500 | 27,500 |
| 5/5-PCL2000 | 50/50 | 44/56 | −14.4 | −20.4 | 88.0 | 120.0 | 1.90 | 139,600 | 33,700 |
| 5/5-PCL2500 | 50/50 | 39/61 | −11.6 | −17.8 | 79.0 | 119.7 | 2.04 | 154,800 | 34,500 |
| 5/5-PCL3000 | 50/50 | 41/59 | −14.7 | −15.4 | 68.3 | 117.6 | 2.35 | 189,400 | 51,800 |
| 5/5-PCL3500 | 50/50 | 40/60 | −12.8 | −12.3 | 83.2 | 116.5 | 1.84 | 132,600 | 31,300 |
| 5/5-PCL4000 | 50/50 | 47/53 | −13.9 | −10.4 | 81.5 | 113.0 | 1.79 | 127,500 | 28,800 |
| 4/6-PCL3000 | 40/60 | 27/73 | −21.1 | −27.5 | 43.8 | 83.3 | 1.43 | 92,800 | 46,200 |
| 6/4-PCL3000 | 60/40 | 57/43 | −0.5 | 0.90 | 48.5 | 152.2 | 1.78 | 132,300 | 35,300 |
| Sample | HS/SS (wt%) | Ultimate Strength (MPa) | Elongation (%) | Hardness | |
|---|---|---|---|---|---|
| Shore A | Shore D | ||||
| 5/5-PTMEG | 50/50 | 23.42 | 1166 | 85 | 29 |
| 5/5-PCL1000 | 50/50 | 33.83 | 1105 | 90 | 35 |
| 5/5-PCL2000 | 50/50 | 36.26 | 1560 | 93 | 38 |
| 5/5-PCL2500 | 50/50 | 38.46 | 1395 | 91 | 37 |
| 5/5-PCL3000 | 50/50 | 39.72 | 1152 | 93 | 39 |
| 5/5-PCL3500 | 50/50 | 37.76 | 1242 | 95 | 39 |
| 5/5-PCL4000 | 50/50 | 24.47 | 856 | 95 | 40 |
| 4/6-PCL3000 | 40/60 | 33.96 | 1638 | 75 | 22 |
| 6/4-PCL3000 | 60/40 | 44.92 | 820 | 98 | 47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-H.; Woo, J.-J.; Kim, I. Sustainable Polycaprolactone Polyol-Based Thermoplastic Poly(ester ester) Elastomers Showing Superior Mechanical Properties and Biodegradability. Polymers 2023, 15, 3209. https://doi.org/10.3390/polym15153209
Choi J-H, Woo J-J, Kim I. Sustainable Polycaprolactone Polyol-Based Thermoplastic Poly(ester ester) Elastomers Showing Superior Mechanical Properties and Biodegradability. Polymers. 2023; 15(15):3209. https://doi.org/10.3390/polym15153209
Chicago/Turabian StyleChoi, Jin-Hyeok, Jeong-Jae Woo, and Il Kim. 2023. "Sustainable Polycaprolactone Polyol-Based Thermoplastic Poly(ester ester) Elastomers Showing Superior Mechanical Properties and Biodegradability" Polymers 15, no. 15: 3209. https://doi.org/10.3390/polym15153209