

Synthesis, Thermogravimetric Analysis, and Kinetic Study of Poly-N-Isopropylacrylamide with Varied Initiator Content

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PNIPA Nanoparticles
2.3. Powder X-ray Diffraction Analysis (PXRD)
2.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.5. Thermogravimetric Analysis (TGA)
3. Results
3.1. Synthesis
3.2. Powder X-ray Diffraction Analysis (XRPD)
3.3. Attenuated Total Reflection Fourier-Transformed Infrared Spectroscopy (ATR-FTIR)
3.4. Thermogravimetric Analysis (TGA)
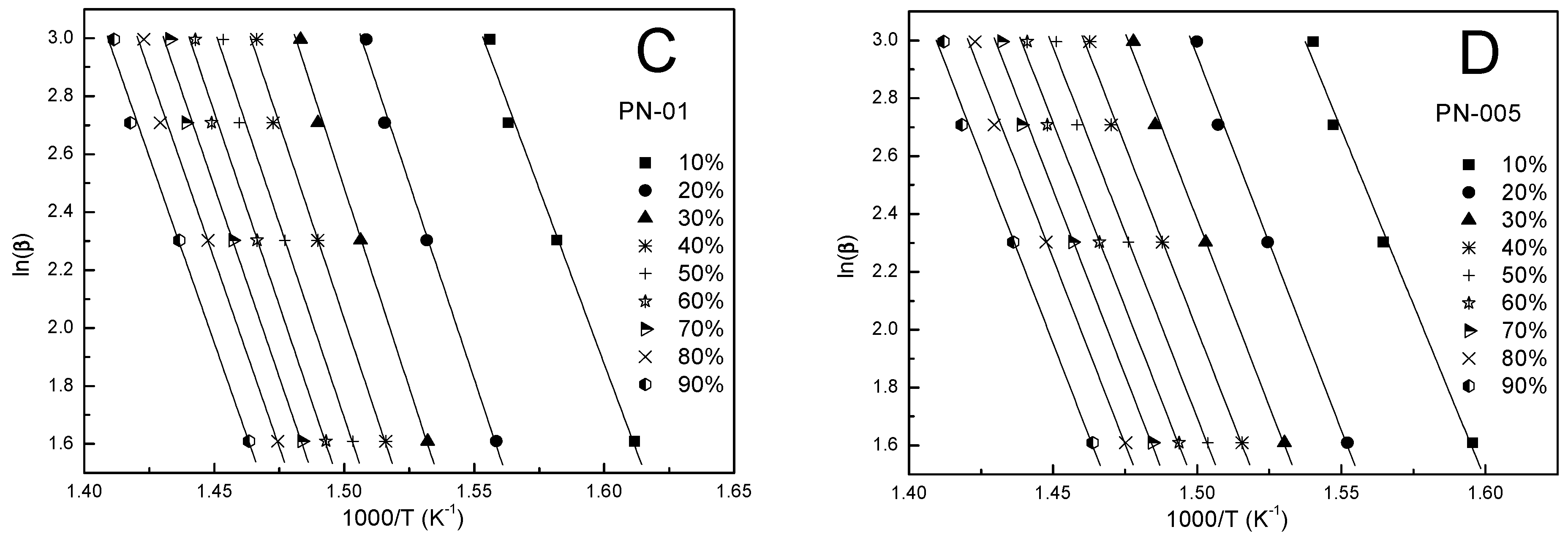
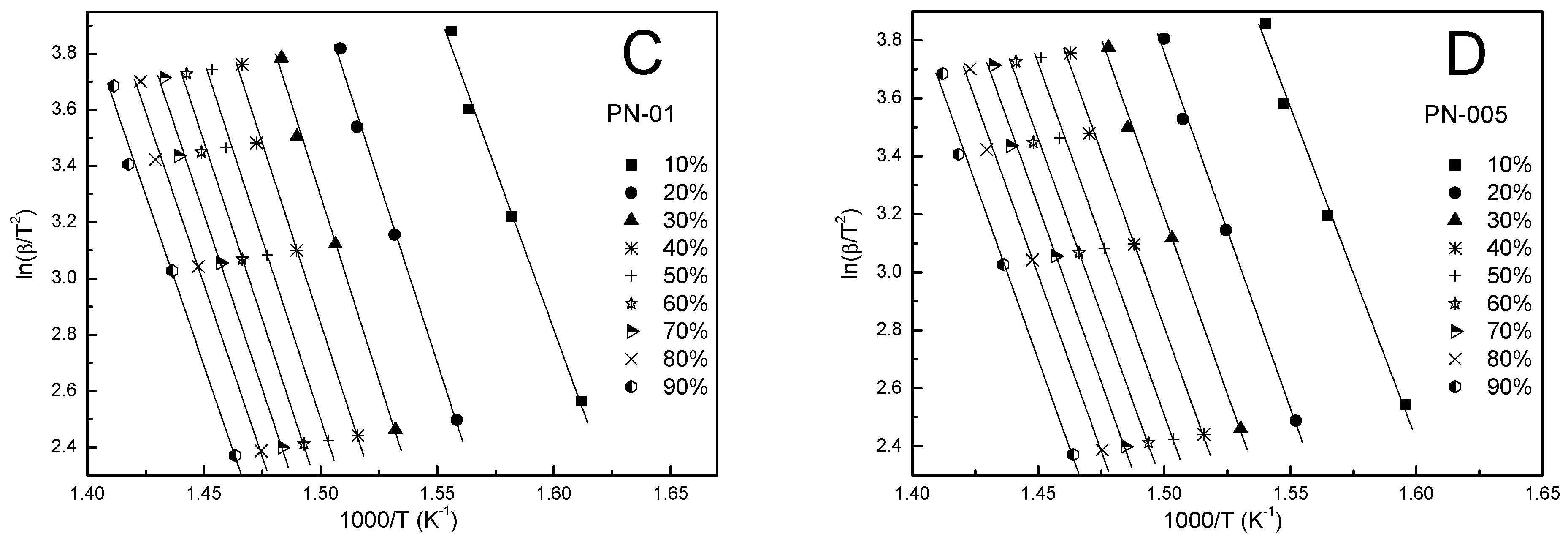
3.5. Kinetic Analysis
4. Discussion
4.1. XRPD
4.2. ATR-FTIR
4.3. TGA
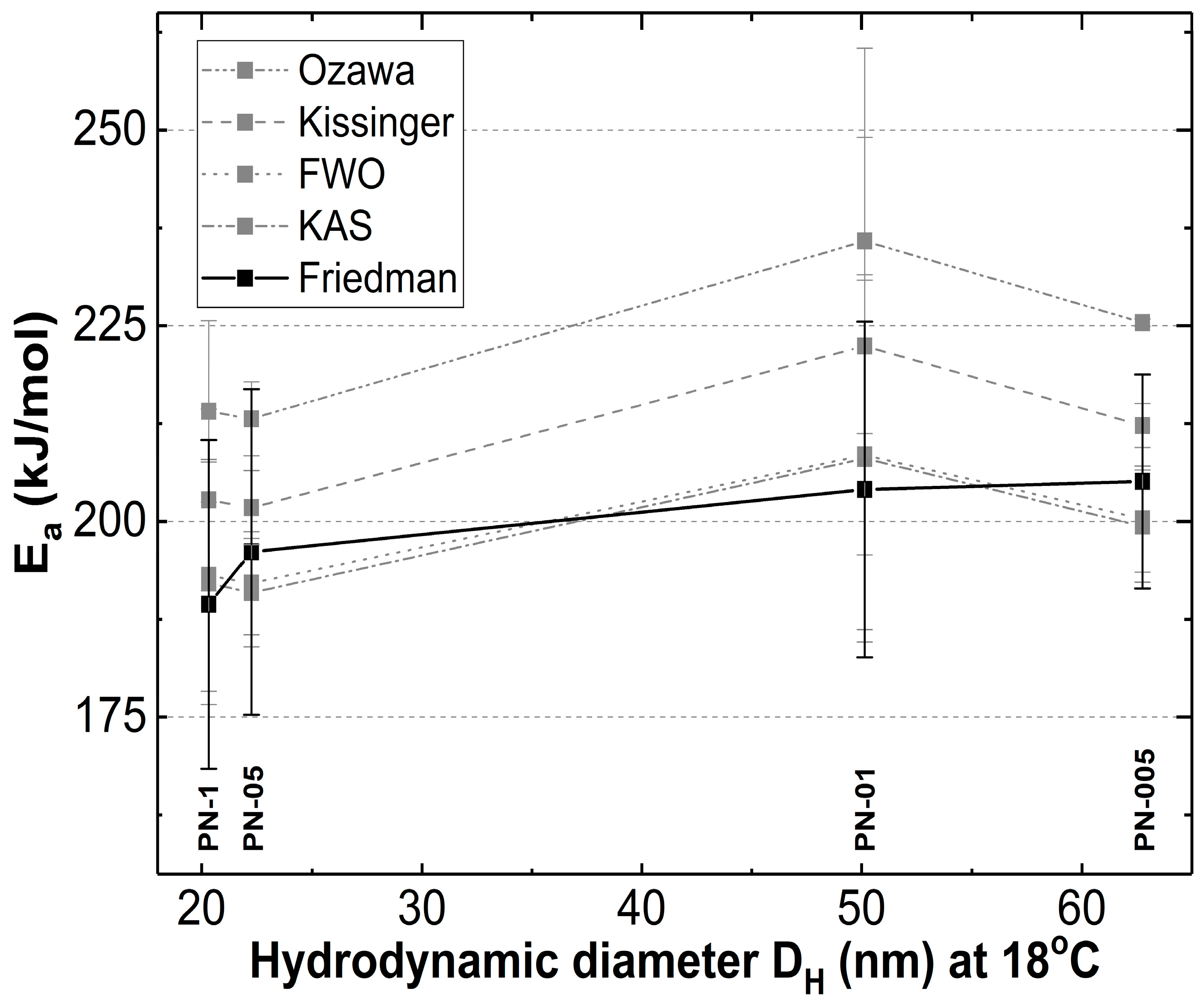
4.4. Kinetic
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sözüdoğru, E.; Clarke, B. Uncertainty in Drug Discovery: Strategies, Heuristics and Technologies. In Uncertainty in Pharmacology: Epistemology, Methods, and Decisions; LaCaze, A., Osimani, B., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 153–171. [Google Scholar] [CrossRef]
- Campora, S.; Mohsen, R.; Passaro, D.; Samir, H.; Ashraf, H.; Al-Mofty, S.E.-D.; Diab, A.A.; El-Sherbiny, I.M.; Snowden, M.J.; Ghersi, G. Functionalized Poly(N-isopropylacrylamide)-Based Microgels in Tumor Targeting and Drug Delivery. Gels 2021, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; He, C.; Xiao, C.; Zhuang, X.; Chen, X. Biodegradable pH-responsive polyacrylic acid derivative hydrogels with tunable swelling behavior for oral delivery of insulin. Polymer 2013, 54, 1786–1793. [Google Scholar] [CrossRef]
- Özkahraman, B.; Acar, I.; Güçlü, G. Synthesis of N-vinylcaprolactam and methacrylic acid based hydrogels and investigation of drug release characteristics. Polym. Bull. 2022, 80, 5149–5181. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Christa, J. Temperature and pH sensitive multi-functional magnetic nanocomposite for the controlled delivery of 5-fluorouracil, an anticancer drug. J. Drug Deliv. Sci. Technol. 2020, 55, 101476. [Google Scholar] [CrossRef]
- Kobryń, J.; Zięba, T.; Sowa, S.K.; Musiał, W. Influence of Acetylated Annealed Starch on the Release of β-Escin from the Anionic and Non-Ionic Hydrophilic Gels. Pharmaceutics 2020, 12, 84. [Google Scholar] [CrossRef]
- Gaware, S.A.; Rokade, K.A.; Kale, S.N. Silica-chitosan nanocomposite mediated pH-sensitive drug delivery. J. Drug Deliv. Sci. Technol. 2019, 49, 345–351. [Google Scholar] [CrossRef]
- Huang, G.; Huang, H. Application of hyaluronic acid as carriers in drug delivery. Drug Deliv. 2018, 25, 766–772. [Google Scholar] [CrossRef]
- Ciolacu, D.E.; Nicu, R.; Ciolacu, F. Cellulose-Based Hydrogels as Sustained Drug-Delivery Systems. Materials 2020, 13, 5270. [Google Scholar] [CrossRef] [PubMed]
- Audureau, N.; Coumes, F.; Veith, C.; Guibert, C.; Guigner, J.-M.; Stoffelbach, F.; Rieger, J. Synthesis and Characterization of Temperature-Responsive N-Cyanomethylacrylamide-Containing Diblock Copolymer Assemblies in Water. Polymers 2021, 13, 4424. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Wang, H. Temperature-responsive polymers: Synthesis, properties, and biomedical applications. Nano Res. 2018, 11, 5400–5423. [Google Scholar] [CrossRef]
- Xu, X.-F.; Pan, C.-Y.; Zhang, W.-J.; Hong, C.-Y. Polymerization-Induced Self-Assembly Generating Vesicles with Adjustable ph-Responsive Release Performance. Macromolecules 2019, 52, 1965–1975. [Google Scholar] [CrossRef]
- Kocak, G.; Tuncer, C.; Bütün, V. pH-Responsive polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, X.; Wen, X.; Xu, Q.; Zeng, H.; Zhao, Y.; Liu, M.; Wang, Z.; Hu, X.; Wang, Y. Bio-responsive smart polymers and biomedical applications. J. Phys. Mater. 2019, 2, 032004. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Costa, R.O.R.; Freitas, R.F.S. Phase behavior of poly(N-isopropylacrylamide) in binary aqueous solutions. Polymer 2002, 43, 5879. [Google Scholar] [CrossRef]
- Ansari, M.J.; Rajendran, R.R.; Mohanto, S.; Agarwal, U.; Panda, K.; Dhotre, K.; Manne, R.; Deepak, A.; Zafar, A.; Yasir, M.; et al. Poly(N-isopropylacrylamide)-Based Hydrogels for Biomedical Applications: A Review of the State-of-the-Art. Gels 2022, 8, 454. [Google Scholar] [CrossRef]
- Halperin, A.; Kröger, M.; Winnik, F.M. Poly(N-isopropylacrylamide) phase diagrams: Fifty years of research. Angew Chem. Int. Ed. 2015, 54, 15342–15367. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, C.; Yang, X.; Shen, B.; Sun, Y.; Chen, Y.; Xu, X.; Sun, H.; Yu, K.; Yang, B.; et al. PH- and Temperature-Sensitive Hydrogel Nanoparticles with Dual Photoluminescence for Bioprobes. ACS Nano 2016, 10, 5856–5863. [Google Scholar] [CrossRef]
- Ziane, S.; Schlaubitz, S.; Miraux, S.; Patwa, A.; Lalande, C.; Bilem, I.; Lepreux, S.; Rousseau, B.; Le Meins, J.F.; Latxague, L. A thermosensitive low molecular weight hydrogel as scaffold for tissue engineering. Eur. Cells Mater. 2012, 23, 147–160. [Google Scholar] [CrossRef]
- Nadour, M.; Boukraa, F.; Ouradi, A.; Benaboura, A. Effects of methylcellulose on the properties and morphology of polysulfone membranes prepared by phase inversion. J. Mater. Res. 2017, 20, 339–348. [Google Scholar] [CrossRef]
- Trivedi, M.K.; Branton, A.; Trivedi, D.; Nayak, G.; Mishra, R.; Jana, S. Characterization of Physicochemical and Thermal Properties of Biofield Treated Ethyl Cellulose and Methyl Cellulose. Int. J. Biomed. Mater. Res. 2015, 3, 83–91. [Google Scholar] [CrossRef]
- Guo, L.; Liu, G.; Hong, R.-Y.; Li, H.-Z. Preparation and Characterization of Chitosan Poly(acrylic acid) Magnetic Microspheres. Mar. Drugs. 2010, 8, 2212–2222. [Google Scholar] [CrossRef] [PubMed]
- Maurer, J.J.; Harvey, G.D. Thermal degradation characteristics of poly(acrylamide-co-acrylic acid) and poly(acrylamide-co-sodium acrylate) copolymers. Thermochim. Acta 1987, 121, 295–306. [Google Scholar] [CrossRef]
- McNeill, I.C.; Sadeghi, S.M.T. Thermal stability and degradation mechanisms of poly(acrylic acid) and its salts: Part 1—Poly(acrylic acid). Polym. Degrad. Stab. 1990, 29, 233–246. [Google Scholar] [CrossRef]
- Ekici, S. Intelligent poly(N-isopropylacrylamide)-carboxymethyl cellulose full interpenetrating polymeric networks for protein adsorption studies. J. Mater. Sci. 2011, 46, 2843–2850. [Google Scholar] [CrossRef]
- Jamrógiewicz, M.; Ciesielski, A. Application of vibrational spectrosco- py, thermal analyses and X-ray diffraction in the rapid evaluation of the stability in solid-state of ranitidine, famotidine and cimetidine. J. Pharm. Biomed. Anal. 2015, 107, 236–243. [Google Scholar] [CrossRef]
- Sabbagh, B.A.; Kumar, P.V.; Chew, Y.L.; Chin, J.H.; Akowuah, G.A. Determination of metformin in fixed-dose combination tablets by ATR-FTIR spectroscopy. Chem. Data Collect. 2022, 39, 100868. [Google Scholar] [CrossRef]
- Azminah, A.; Ahmad, I.; Fikri, J.A.N.; Jumadil, M.I.; Erza, N.A.F.; Abdullah, S.; Simamora, A.; Abdul, M.I. Rapid detection of synthetic adulterants in Indonesian herbal medicines using ATR-FTIR spectroscopy combined with chemometrics. J. Res. Pharm. 2023, 27, 184–195. [Google Scholar] [CrossRef]
- de Mendonça, C.M.S.; de Barros Lima, I.P.; Aragão, C.F.S.; Gomes, A.P.B. Thermal compatibility between hydroquinone and retinoic acid in pharmaceutical formulations. J. Therm. Anal. Calorim. 2014, 115, 2277–2285. [Google Scholar] [CrossRef]
- Tita, D.; Fulias, A.; Tita, B. Thermal stability of ketoprofen-active substance and tablets: Part 1. Kinetic study of the active substance under non-isothermal conditions. J. Therm. Anal. Calorim. 2011, 105, 501–508. [Google Scholar] [CrossRef]
- Al-Nahary, T.T.; El-Ries, M.A.; Sultan, M.; Mabkhot, Y.N.; Al-Hussam, A.M. Thermal stability of anti-rheumatic pharmaceutical drugs parecoxib sodium and valdecoxib. J. Saudi Chem. Soc. 2012, 16, 177–182. [Google Scholar] [CrossRef]
- Radha, S.; Gutch, P.K.; Ganesan, K.; Vijayaraghavan, R.; Suman, J.; Subodh, D. Thermal analysis of interactions between an oxime and excipients in some binary mixtures by differential scanning calorimetry and thermagravimetric analysis. J. Pharm. Res. 2010, 3, 590–595. [Google Scholar]
- Ozawa, T. A New method of analyzing thermogravimetric data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Flynn, H.J.; Wall, L.A. General treatment of the thermogravimetry of polymers. J. Res. Natl. Bur. Stand. A Phys. Chem. 1966, 70, 487–523. [Google Scholar] [CrossRef] [PubMed]
- Akahira, T.; Sunose, T. Method of determining activation deterioration constant of electrical insulating materials. Rep. Res. Inst. Chiba Inst. Technol. (Sci. Technol.) 1971, 16, 22–31. [Google Scholar]
- Friedman, H.L. Kinetics of Thermal Degradation of Char- forming Plastics from Thermo-gravimetry. Application to a Phenolic Plastic. J. Polym. Sci. C 1965, 50, 183–195. [Google Scholar]
- Málek, J. Kinetic analysis of crystallization processes in amorphous materials. Thermochim. Acta 2000, 355, 239–253. [Google Scholar] [CrossRef]
- Doyle, C. Kinetic analysis of thermogravimetric data. J. Appl. Polym. Sci. 1961, 5, 285–292. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J. Kinetic parameters from thermogravimetric data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Sousa, R.G.; Magalhaes, W.F.; Freitas, R.F.S. Glass transition and thermal stability of poly(N-isopropylacrylamide) gels and some of their copolymers with acrylamide. Polym. Degrad. Stab. 1998, 61, 275–281. [Google Scholar] [CrossRef]
- Boutris, C.; Chatzi, E.G.; Kiparissides, C. Characterization of the LCST behaviour of aqueous poly(N-isopropylacrylamide) solutions by thermal and cloud point techniques. Polymer 1997, 38, 2567–2570. [Google Scholar] [CrossRef]
- Bauri, K.; Roy, S.G.; Arora, S.; Dey, R.K.; Goswami, A.; Madras, G.; De, P. Thermal degradation kinetics of thermoresponsive poly(N-isopropylacrylamide-co-N,N-dimethylacrylamide) copolymers prepared via RAFT polymerization. J. Therm. Anal. Calorim. 2013, 111, 753–761. [Google Scholar] [CrossRef]
- Stanescu, P.O.; Turturica, G.; Andrei, M.; Draghici, C.; Vuluga, D.M.; Zaharia, A.; Sarbu, A.; Teodorescu, M. Kinetic Study upon the Thermal Degradation of Poly(Nisopropylacrylamide-co-5,6-benzo-2-methylene-1,3-dioxepane) Statistical Copolymers. Mater. Plast. 2015, 52, 193–197. [Google Scholar] [CrossRef]
- Patra, P.; Rameshbabu, A.P.; Das, D.; Dhara, S.; Panda, A.B.; Pal, S. Stimuli-responsive, biocompatible hydrogel derived from glycogen and poly(N-isopropylacrylamide) for colon targeted delivery of ornidazole and 5-amino salicylic acid. Polym. Chem. 2016, 7, 5426–5435. [Google Scholar] [CrossRef]
- Gola, A.; Knysak, T.; Musiał, W. The Influence of Anionic Initiator on the Selected Properties of Poly-N-Isopropyl Acrylamide Evaluated for Controlled Drug Delivery. Molecules 2017, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-M.; Pramoda, K.P.; Yang, Y.-Y.; Chow, S.Y.; He, C. Cholesteryl-grafted functional amphiphilic poly(N-isopropylacryla-mide-co-N-hydroxylmethylacrylamide): Synthesis, temperature-sensitivity, self-assembly and encapsulation of a hydrophobic agent. Biomaterials 2004, 25, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Ilić-Stojanović, S.; Nikolić, L.; Nikolić, V.; Petrović, S.; Oro, V.; Mitić, Ž.; Najman, S. Semi-Crystalline Copolymer Hydrogels as Smart Drug Carriers: In Vitro Thermo-Responsive Naproxen Release Study. Pharmaceutics 2021, 13, 158. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of infrared spectra, a practical approach. In Encyclopedia ofAnalytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2000; pp. 10815–10837. ISBN 0471976709. [Google Scholar]
- Ilić-Stojanović, S.; Nikolić, L.; Nikolić, V.; Ristić, I.; Budinski-Simendić, J.; Kapor, A.; Nikolić, G.M. The structure characterization of thermosensitive poly(N-isopropylacrylamide-co-2-hydroxypropyl methacrylate) hydrogel. Polym. Int. 2014, 63, 973–981. [Google Scholar] [CrossRef]
- Hirashima, Y.; Sato, H.; Miyashita, Y.; Suzuki, A. ATR-FTIR spcctroscopic study on hydrogen bonding of Poly(N-isopropylacrylamide-co-sodium acrylate) gel. Macromolecules 2005, 38, 9280–9286. [Google Scholar] [CrossRef]
- Mikawa, Y. Characteristic absorption bands of vinyl ethers. Bull. Chem. Soc. Jpn. 1956, 29, 110–115. [Google Scholar] [CrossRef]
- McManis, G.E.; Gast, L.E. IR spectra of long chain vinyl derivatives. J. Am. Oil Chem. Soc. 1971, 48, 668–673. [Google Scholar] [CrossRef]
- Yang, J.; Miranda, R.; Roy, C. Using the DTG curve fitting method to determine the apparent kinetic parameters of thermal decomposition of polymers. Polym. Degrad. Stab. 2001, 73, 455–461. [Google Scholar] [CrossRef]
- Aboulkas, A.; El Harfi, K.; El Bouadili, A. Thermal degradation behaviors of polyethylene and polypropylene. Part I: Pyrolysis kinetics and mechanisms. Energy Convers. Manag. 2010, 51, 1363–1369. [Google Scholar] [CrossRef]
- Heydari, M.; Rahman, M.; Gupta, R. Kinetic Study and Thermal Decomposition Behavior of Lignite Coal. Int. J. Chem. Eng. 2015, 2015, 9. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Sbirrazzuoli, N. Isoconversional kinetic analysis of thermally stimulated processes in polymers. Macromol. Rapid Commun. 2006, 27, 1515–1532. [Google Scholar] [CrossRef]
- Wu, W.; Cai, J.; Liu, R. Isoconversional kineticanalysisof distributed activation energy model processes for pyrolysis of solid fuels. Ind. Eng. Chem. Res. 2013, 52, 14376–14383. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Nanosphere * | Monomer NIPA [g] | Initiator KPS [g] |
|---|---|---|
| PN-1 | 5.002 | 5.972 |
| PN-05 | 5.004 | 2.986 |
| PN-01 | 5.006 | 0.598 |
| PN-005 | 5.002 | 0.300 |
| Sample | Heating Rate [°C min−1] | Tm [°C] | Rate of Mass Loss [% min−1] | TOnset [°C] | TEndset [°C] | Res [%] | T0.5wt.% [°C] |
|---|---|---|---|---|---|---|---|
| PN-005 | 5 | 400.6 | 10.69 | 372.6 | 394.5 | 4.79 | 269.0 |
| 10 | 412.1 | 19.55 | 380.6 | 427.2 | 5.63 | 275.0 | |
| 15 | 420.4 | 27.51 | 387.0 | 436.2 | 5.42 | 253.0 | |
| 20 | 424.8 | 35.97 | 395.1 | 440.8 | 5.23 | 193.0 | |
| PN-01 | 5 | 400.5 | 10.40 | 369.2 | 413.1 | 4.58 | 260.0 |
| 10 | 411.9 | 19.87 | 380.7 | 425.4 | 6.88 | 272.0 | |
| 15 | 421.8 | 26.92 | 387.7 | 436.2 | 3.99 | 239.0 | |
| 20 | 424.5 | 34.50 | 394.1 | 440.8 | 4.25 | 201.0 | |
| PN-05 | 5 | 397.7 | 11.04 | 372.8 | 392.5 | 4.60 | 260.0 |
| 10 | 411.3 | 19.61 | 380.7 | 424.6 | 6.74 | 244.0 | |
| 15 | 418.5 | 28.09 | 389.1 | 432.7 | 4.94 | 194.0 | |
| 20 | 423.2 | 34.88 | 393.8 | 438.6 | 5.26 | 175.0 | |
| PN-1 | 5 | 395.8 | 8.92 | 354.9 | 417.2 | 9.43 | 209.0 |
| 10 | 408.9 | 16.44 | 374.6 | 423.8 | 10.07 | 198.0 | |
| 15 | 416.0 | 23.95 | 381.4 | 432.6 | 6.44 | 127.0 | |
| 20 | 422.7 | 29.0 | 390.6 | 437.8 | 7.81 | 151.1 |
| PN-1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Conversion, α | |||||||||
| 0.10 | 0.20 | 0.30 | 0.40 | 0.50 | 0.60 | 0.70 | 0.80 | 0.90 | ||
| FWO | Ea kJ mol−1 | 201.49 ± 24.21 | 194.05 ± 13.24 | 193.49 ± 13.41 | 194.07 ± 14.15 | 191.21 ± 13.28 | 191.12 ± 13.87 | 190.50 ± 13.24 | 190.42 ± 13.02 | 191.81 ± 13.95 |
| A | 5.55 × 1021 | 3.89 × 1018 | 8.58 × 1017 | 3.96 × 1017 | 1.31 × 1017 | 1.02 × 1017 | 6.58 × 1016 | 4.82 × 1016 | 4.78 × 1016 | |
| r2 | 0.8976 | 0.9963 | 0.9987 | 0.9987 | 0.9978 | 0.9971 | 0.9969 | 0.9963 | 0.9962 | |
| KAS | Ea kJ mol−1 | 201.99 ± 25.47 | 193.51 ± 13.95 | 192.63 ± 14.13 | 193.05 ± 14.90 | 189.93 ± 14.00 | 189.74 ± 14.61 | 189.00 ± 13.95 | 188.85 ± 13.72 | 190.19 ± 14.70 |
| A | 2.13 × 1021 | 1.31 × 1018 | 2.74 × 1017 | 1.22 × 1017 | 3.97 × 1016 | 3.02 × 1016 | 1.93 × 1016 | 1.39 × 1016 | 1.35 × 1016 | |
| r2 | 0.8887 | 0.9959 | 0.9985 | 0.9986 | 0.9975 | 0.9968 | 0.9966 | 0.9959 | 0.9957 | |
| Friedman | Ea kJ mol−1 | 137.01 ± 16.97 | 230.51 ± 42.59 | 189.98 ± 30.55 | 191.94 ± 13.90 | 193.47 ± 26.97 | 188.39 ± 18.20 | 199.13 ± 12.04 | 187.37 ± 9.06 | 187.16 ± 18.22 |
| A | 2.97 × 1011 | 4.49 × 1020 | 2.40 × 1015 | 7.7 × 1014 | 4.73 × 1015 | 6.43 × 1014 | 9.2 × 1014 | 7.25 × 1013 | 6.92 × 1013 | |
| r2 | 0.8917 | 0.9324 | 0.9367 | 0.9952 | 0.9948 | 0.9906 | 0.9958 | 0.9934 | 0.9812 | |
| Kissinger | Ea kJ mol−1 | 202.75 ± 11.61 | ||||||||
| A | 2.06 × 1011 | |||||||||
| r2 | 0.9899 | |||||||||
| Ozawa | Ea kJ mol−1 | 214.06 ± 11.60 | ||||||||
| A | 7.02 × 1017 | |||||||||
| r2 | 0.9910 | |||||||||
| PN-05 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Conversion, α | |||||||||
| 0.10 | 0.20 | 0.30 | 0.40 | 0.50 | 0.60 | 0.70 | 0.80 | 0.90 | ||
| FWO | Ea kJ mol−1 | 193.70 ± 2.17 | 193.15 ± 0.71 | 192.16 ± 4.69 | 190.55 ± 5.71 | 190.67 ± 7.48 | 192.12 ± 9.04 | 191.66 ± 8.92 | 192.19 ± 9.68 | 192.66 ± 10.84 |
| A | 1.12 × 1018 | 2.17 × 1017 | 1.12 × 1017 | 5.91 × 1016 | 5.17 × 1016 | 6.11 × 1016 | 4.39 × 1016 | 4.16 × 1016 | 3.86 × 1016 | |
| r2 | 0.9936 | 0.9945 | 0.9956 | 0.9949 | 0.9950 | 0.9951 | 0.9950 | 0.9940 | 0.9945 | |
| KAS | Ea kJ mol−1 | 193.31 ± 2.27 | 192.33 ± 0.76 | 191.07 ± 4.95 | 189.26 ± 6.02 | 189.29 ± 7.88 | 190.74 ± 9.52 | 190.19 ± 9.39 | 190.67 ± 10.20 | 191.08 ± 11.41 |
| A | 3.90 × 1017 | 7.00 × 1016 | 3.50 × 1016 | 1.79 × 1016 | 1.54 × 1016 | 1.79 × 1016 | 1.27 × 1016 | 1.19 × 1016 | 1.09 × 1016 | |
| r2 | 0.9929 | 0.9939 | 0.9951 | 0.9943 | 0.9946 | 0.9945 | 0.9944 | 0.9933 | 0.9939 | |
| Friedman | Ea kJ mol−1 | 185.26 ± 38.41 | 189.83 ± 13.17 | 190.02 ± 10.42 | 185.28 ± 8.85 | 190.16 ± 12.74 | 196.12 ± 36.42 | 192.53 ± 12.99 | 202.90 ± 6.64 | 232.59 ± 47.57 |
| A | 1.03 × 1017 | 6.03 × 1014 | 2.35 × 1014 | 7.73 × 1013 | 2.72 × 1014 | 6.52 × 1016 | 4.31 × 1014 | 8.43 × 1014 | 5.96 × 1019 | |
| r2 | 0.9881 | 0.9879 | 0.9907 | 0.9960 | 0.9955 | 0.9773 | 0.9926 | 0.9799 | 0.9954 | |
| Kissinger | Ea kJ mol−1 | 201.76 ± 4.74 | ||||||||
| A | 7.21 × 1010 | |||||||||
| r2 | 0.9926 | |||||||||
| Ozawa | Ea kJ mol−1 | 213.11 ± 4.73 | ||||||||
| A | 2.48 × 1017 | |||||||||
| r2 | 0.9933 | |||||||||
| PN-01 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Conversion, α | |||||||||
| 0.10 | 0.20 | 0.30 | 0.40 | 0.50 | 0.60 | 0.70 | 0.80 | 0.90 | ||
| FWO | Ea kJ mol−1 | 193.03 ± 30.66 | 214.48 ± 28.95 | 218.03 ± 26.60 | 213.15 ± 23.87 | 211.92 ± 22.19 | 210.63 ± 19.57 | 207.61 ± 17.77 | 204.59 ± 16.14 | 202.68 ± 15.03 |
| A | 1.65 × 1020 | 1.76 × 1021 | 9.10× 1020 | 1.17 × 1020 | 3.99 × 1019 | 1.26 × 1019 | 3.95 × 1018 | 1.02 × 1018 | 4.18 × 1017 | |
| r2 | 0.9935 | 0.9955 | 0.9947 | 0.9935 | 0.9927 | 0.9931 | 0.9930 | 0.9929 | 0.9928 | |
| KAS | Ea kJ mol−1 | 192.49 ± 32.23 | 214.71 ± 30.43 | 218.26 ± 27.97 | 213.00 ± 25.10 | 211.61 ± 23.33 | 210.17 ± 20.58 | 206.92 ± 18.69 | 203.67 ± 16.98 | 201.57 ± 15.81 |
| A | 5.59 × 1019 | 5.59 × 1020 | 2.80 × 1020 | 3.5 × 1019 | 1.18 × 1019 | 3.67 × 1018 | 1.14 × 1018 | 2.88 × 1017 | 1.17 × 1017 | |
| r2 | 0.9928 | 0.9950 | 0.9942 | 0.9928 | 0.9919 | 0.9924 | 0.9923 | 0.9921 | 0.9919 | |
| Friedman | Ea kJ mol−1 | 175.17 ± 50.77 | 238.91 ± 28.01 | 212.82 ± 23.94 | 206.19 ± 3.83 | 208.49 ± 15.94 | 203.43 ± 16.74 | 202.50 ± 16.73 | 192.75 ± 18.60 | 196.44 ± 18.47 |
| A | 5.11 × 1016 | 8.94 × 1019 | 2.85 × 1017 | 2.02 × 1015 | 8.36 × 1015 | 4.01 × 1015 | 2.3 × 1015 | 4.46 × 1014 | 4.77 × 1014 | |
| r2 | 0.9799 | 0.9378 | 0.9845 | 0.9945 | 0.9929 | 0.9981 | 0.9934 | 0.9825 | 0.9828 | |
| Kissinger | Ea kJ mol−1 | 222.41 ± 26.68 | ||||||||
| A | 1.20 × 1014 | |||||||||
| r2 | 0.9853 | |||||||||
| Ozawa | Ea kJ mol−1 | 235.84 ± 24.62 | ||||||||
| A | 4.19 × 1020 | |||||||||
| r2 | 0.9900 | |||||||||
| PN-005 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Conversion, α | |||||||||
| 0.10 | 0.20 | 0.30 | 0.40 | 0.50 | 0.60 | 0.70 | 0.80 | 0.90 | ||
| FWO | Ea kJ mol−1 | 188.80 ± 6.98 | 201.07 ± 2.19 | 202.16 ± 6.51 | 200.30 ± 5.03 | 201.36 ± 7.57 | 200.98 ± 6.81 | 202.08 ± 8.22 | 202.00 ± 8.95 | 203.87 ± 8.91 |
| A | 3.10 × 1017 | 7.37 × 1017 | 7.82 × 1017 | 3.10 × 1017 | 3.70 × 1017 | 2.31 × 1017 | 2.58 × 1017 | 2.18 × 1017 | 2.16 × 1017 | |
| r2 | 0.9917 | 0.9953 | 0.9960 | 0.9954 | 0.9950 | 0.9942 | 0.9940 | 0.9934 | 0.9924 | |
| KAS | Ea kJ mol−1 | 187.94 ± 7.32 | 200.55 ± 2.32 | 201.54 ± 6.85 | 199.47 ± 5.30 | 200.49 ± 7.97 | 200.02 ± 7.17 | 201.11 ± 8.66 | 200.95 ± 9.42 | 202.83 ± 9.38 |
| A | 1.03 × 1017 | 2.32 × 1017 | 2.40 × 1017 | 9.33 × 1016 | 1.09 × 1017 | 6.75 × 1016 | 7.44 × 1016 | 6.21 × 1016 | 6.06 × 1016 | |
| r2 | 0.9908 | 0.9950 | 0.9956 | 0.9950 | 0.9943 | 0.9936 | 0.9934 | 0.9927 | 0.9916 | |
| Friedman | Ea kJ mol−1 | 217.46 ± 6.93 | 206.81 ± 7.16 | 227.40 ± 17.25 | 188.00 ± 15.80 | 200.52 ± 9.37 | 199.87 ± 7.48 | 201.58 ± 9.89 | 208.03 ± 12.31 | 203.34 ± 36.58 |
| A | 3.63 × 1016 | 4.51 × 1015 | 4.42 × 1017 | 2.48 × 1014 | 1.29 × 1015 | 8.41 × 1014 | 9.75 × 1014 | 3.22 × 1015 | 1.7 × 1016 | |
| r2 | 0.9954 | 0.9944 | 0.9893 | 0.9893 | 0.9936 | 0.9910 | 0.9801 | 0.9879 | 0.9816 | |
| Kissinger | Ea kJ mol−1 | 212.25 ± 2.80 | ||||||||
| A | 3.45 × 1011 | |||||||||
| r2 | 0.9899 | |||||||||
| Ozawa | Ea kJ mol−1 | 225.37 ± 0.49 | ||||||||
| A | 1.51 × 1018 | |||||||||
| r2 | 0.9857 | |||||||||
| Ozawa | Kissinger | FWO | KAS | FD | ||
|---|---|---|---|---|---|---|
| PN-1 | Ea kJ mol−1 | 214.06 ± 11.60 | 202.75 ± 11.61 | 193.10 ± 14.80 | 192.10 ± 15.50 | 189.40 ± 21.00 |
| A | 7.02 × 1017 | 2.06 × 1011 | 6.18 × 1020 | 2.36 × 1020 | 4.98 × 1019 | |
| r2 | 0.9910 | 0.9899 | 0.9862 | 0.9849 | 0.9680 | |
| PN-05 | Ea kJ mol−1 | 213.11 ± 4.73 | 201.76 ± 4.74 | 192.10 ± 6.60 | 190.90 ± 6.93 | 196.10 ± 20.8 |
| A | 2.48 × 1017 | 7.21 × 1010 | 1.97 × 1017 | 6.50 × 1016 | 6.64 × 1018 | |
| r2 | 0.9933 | 0.9926 | 0.9947 | 0.9941 | 0.9892 | |
| PN-01 | Ea kJ mol−1 | 235.84 ± 24.62 | 222.41 ± 26.68 | 208.50 ± 22.31 | 208.05 ± 23.46 | 204.08 ± 21.45 |
| A | 4.19 × 1020 | 1.20 × 1014 | 3.35 × 1020 | 1.05 × 1020 | 9.97 × 1018 | |
| r2 | 0.9900 | 0.9853 | 0.9935 | 0.9928 | 0.9829 | |
| PN-005 | Ea kJ mol−1 | 225.40 ± 0.49 | 212.25 ± 2.80 | 200.3 ± 6.78 | 199.40 ± 7.16 | 205.10 ± 13.65 |
| A | 1.51 × 10 18 | 3.45 × 1011 | 3.82 × 1017 | 1.16 × 1017 | 5.63 × 1016 | |
| r2 | 0.9857 | 0.9899 | 0.9942 | 0.9899 | 0.9893 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gola, A.; Knysak, T.; Mucha, I.; Musiał, W. Synthesis, Thermogravimetric Analysis, and Kinetic Study of Poly-N-Isopropylacrylamide with Varied Initiator Content. Polymers 2023, 15, 2427. https://doi.org/10.3390/polym15112427
Gola A, Knysak T, Mucha I, Musiał W. Synthesis, Thermogravimetric Analysis, and Kinetic Study of Poly-N-Isopropylacrylamide with Varied Initiator Content. Polymers. 2023; 15(11):2427. https://doi.org/10.3390/polym15112427
Chicago/Turabian StyleGola, Agnieszka, Tomasz Knysak, Igor Mucha, and Witold Musiał. 2023. "Synthesis, Thermogravimetric Analysis, and Kinetic Study of Poly-N-Isopropylacrylamide with Varied Initiator Content" Polymers 15, no. 11: 2427. https://doi.org/10.3390/polym15112427