
Formulation of Self-Nanoemulsifying Drug Delivery System of Cephalexin: Physiochemical Characterization and Antibacterial Evaluation

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preliminary Screening
2.2.2. Pseudo Ternary Diagram
2.2.3. Optimization
2.2.4. Preparation of CEP-Loaded SNEDDS
2.2.5. Evaluation of CEP SNEDDS
Droplet Characterization
Detection of Self-Nanoemulsification Time
Percentage Transmittance
2.2.6. Evaluation of Optimized Formulation
Thermodynamic Stability
Viscosity and Refractive Index
DSC Analysis
2.2.7. Drug Release Study
2.2.8. Ex-Vivo Permeation Study
2.2.9. Antimicrobial Study
2.2.10. Bioavailability Study
2.2.11. Extraction of CEP from Plasma
2.2.12. Pharmacokinetic Study
2.2.13. Statistical Analysis
3. Results and Discussion
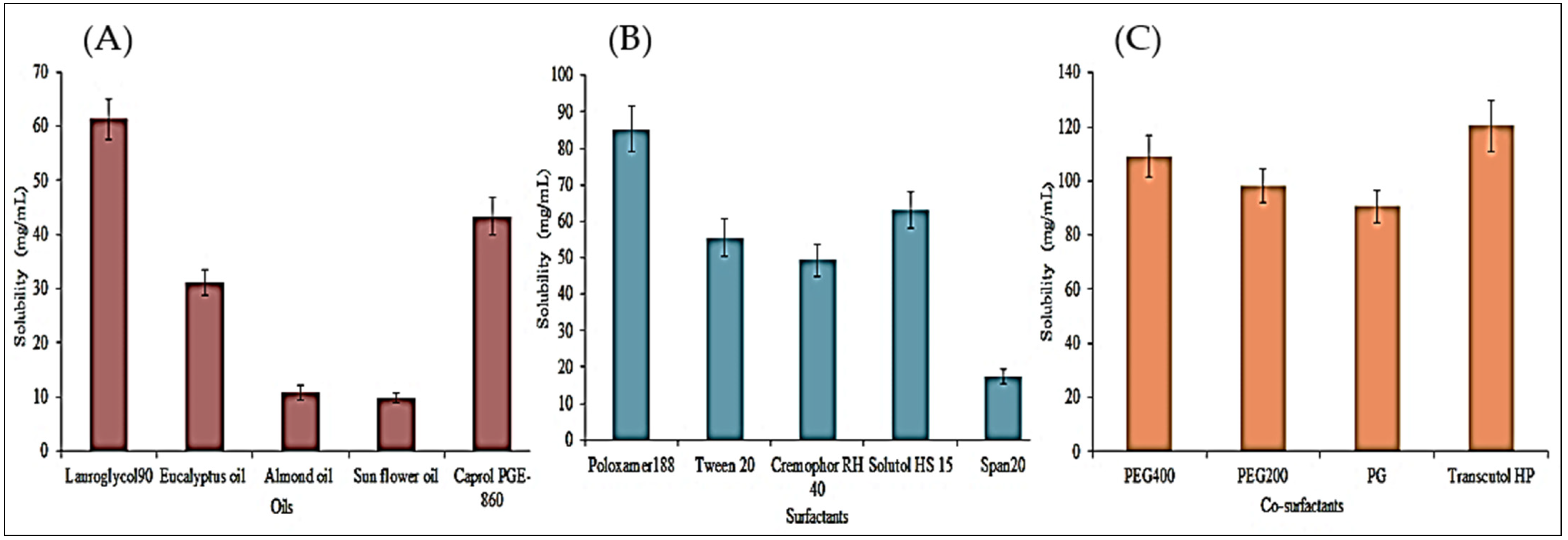
3.1. Preliminary Screening
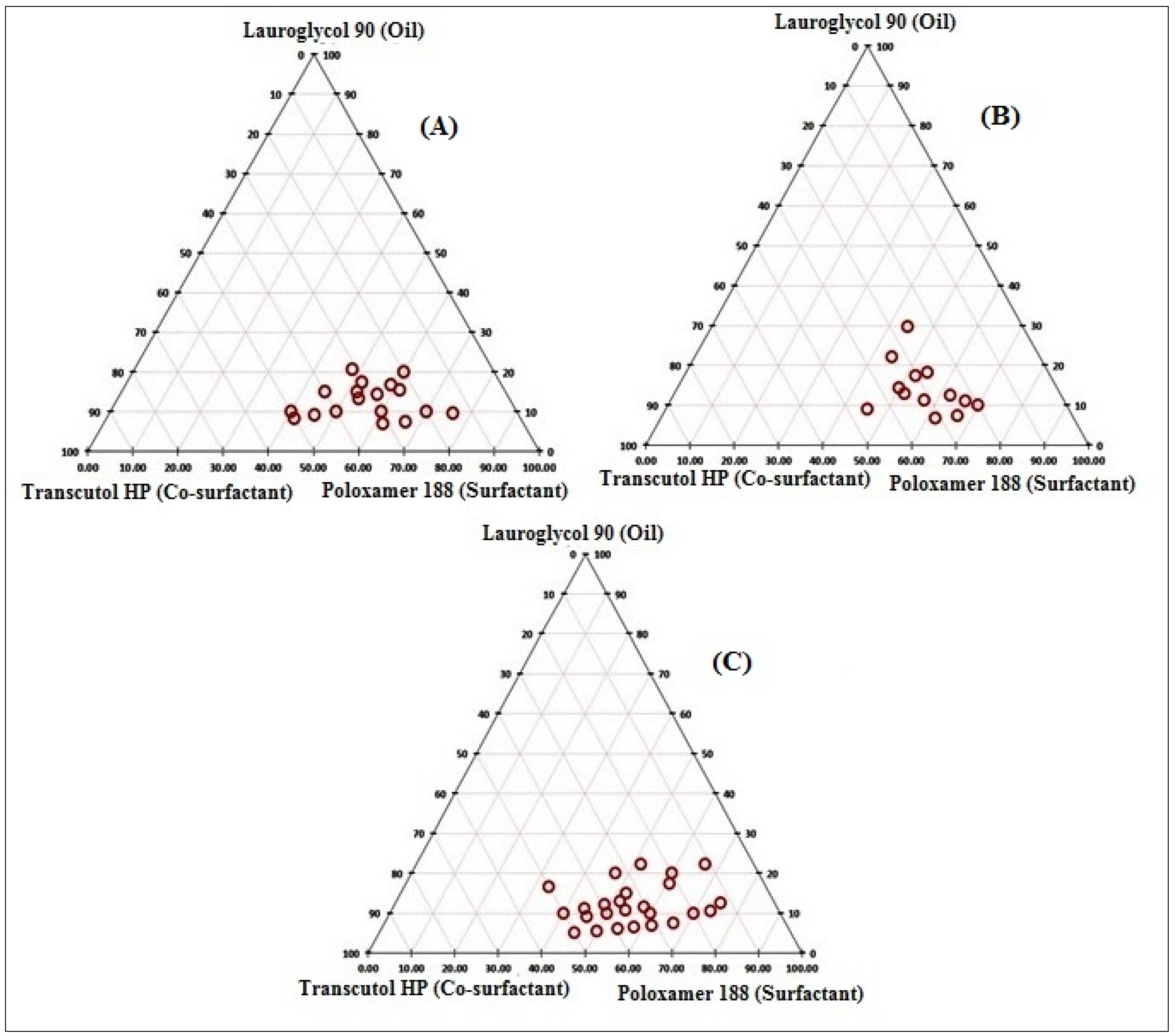
3.2. Preparation of the Pseudo Ternary Diagram
3.3. Optimization
3.4. Effect of Variables on Globule Size (Y1, nm)
3.5. Effect of Variables on Transmittance (Y2, %)
3.6. Effect of Variables on Emulsification Time (Y3, s)
3.7. Point Prediction for Optimization
3.8. Evaluation of CEP SNEDDS
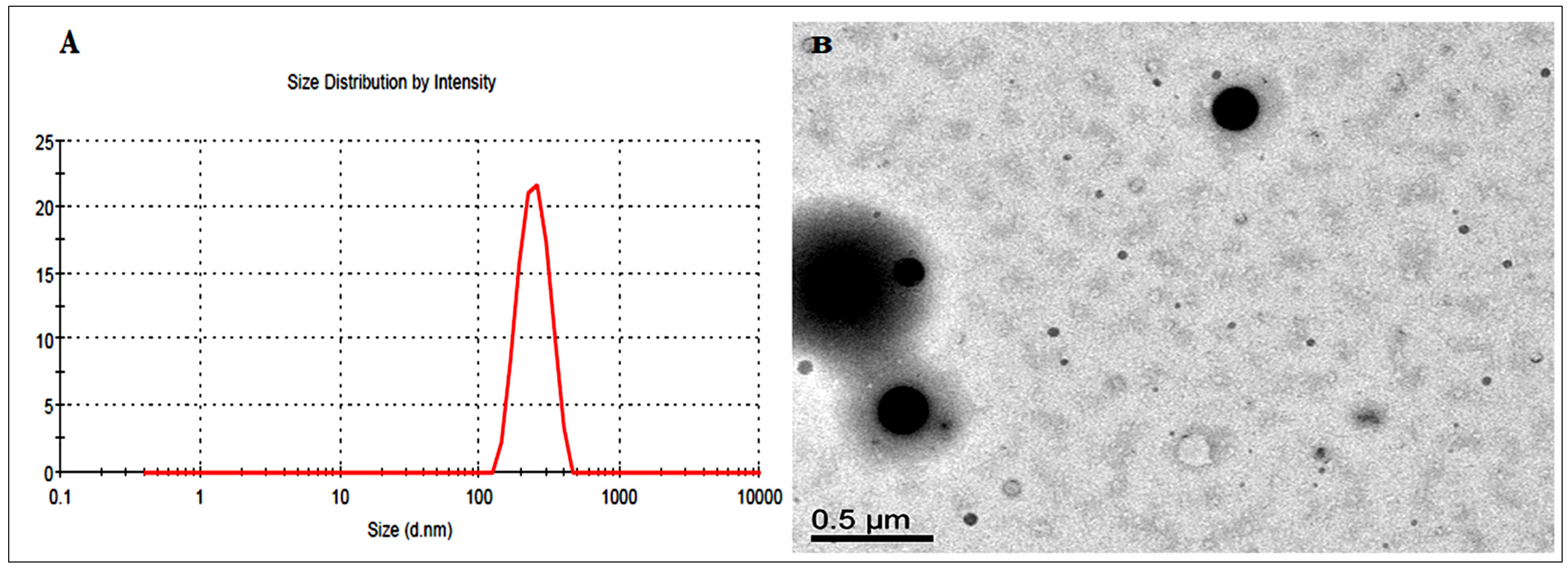
3.8.1. Droplet Characterization
3.8.2. Characterization of Optimized SNEDDS (Opt-F3)
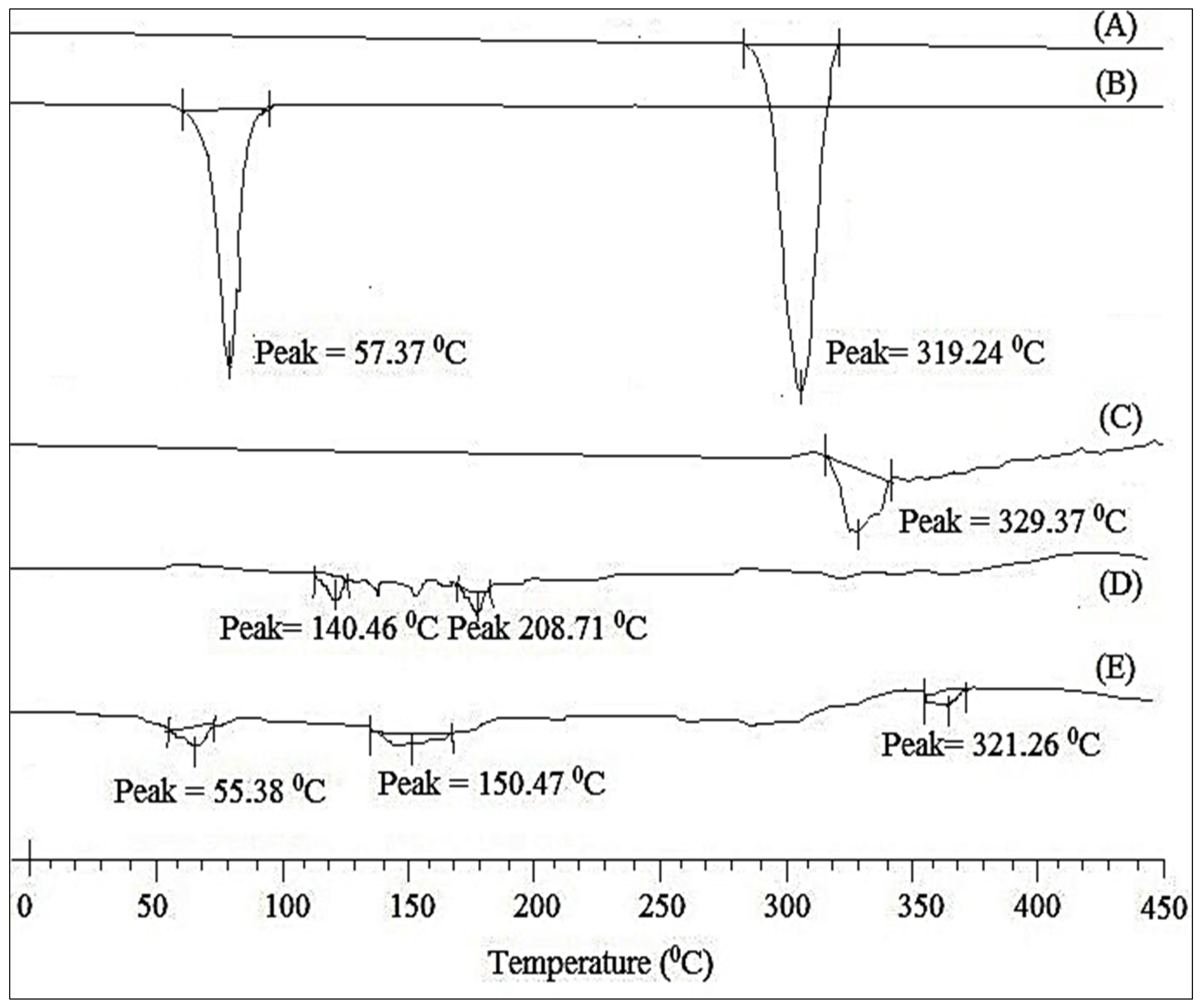
3.8.3. DSC Analysis
3.8.4. In-Vitro Drug Release
3.8.5. Ex-Vivo Permeation Study
3.9. Antimicrobial Study
3.10. Bioavailability Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hezave, A.Z.; Esmaeilzadeh, F. Investigation of the rapid expansion of supercritical solution parameters effects on size and morphology of cephalexin particles. J. Aerosol Sci. 2010, 41, 1090–1102. [Google Scholar] [CrossRef]
- Basaldella, E.I.; Legnoverde, M.S. Functionalized silica matrices for controlled delivery of cephalexin. J. Sol-Gel Sci. Technol. 2010, 56, 191–196. [Google Scholar] [CrossRef]
- Kreil, V.; Ambros, L.; Prados, A.P.; Tarragona, L.; Monfrinotti, A.; Bramuglia, G.; Rebuelto, M. Pharmacokinetics of Immediate and Sustained Release Cephalexin Administered by Different Routes to Llamas (Lama glama). Adv. Pharmacol. Sci. 2016, 2016, 4621039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plöger, G.F.; Quizon, P.M.; Abrahamsson, B.; Cristofoletti, R.; Groot, D.W.; Parr, A.; Langguth, P.; Polli, J.E.; Shah, V.P.; Tajiri, T.; et al. Biowaiver Monographs for Immediate Release Solid Oral Dosage Forms: Cephalexin Monohydrate. J. Pharm. Sci. 2020, 109, 1846–1862. [Google Scholar] [CrossRef] [PubMed]
- Fanun, M.; Papadimitriou, V.; Xenakis, A. Characterization of cephalexin loaded nonionic microemulsions. J. Colloid Interface Sci. 2011, 361, 115–121. [Google Scholar] [CrossRef]
- Yin, L.; Qin, C.; Chen, K.; Zhu, C.; Cao, H.; Zhou, J.; He, W.; Zhang, Q. Gastro-floating tablets of cephalexin: Preparation and in vitro/in vivo evaluation. Int. J. Pharm. 2013, 452, 241–248. [Google Scholar] [CrossRef]
- Jishnu, V.; Prabhakaran, R.; Gilhotra, R. Formulation and Evaluation of Cephalexin Extended Release Matrix Tablets Using 32 Factorial Design. J. Young-Pharm. 2011, 3, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.A.; Jawalkar, S.S.; Aminabhavi, T.M. Controlled release of cephalexin through gellan gum beads: Effect of formulation parameters on entrapment efficiency, size, and drug release. Eur. J. Pharm. Biopharm. 2006, 63, 249–261. [Google Scholar] [CrossRef]
- Rahim, S.; Bhayo, A.M.; Shah, M.R.; Malik, M.I. Star-shaped poly(ethylene oxide)-block-poly(caprolactone) conjugated silver nanoparticles: A colorimetric probe for cephalexin in environmental, biological and pharmaceutical samples. Microchem. J. 2019, 149, 104048. [Google Scholar] [CrossRef]
- Krstić, M.; Medarević, Đ.; Đuriš, J.; Ibrić, S. Self-nanoemulsifying drug delivery systems (SNEDDS) and self-microemulsifying drug delivery systems (SMEDDS) as lipid nanocarriers for improving dissolution rate and bioavailability of poorly soluble drugs. In Lipid Nanocarriers for Drug Targeting; Grumzescu, A.M., Ed.; William Andrew: Oxford, UK, 2018; pp. 473–508. [Google Scholar]
- Zupančič, O.; Grieβinger, J.A.; Rohrer, J.; de Sousa, I.P.; Danninger, L.; Partenhauser, A.; Sündermann, N.E.; Laffleur, F.; Bernkop-Schnürch, A. Development, in vitro and in vivo evaluation of a self-emulsifying drug delivery system (SEDDS) for oral enoxaparin administration. Eur. J. Pharm. Biopharm. 2016, 109, 113–121. [Google Scholar] [CrossRef]
- Kanwal, T.; Saifullah, S.; Rehman, J.U.; Kawish, M.; Razzak, A.; Maharjan, R.; Imran, M.; Ali, I.; Roome, T.; Simjee, S.U.; et al. Design of absorption enhancer containing self-nanoemulsifying drug delivery system (SNEDDS) for curcumin improved anti-cancer activity and oral bioavailability. J. Mol. Liq. 2021, 324, 114774. [Google Scholar] [CrossRef]
- Kanwal, T.; Kawish, M.; Maharjan, R.; Ghaffar, I.; Ali, H.S.; Imran, M.; Perveen, S.; Saifullah, S.; Simjee, S.U.; Shah, M.R. Design and development of permeation enhancer containing self-nanoemulsifying drug delivery system (SNEDDS) for ceftriaxone sodium improved oral pharmacokinetics. J. Mol. Liq. 2019, 289, 111098. [Google Scholar] [CrossRef]
- Agrawal, A.G.; Kumar, A.; Gide, P.S. Self emulsifying drug delivery system for enhanced solubility and dissolution of glipizide. Colloids Surf. B Biointerfaces 2015, 126, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Anwer, K.; Iqbal, M.; Aldawsari, M.F.; Alalaiwe, A.; Ahmed, M.M.; Muharram, M.M.; Ezzeldin, E.; Mahmoud, M.A.; Imam, F.; Ali, R. Improved antimicrobial activity and oral bioavailability of delafloxacin by self-nanoemulsifying drug delivery system (SNEDDS). J. Drug Deliv. Sci. Technol. 2021, 64, 102572. [Google Scholar] [CrossRef]
- Kamble, R.N.; Mehta, P.; Kumar, A. Efavirenz Self-Nano-Emulsifying Drug Delivery System: In Vitro and In Vivo Evaluation. AAPS PharmSciTech 2015, 17, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, N.; Mohammed, H.; Khan, R.A.; Yusuf, M.; Singh, V.; Mohammad, H.A.; Al-Omar, M.S.; Abdellatif, A.A.; Naz, M.; Khadri, H. Self-Generating nano-emulsification techniques for alternatively-routed, bioavailability enhanced delivery, especially for anti-cancers, anti-diabetics, and miscellaneous drugs of natural, and synthetic origins. J. Drug Deliv. Sci. Technol. 2020, 58, 101808. [Google Scholar] [CrossRef]
- Kumar, R.; Khursheed, R.; Kumar, R.; Awasthi, A.; Sharma, N.; Gowthamarajan, K.; Wadhwani, A. Self-nanoemulsifying drug delivery system of fisetin: Formulation, optimization, characterization and cytotoxicity assessment. J. Drug Deliv. Sci. Technol. 2019, 54, 101252. [Google Scholar] [CrossRef]
- Bhaskara Rao, B.V.; Mukherji, R.; Shitre, G.; Alam, F.; Prabhune, A.; Kale, S. Controlled release of antimicrobial Cephalexin drug from silica microparticles. Mater. Sci. Eng. C 2014, 34, 9–14. [Google Scholar] [CrossRef]
- Sahadevan, J.; Prabhakaran, R.; Vijay, J.; Gilhotra, R.M. Formulation and Evaluation of Cephalexin Extended-release Matrix Tablets Using Hydroxy Propyl Methyl Cellulose as Rate-controlling Polymer. J. Young-Pharm. 2012, 4, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Gao, C.; Tian, X.; Chao, B.; Wang, F.; Zhang, Y.; Zou, J.; Liu, D. Pharmacokinetics, tissue distribution and excretion of luteolin and its major metabolites in rats: Metabolites predominate in blood, tissues and are mainly excreted via bile. J. Funct. Foods 2017, 35, 332–340. [Google Scholar] [CrossRef]
- Zafar, A.; Imam, S.S.; Alruwaili, N.K.; Alsaidan, O.A.; Elkomy, M.H.; Ghoneim, M.M.; Alshehri, S.; Mahmoud, A.; Ali, A.; Alharbi, K.S.; et al. Development of Piperine-Loaded Solid Self-Nanoemulsifying Drug Delivery System: Optimization, In-Vitro, Ex-Vivo, and In-Vivo Evaluation. Nanomaterials 2021, 11, 2920. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.M.; Ibrahim, H.M.; Samy, A.M. Development and optimisation of atorvastatin calcium loaded self-nanoemulsifying drug delivery system (SNEDDS) for enhancing oral bioavailability: In vitro and in vivo evaluation. J. Microencapsul. 2017, 34, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Sara, U.V.S. Solid lipid nanoparticles for nose to brain delivery of haloperidol: In vitro drug release and pharmacokinetics evaluation. Acta Pharm. Sin. B 2014, 4, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-Y.; Kang, J.-H.; Ngo, T.L.; Tran, P.; Lee, Y.-B. Preparation and Evaluation of Solid-Self-Emulsifying Drug Delivery System Containing Paclitaxel for Lymphatic Delivery. J. Nanomater. 2016, 2016, 3642418. [Google Scholar] [CrossRef]
- Shanmugam, S.; Baskaran, R.; Balakrishnan, P.; Thapa, P.; Yong, C.S.; Yoo, B.K. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid lutein. Eur. J. Pharm. Biopharm. 2011, 79, 250–257. [Google Scholar] [CrossRef]
- Wu, B.; Deng, D.; Lu, Y.; Wu, W. Biphasic release of indomethacin from HPMC/pectin/calcium matrix tablet: II. Influencing variables, stability and pharmacokinetics in dogs. Eur. J. Pharm. Biopharm. 2008, 69, 294–302. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Beg, S.; Swain, S.; Singh, H.P.; Patra, C.N.; Rao, M.B. Development, Optimization, and Characterization of Solid Self-Nanoemulsifying Drug Delivery Systems of Valsartan Using Porous Carriers. AAPS PharmSciTech 2012, 13, 1416–1427. [Google Scholar] [CrossRef] [Green Version]
- Yasir, M.; Chauhan, I.; Zafar, A.; Verma, M.; Noorulla, K.; Tura, A.J.; Alruwaili, N.K.; Haji, M.J.; Puri, D.; Gobena, W.G.; et al. Buspirone loaded solid lipid nanoparticles for amplification of nose to brain efficacy: Formulation development, optimization by Box-Behnken design, in-vitro characterization and in-vivo biological evaluation. J. Drug Deliv. Sci. Technol. 2020, 61, 102164. [Google Scholar] [CrossRef]
- Rai, A.; Prabhune, A.; Perry, C.C. Antibiotic mediated synthesis of gold nanoparticles with potent antimicrobial activity and their application in antimicrobial coatings. J. Mater. Chem. 2010, 20, 6789–6798. [Google Scholar] [CrossRef] [Green Version]
- Shazly, G.A. Corrigendum to “Ciprofloxacin Controlled-Solid Lipid Nanoparticles: Characterization, In Vitro Release, and Antibacterial Activity Assessment”. BioMed Res. Int. 2017, 2017, 6761452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Cho, J.H.; Park, J.H.; Kim, J.S.; Song, E.S.; Kwon, J.; Giri, B.R.; Jin, S.G.; Kim, K.S.; Choi, H.-G. Self-microemulsifying drug delivery system (SMEDDS) for improved oral delivery and photostability of methotrexate. Int. J. Nanomed. 2019, 14, 4949–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anton, N.; Vandamme, T.F. The universality of low-energy nano-emulsification. Int. J. Pharm. 2009, 377, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Dhingani, A.; Tilala, J.; Raval, M.; Sheth, N. Formulation and development of self-nanoemulsifying granules of olmesartan medoxomil for bioavailability enhancement. Part. Sci. Technol. 2014, 32, 274–290. [Google Scholar] [CrossRef]
- Ahmad, N.; Ahmad, R.; Al-Qudaihi, A.; Alaseel, S.E.; Fita, I.Z.; Khalid, M.S.; Pottoo, F.H.; Bolla, S.R. A novel self-nanoemulsifying drug delivery system for curcumin used in the treatment of wound healing and inflammation. 3 Biotech 2019, 9, 360. [Google Scholar] [CrossRef]
- Ige, P.P.; Baria, R.K.; Gattani, S.G. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf. B Biointerfaces 2013, 108, 366–373. [Google Scholar] [CrossRef]
- Cui, H.; Zhou, H.; Lin, L. The specific antibacterial effect of the Salvia oil nanoliposomes against Staphylococcus aureus biofilms on milk container. Food Control 2016, 61, 92–98. [Google Scholar] [CrossRef]
- Fisher, J.F.; Meroueh, S.O.; Mobashery, S. Bacterial resistance to beta-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Levels | ||
|---|---|---|---|
| Low (−) | Medium (0) | High (+) | |
| A = Oil (%, w/w) | 5 | 16.5 | 28 |
| B = Surfactant (%, w/w) | 32 | 55 | 78 |
| C = Co-surfactant (%, w/w) | 8 | 30 | 52 |
| Responses | Goals | ||
| Y1 = Globule size (nm) | |||
| Y2 = Transmittance (%) | |||
| Y3 = Emulsification time (s) | |||
| S.No | Oil Conc. (%) | Surfactant Conc. (%) | Co-Surfactant Conc. (%) | Globule Size (nm) | Transmittance (%) | Emulsification Time (s) |
|---|---|---|---|---|---|---|
| F1 | 5.0 | 32 | 30 | 67.53 | 94.78 | 61 |
| F2 | 28.0 | 32 | 30 | 205.84 | 83.73 | 146 |
| F3 | 5.0 | 78 | 30 | 36.02 | 96.75 | 48 |
| F4 | 28.0 | 78 | 30 | 123.41 | 94.65 | 83 |
| F5 | 5.0 | 55 | 8 | 71.04 | 91.8 | 64 |
| F6 | 28.0 | 55 | 8 | 184.28 | 87.6 | 137 |
| F7 | 5.0 | 55 | 52 | 30.66 | 99.39 | 35 |
| F8 | 28.0 | 55 | 52 | 146.04 | 89.77 | 77 |
| F 9 | 16.5 | 32 | 8 | 176.26 | 87.04 | 108 |
| F10 | 16.5 | 78 | 8 | 100.73 | 94.73 | 44 |
| F11 | 16.5 | 32 | 52 | 120.93 | 92.62 | 37 |
| F12 | 16.5 | 78 | 52 | 71.15 | 97.06 | 25 |
| F13 * | 16.5 | 55 | 30 | 114.71 | 96.06 | 64 |
| F14 * | 16.5 | 55 | 30 | 107.04 | 96.48 | 63 |
| F15 * | 16.5 | 55 | 30 | 109.74 | 96.26 | 65 |
| F16 * | 16.5 | 55 | 30 | 106.26 | 96.26 | 65 |
| F17 * | 16.5 | 55 | 30 | 107.21 | 96.39 | 64 |
| Terms | Globule Size (Y1, nm) | Transmittance (Y2, %) | Emulsification Time (Y3, s) |
|---|---|---|---|
| R2 | 0.9981 | 0.9995 | 0.9997 |
| Adjusted R2 | 0.9957 | 0.9988 | 0.9992 |
| Prediced R2 | 0.9885 | 0.9966 | 0.9968 |
| Model F-Value | 415.79 | 1475.42 | 2276.38 |
| Model p-value | <0.0001 | <0.0001 | <0.0001 |
| Lack of fit F-value | 0.62 | 0.67 | 1.55 |
| Lack of fit p-value | 0.6382 * | 0.6121 * | 0.3329 * |
| Adequate Precision | 73.204 | 130.229 | 168.648 |
| S.No | Composition O: Sur: Co-Sur | Actual value | Predicted Value | ||||
|---|---|---|---|---|---|---|---|
| Y1 (nm) | Y2 (%) | Y3 (s) | Y1 (nm) | Y2 (%) | Y3 (s) | ||
| Opt-F1 | 16.5:55.0:30 | 114.71 ± 3.27 | 96.06 ± 1.25 | 64 ± 2 | 108.99 | 96.29 | 64.20 |
| Opt-F2 | 14.0:58.0:30 | 97.27 ± 2.35 | 96.38 ± 1.93 | 57.26 ± 2 | 91.33 | 97.28 | 56.19 |
| Opt-F3 | 14.0:59.0:32 | 87.25 ± 3.16 | 99.13 ± 0.5 | 51.82 ± 1 | 84.39 | 98.84 | 49.44 |
| Pharmacokinetic Parameters | CEP-SNEDDS-opt (Opt-F3) | CEP Dispersion |
|---|---|---|
| Cmax (ng/mL) | 7.69 ± 0.72 | 4.69 ± 0.57 |
| Tmax (h) | 4 | 2 |
| AUC0–24 (µ·h/mL) | 71.37 ± 4.28 | 20.50 ± 3.76 |
| AUC0–∞ (µ·h/mL) | 77.31 ± 4.87 | 20.73 ± 3.76 |
| AUMC0–24 (µg·h2/mL) | 534.50 | 86.90 |
| AUMC0–∞ (µg·h2/mL) | 727.70 | 93.33 |
| Half life (t1/2 h) | 5.91 ± 0.15 | 1.24 ± 0.12 |
| Elimination rate constant (h−1) | 0.12± 0.05 | 0.21 ± 0.03 |
| MRT (h) | 9.41 ± 0.45 | 4.50 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafar, A.; Yasir, M.; Alruwaili, N.K.; Imam, S.S.; Alsaidan, O.A.; Alshehri, S.; Ghoneim, M.M.; Alquraini, A.; Rawaf, A.; Ansari, M.J.; et al. Formulation of Self-Nanoemulsifying Drug Delivery System of Cephalexin: Physiochemical Characterization and Antibacterial Evaluation. Polymers 2022, 14, 1055. https://doi.org/10.3390/polym14051055
Zafar A, Yasir M, Alruwaili NK, Imam SS, Alsaidan OA, Alshehri S, Ghoneim MM, Alquraini A, Rawaf A, Ansari MJ, et al. Formulation of Self-Nanoemulsifying Drug Delivery System of Cephalexin: Physiochemical Characterization and Antibacterial Evaluation. Polymers. 2022; 14(5):1055. https://doi.org/10.3390/polym14051055
Chicago/Turabian StyleZafar, Ameeduzzafar, Mohd Yasir, Nabil K. Alruwaili, Syed Sarim Imam, Omar Awad Alsaidan, Sultan Alshehri, Mohammed M. Ghoneim, Ali Alquraini, Alenazy Rawaf, Mohammad Javed Ansari, and et al. 2022. "Formulation of Self-Nanoemulsifying Drug Delivery System of Cephalexin: Physiochemical Characterization and Antibacterial Evaluation" Polymers 14, no. 5: 1055. https://doi.org/10.3390/polym14051055