
In Situ Chemical Modification of Thermoplastic Starch with Poly(L-lactide) and Poly(butylene succinate) for an Effectively Miscible Ternary Blend


Abstract
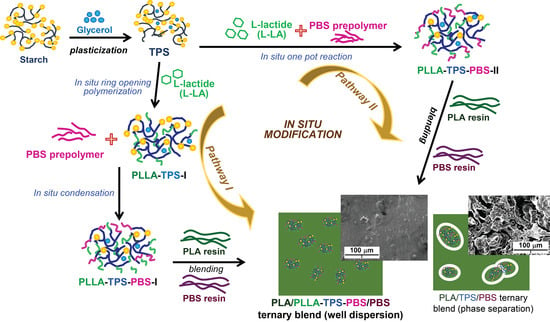
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PLLA-TPS-PBS Copolymers
2.3. Preparation of PLA/PLLA-TPS-PBS/PBS Ternary Blend
2.4. Characterization
2.4.1. Structural Analyses
2.4.2. Contact Angle Measurement and Morphological Observation
2.4.3. Thermal Properties
2.4.4. Dynamic Mechanical Analysis
3. Results and Discussion
3.1. Structural Analyses of the PLLA-TPS and PLLA-TPS-PBS Copolymers
3.2. Cross-Sectional Morphologies
3.2.1. PLLA-TPS-PBS Copolymers
3.2.2. PLA/PLLA-TPS-PBS/PBS Ternary Blends
3.3. Thermal Properties of the PLA/PLLA-TPS-PBS/PBS Blends
3.4. Dynamic Mechanical Analysis of of the PLA/PLLA-TPS-PBS/PBS Blends
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Philp, J.C.; Bartsev, A.; Ritchie, R.J.; Baucher, M.-A.; Guy, K. Bioplastics science from a policy vantage point. N. Biotechnol. 2013, 30, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Garlotta, D.A. Literature review of poly(lactic acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Sin, L.T.; Rahmat, A.R.; Rahman, W.A.W.A. Applications of Poly(lactic Acid). In Handbook of Biopolymers and Biodegradable Plastics; Ebnesajjad, S., Ed.; William Andrew Publishing: Boston, MA, USA, 2013; pp. 55–69. [Google Scholar]
- Kanemura, C.; Nakashima, S.; Hotta, A. Mechanical properties and chemical structures of biodegradable poly(butylene-succinate) for material reprocessing. Polym. Degrad. Stab. 2012, 97, 972–980. [Google Scholar] [CrossRef]
- Xu, J.; Guo, B.-H. Poly(butylene succinate) and its copolymers: Research, development and industrialization. Biotechnol. J. 2010, 5, 1149–1163. [Google Scholar] [CrossRef]
- Teixeira, E.M.; Da Róz, A.L.; Carvalho, A.J.F.; Curvelo, A.A.S. The effect of glycerol/sugar/water and sugar/water mixtures on the plasticization of thermoplastic cassava starch. Carbohydr. Polym. 2007, 69, 619–624. [Google Scholar] [CrossRef]
- Yeh, J.-T.; Hou, Y.-J.; Cheng, L.; Wang, Y.-Z.; Yang, L.; Wang, C.-K. Water proof and strength retention properties of thermoplastic starch based biocomposites modified with glutaraldehyde. Carbohydr. Polym. 2015, 127, 135–144. [Google Scholar] [CrossRef]
- Wang, N.; Yu, J.; Chang, P.R.; Ma, X. Influence of formamide and water on the properties of thermoplastic starch/poly(lactic acid) blends. Carbohydr. Polym. 2008, 71, 109–118. [Google Scholar] [CrossRef]
- Salehiyan, R.; Sinha Ray, S. Processing of polymer blends, Emphasizing: Melt compounding; Influence of nanoparticles on blend morphology and rheology; Reactive processing in ternary systems; Morphology-property relationships; Performance and application challenges; and Opportunities and future trends. In Processing of Polymer-Based Nanocomposites: Processing-Structure-Property-Performance Relationships; Sinha Ray, S., Ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 167–197. [Google Scholar]
- Nofar, M.; Sacligil, D.; Carreau, P.J.; Kamal, M.R.; Heuzey, M.-C. Poly (lactic acid) blends: Processing, properties and applications. Int. J. Biol. Macromol. 2019, 125, 307–360. [Google Scholar] [CrossRef]
- Palai, B.; Biswal, M.; Mohanty, S.; Nayak, S.K. In situ reactive compatibilization of polylactic acid (PLA) and thermoplastic starch (TPS) blends; synthesis and evaluation of extrusion blown films thereof. Ind. Crops. Prod. 2019, 141, 111748. [Google Scholar] [CrossRef]
- Soares, F.C.; Yamashita, F.; Müller, C.M.O.; Pires, A.T.N. Thermoplastic starch/poly(lactic acid) sheets coated with cross-linked chitosan. Polym. Test. 2013, 32, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Noivoil, N.; Yoksan, R. Compatibility improvement of poly(lactic acid)/thermoplastic starch blown films using acetylated starch. J. Appl. Polym. Sci. 2021, 138, 49675. [Google Scholar] [CrossRef]
- Liao, H.-T.; Wu, C.-S. Preparation and characterization of ternary blends composed of polylactide, poly(ɛ-caprolactone) and starch. Mater. Sci. Eng. A. 2009, 515, 207–214. [Google Scholar] [CrossRef]
- Mittal, V.; Akhtar, T.; Matsko, N. Mechanical, Thermal, Rheological and Morphological Properties of Binary and Ternary Blends of PLA, TPS and PCL. Macromol. Mater. Eng. 2015, 300, 423–435. [Google Scholar] [CrossRef]
- Sarazin, P.; Li, G.; Orts, W.J.; Favis, B.D. Binary and ternary blends of polylactide, polycaprolactone and thermoplastic starch. Polymer 2008, 49, 599–609. [Google Scholar] [CrossRef]
- Bulatović, V.O.; Mandić, V.; Kučić Grgić, D.; Ivančić, A. Biodegradable polymer blends based on thermoplastic starch. J. Polym. Environ. 2021, 29, 492–508. [Google Scholar] [CrossRef]
- Koh, J.J.; Zhang, X.; Kong, J.; He, C. Compatibilization of multicomponent composites through a transitioning phase: Interfacial tensions considerations. Compos. Sci. Technol. 2018, 164, 34–43. [Google Scholar] [CrossRef]
- Quiles-Carrillo, L.; Montanes, N.; Pineiro, F.; Jorda-Vilaplana, A.; Torres-Giner, S. Ductility and toughness improvement of injection-molded compostable pieces of polylactide by melt blending with poly(ε-caprolactone) and thermoplastic starch. Materials 2018, 11, 2138. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Z.; Ying, S.; Hongye, F.; Jie, R.; Ren, T. Preparation, characterization and properties of binary and ternary blends with thermoplastic starch, poly(lactic acid) and poly(butylene succinate). Polym. Renew. Resour. 2011, 2, 49–62. [Google Scholar] [CrossRef]
- Ren, J.; Fu, H.; Ren, T.; Yuan, W. Preparation, characterization and properties of binary and ternary blends with thermoplastic starch, poly(lactic acid) and poly(butylene adipate-co-terephthalate). Carbohydr. Polym. 2009, 77, 576–582. [Google Scholar] [CrossRef]
- Phattarateera, S.; Junsook, N.; Kumsang, P.; Aontee, A.; Kerddonfag, N. The ternary blends of TPS/PBAT/PLA films: A study on the morphological and mechanical properties. Key Eng. Mater. 2020, 861, 170–175. [Google Scholar] [CrossRef]
- Shirai, M.A.; Olivato, J.B.; Garcia, P.S.; Müller, C.M.O.; Grossmann, M.V.E.; Yamashita, F. Thermoplastic starch/polyester films: Effects of extrusion process and poly (lactic acid) addition. Mater. Sci. Eng. C 2013, 33, 4112–4117. [Google Scholar] [CrossRef] [PubMed]
- Carmona, V.B.; Corrêa, A.C.; Marconcini, J.M.; Mattoso, L.H.C. Properties of a biodegradable ternary blend of thermoplastic starch (TPS), poly(ε-caprolactone) (PCL) and poly(lactic acid) (PLA). J. Polym. Environ. 2015, 23, 83–89. [Google Scholar] [CrossRef]
- Lipsa, R.; Tudorachi, N.; Vasile, C.; Chiriac, A.; Grigoras, A. Novel environmentally friendly copolymers carboxymethyl starch grafted poly(lactic acid). J. Polym. Environ. 2013, 21, 461–471. [Google Scholar] [CrossRef]
- Wootthikanokkhan, J.; Kasemwananimit, P.; Sombatsompop, N.; Kositchaiyong, A.; Isarankura na Ayutthaya, S.; Kaabbuathong, N. Preparation of modified starch-grafted poly(lactic acid) and a study on compatibilizing efficacy of the copolymers in poly(lactic acid)/thermoplastic starch blends. J. Appl. Polym. Sci. 2012, 126, E389–E396. [Google Scholar] [CrossRef]
- Fischer, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Kolloid-Z.u.Z. Polymere. 1973, 251, 980–990. [Google Scholar] [CrossRef]
- Gigli, M.; Negroni, A.; Zanaroli, G.; Lotti, N.; Fava, F.; Munari, A. Environmentally friendly PBS-based copolyesters containing PEG-like subunit: Effect of block length on solid-state properties and enzymatic degradation. React. Funct. Polym. 2013, 73, 764–771. [Google Scholar] [CrossRef]
- Teixeira, E.d.M.; Curvelo, A.A.S.; Corrêa, A.C.; Marconcini, J.M.; Glenn, G.M.; Mattoso, L.H.C. Properties of thermoplastic starch from cassava bagasse and cassava starch and their blends with poly (lactic acid). Ind. Crops. Prod. 2012, 37, 61–68. [Google Scholar] [CrossRef]
- Biliaderis, C.G. Structural Transitions and Related Physical Properties of Starch In Starch, 3rd ed.; BeMiller, J., Whistler, R., Eds.; Academic Press: Cambridge, MA, USA, 2009; pp. 293–372. [Google Scholar]
- Chen, L.; Qiu, X.; Deng, M.; Hong, Z.; Luo, R.; Chen, X.; Jing, X. The starch grafted poly(l-lactide) and the physical properties of its blending composites. Polymer 2005, 46, 5723–5729. [Google Scholar] [CrossRef]
- Zeng, J.-B.; Li, K.-A.; Du, A.-K. Compatibilization strategies in poly(lactic acid)-based blends. RSC Adv. 2015, 5, 32546–32565. [Google Scholar] [CrossRef]
- Choi, E.-J.; Park, J.-K.; Chang, H.-N. Effect of polymerization catalysts on the microstructure of P(LLA-co-εCL). J. Polym. Sci. B Polym. Phys. 1994, 32, 2481–2489. [Google Scholar] [CrossRef]
- Canché-Escamilla, G.; Canché-Canché, M.; Duarte-Aranda, S.; Cáceres-Farfán, M.; Borges-Argáez, R. Mechanical properties and biodegradation of thermoplastic starches obtained from grafted starches with acrylics. Carbohydr. Polym. 2011, 86, 1501–1508. [Google Scholar] [CrossRef]
- Rutot, D.; Degée, P.; Narayan, R.; Dubois, P. Aliphatic polyester-grafted starch composites by in situ ring opening polymerization. Compos. Interfaces 2000, 7, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Carballo, Z.B.; Duarte-Aranda, S.; Canché-Escamilla, G. Properties and biodegradation of thermoplastic starch obtained from grafted starches with poly(lactic acid). J. Polym. Environ. 2019, 27, 2607–2617. [Google Scholar] [CrossRef]
- Noivoil, N.; Yoksan, R. Oligo(lactic acid)-grafted starch: A compatibilizer for poly(lactic acid)/thermoplastic starch blend. Int. J. Biol. Macromol. 2020, 160, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yuan, L.; Laredo, E.; Zhang, M.; Zhou, W. Interfacial properties, viscoelasticity, and thermal behaviors of poly(butylene succinate)/polylactide blend. Ind. Eng. Chem. Res. 2012, 51, 2290–2298. [Google Scholar] [CrossRef]
- Wang, R.; Wang, S.; Zhang, Y.; Wan, C.; Ma, P. Toughening modification of PLLA/PBS blends via in situ compatibilization. Polym. Eng. Sci. 2009, 49, 26–33. [Google Scholar] [CrossRef]
- Deng, Y.; Thomas, N.L. Blending poly(butylene succinate) with poly(lactic acid): Ductility and phase inversion effects. Eur. Polym. J. 2015, 71, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Supthanyakul, R.; Kaabbuathong, N.; Chirachanchai, S. Poly(l-lactide-b-butylene succinate-b-l-lactide) triblock copolymer: A multi-functional additive for PLA/PBS blend with a key performance on film clarity. Polym. Degrad. Stab. 2017, 142, 160–168. [Google Scholar] [CrossRef]
- Supthanyakul, R.; Kaabbuathong, N.; Chirachanchai, S. Random poly(butylene succinate-co-lactic acid) as a multi-functional additive for miscibility, toughness, and clarity of PLA/PBS blends. Polymer 2016, 105, 1–9. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Feeding Molar Ratio | PBS with Different MWs | Sample Code | |
|---|---|---|---|---|
| L-LA | Starch | |||
| I | 0.05 | 1 | PBS1 | PLLA0.05-TPS-PBS1–I |
| I | 0.05 | 1 | PBS2 | PLLA0.05-TPS-PBS2–I |
| I | 0.2 | 1 | PBS1 | PLLA0.2-TPS-PBS1–I |
| I | 0.2 | 1 | PBS2 | PLLA0.2-TPS-PBS2–I |
| II | 0.05 | 1 | PBS1 | PLLA0.05-TPS-PBS1–II |
| II | 0.05 | 1 | PBS2 | PLLA0.05-TPS-PBS2–II |
| II | 0.2 | 1 | PBS1 | PLLA0.2-TPS-PBS1–II |
| II | 0.2 | 1 | PBS2 | PLLA0.2-TPS-PBS2–II |
| Sample | Tg (°C) | Tcc,PLA (°C) | Tm,PBS (°C) | Tm,PLA (°C) | Xc,PLA (%) | Xc,PBS (%) |
|---|---|---|---|---|---|---|
| PLA | 53.9 | 124.5 | - | 152.2, 156.2 | 3.70 | - |
| PBS | - | - | 115.9 | - | - | 66.80 |
| PLA/PBS (50/50) | 54.8 | 104.2 | 114.5 | 145.2, 152.6 | 25.03 | 35.72 |
| PLA/TPS/PBS (35/30/35) | 53.9 | 98.6 | 112.4 | 141.1, 147.5 | 24.66 | 24.47 |
| PLA/PLLA0.05-TPS-PBS1–I/PBS | 50.4 | 93.6 | 111.2 | 139.8, 149.3 | 16.72 | 57.08 |
| PLA/PLLA0.05-TPS-PBS2–I/PBS | 51.9 | 92.5 | 103.9, 111.3 | 138.7, 148.7 | 19.55 | 61.58 |
| PLA/PLLA0.2-TPS-PBS1–I/PBS | 50.4 | 89.7, 94.8 | 104.2, 111.2 | 137.9, 148.8 | 22.60 | 61.65 |
| PLA/PLLA0.2-TPS-PBS2–I/PBS | 48.6 | 89.5, 92.3 | 103.8, 111.3 | 138.7, 148.5 | 20.70 | 63.26 |
| PLA/PLLA0.05-TPS-PBS1–II/PBS | 52.5 | 92.0 | 110.1 | 138.4, 147.7 | 28.94 | 63.40 |
| PLA/PLLA0.05-TPS-PBS2–II/PBS | 53.5 | 96.9 | 113.0 | 142.3, 150.1 | 33.25 | 53.52 |
| PLA/PLLA0.2-TPS-PBS1–II/PBS | 44.2 | 81.8, 89.1 | 109.1 | 135.0, 144.8 | 30.18 | 63.34 |
| PLA/PLLA0.2-TPS-PBS2–II/PBS | 49.7 | 93.0 | 112.0 | 138.4, 149.1 | 23.92 | 62.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jariyasakoolroj, P.; Chirachanchai, S. In Situ Chemical Modification of Thermoplastic Starch with Poly(L-lactide) and Poly(butylene succinate) for an Effectively Miscible Ternary Blend. Polymers 2022, 14, 825. https://doi.org/10.3390/polym14040825
Jariyasakoolroj P, Chirachanchai S. In Situ Chemical Modification of Thermoplastic Starch with Poly(L-lactide) and Poly(butylene succinate) for an Effectively Miscible Ternary Blend. Polymers. 2022; 14(4):825. https://doi.org/10.3390/polym14040825
Chicago/Turabian StyleJariyasakoolroj, Piyawanee, and Suwabun Chirachanchai. 2022. "In Situ Chemical Modification of Thermoplastic Starch with Poly(L-lactide) and Poly(butylene succinate) for an Effectively Miscible Ternary Blend" Polymers 14, no. 4: 825. https://doi.org/10.3390/polym14040825