
Phase Diagrams of Polymerization-Induced Self-Assembly Are Largely Determined by Polymer Recombination



Abstract
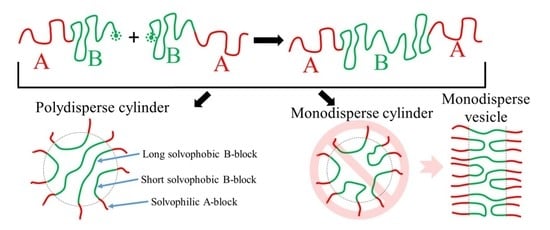
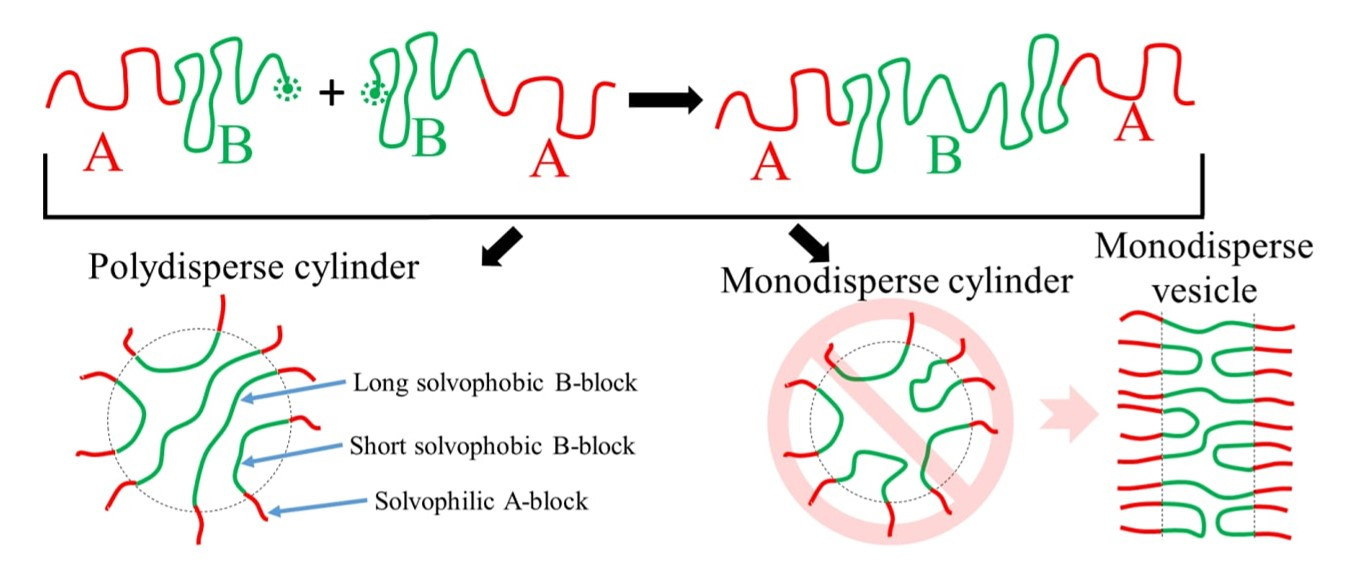
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
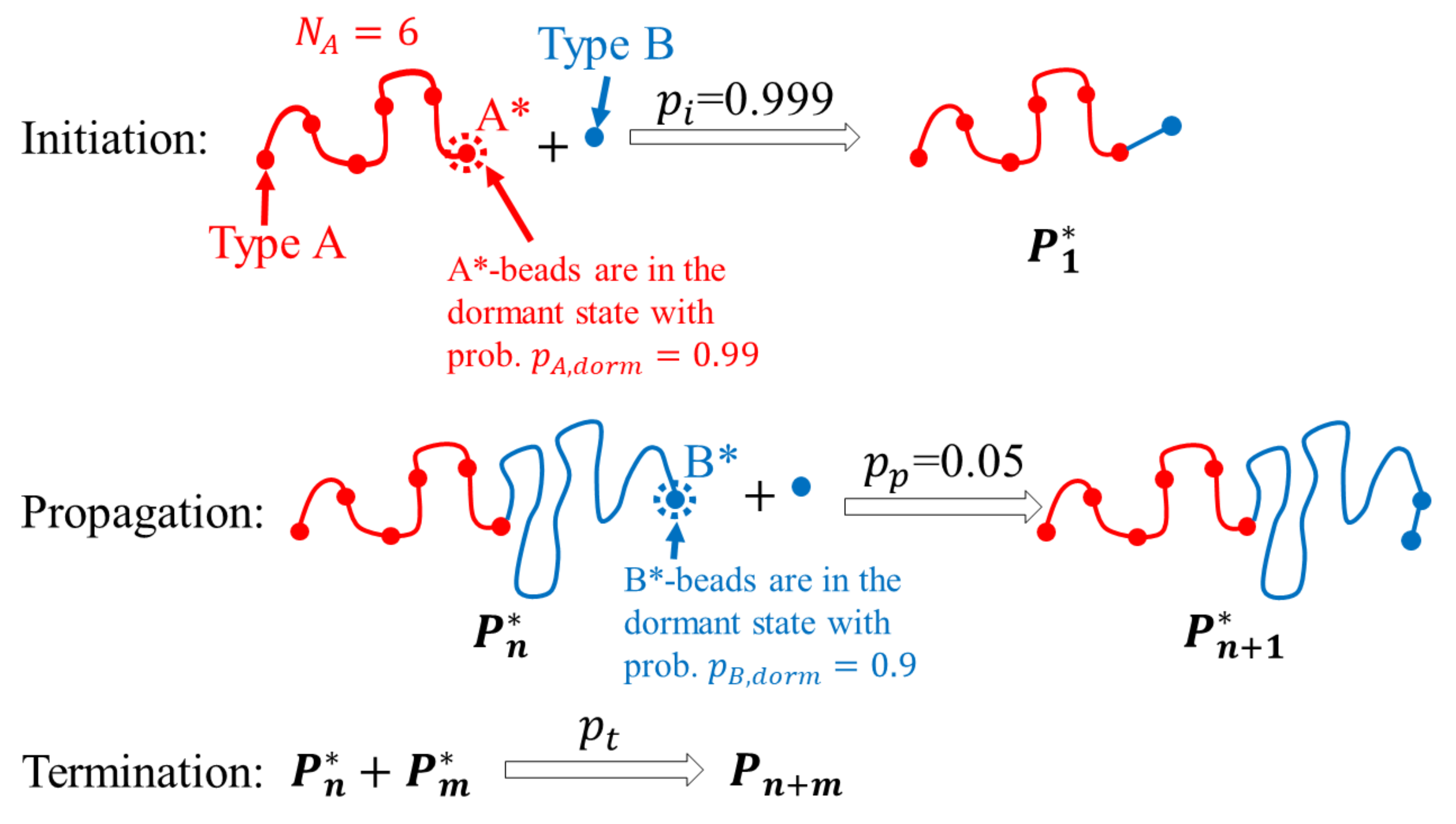
2. Methods
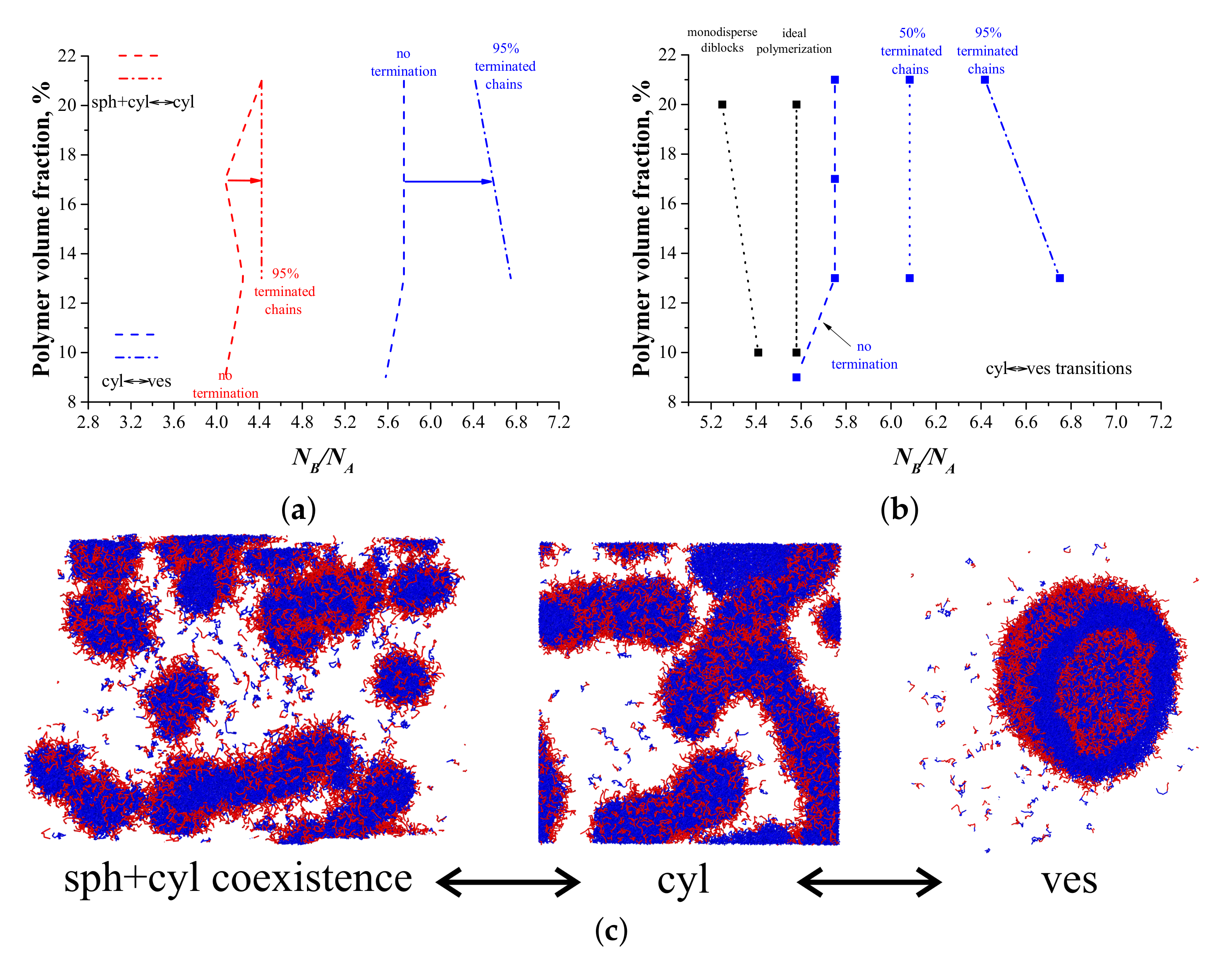
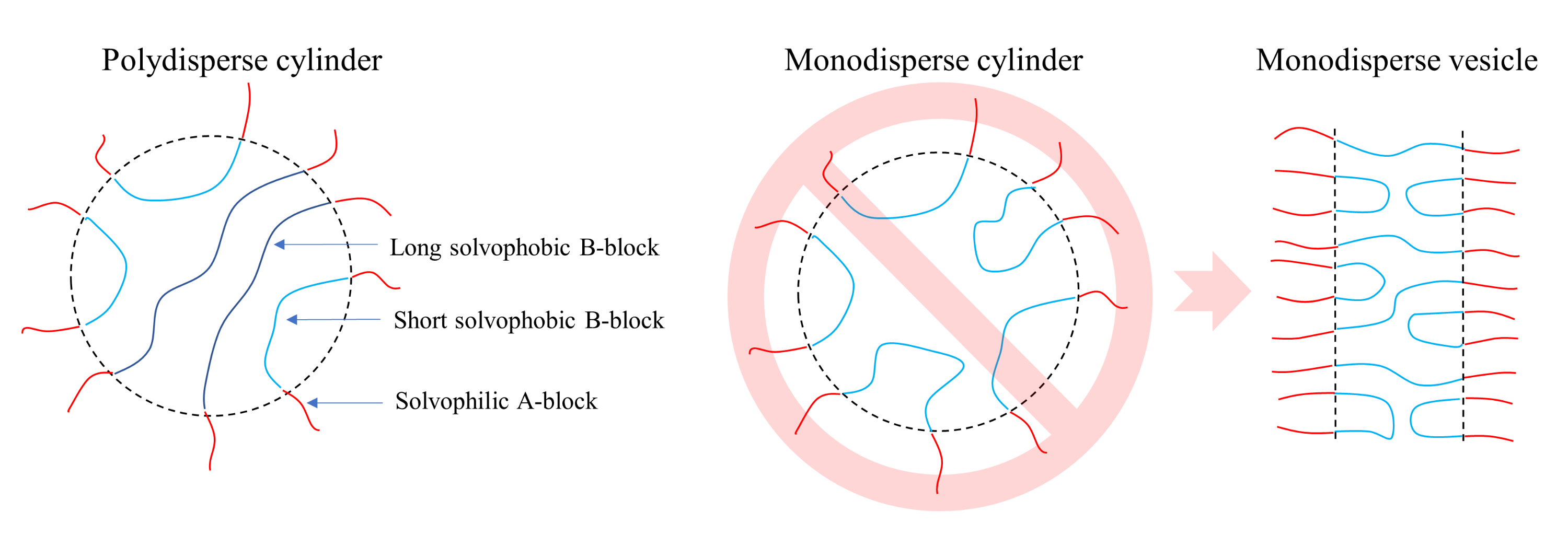
3. Results and Discussion
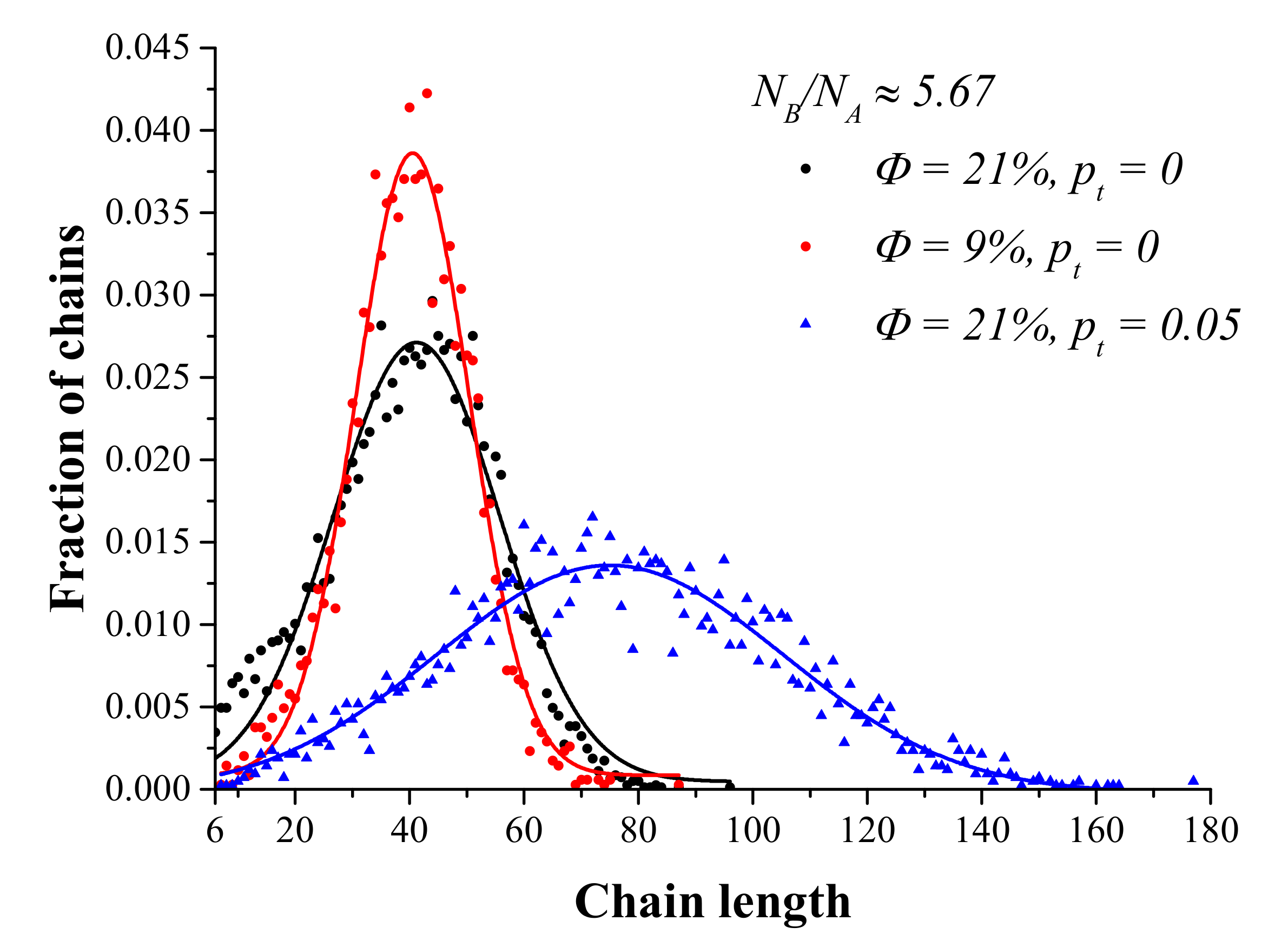
3.1. ATRP without Recombination
3.2. ATRP with Recombination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Tan, Y.; Chen, Y.; Zhang, L.; Tan, J. Expanding the Scope of Polymerization-Induced Self-Assembly: Recent Advances and New Horizons. Macromol. Rapid Commun. 2021, 42, 2100498. [Google Scholar] [CrossRef] [PubMed]
- Blanazs, A.; Madsen, J.; Battaglia, G.; Ryan, A.J.; Armes, S.P. Mechanistic insights for block copolymer morphologies: How do worms form vesicles? J. Am. Chem. Soc. 2011, 133, 16581–16587. [Google Scholar] [CrossRef]
- Tritschler, U.; Pearce, S.; Gwyther, J.; Whittell, G.R.; Manners, I. 50th anniversary perspective: Functional nanoparticles from the solution self-assembly of block copolymers. Macromolecules 2017, 50, 3439–3463. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, S.; Blanazs, A.; Armes, S.P.; Ryan, A.J.; Lewis, A.L. Aqueous dispersion polymerization: A new paradigm for in situ block copolymer self-assembly in concentrated solution. J. Am. Chem. Soc. 2011, 133, 15707–15713. [Google Scholar] [CrossRef]
- Warren, N.J.; Mykhaylyk, O.O.; Mahmood, D.; Ryan, A.J.; Armes, S.P. RAFT aqueous dispersion polymerization yields poly (ethylene glycol)-based diblock copolymer nano-objects with predictable single phase morphologies. J. Am. Chem. Soc. 2014, 136, 1023–1033. [Google Scholar] [CrossRef]
- Wan, W.M.; Pan, C.Y. Atom transfer radical dispersion polymerization in an ethanol/water mixture. Macromolecules 2007, 40, 8897–8905. [Google Scholar] [CrossRef]
- Wang, G.; Schmitt, M.; Wang, Z.; Lee, B.; Pan, X.; Fu, L.; Yan, J.; Li, S.; Xie, G.; Bockstaller, M.R.; et al. Polymerization-induced self-assembly (PISA) using ICAR ATRP at low catalyst concentration. Macromolecules 2016, 49, 8605–8615. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Zhang, W. Synthesis of diblock copolymer nano-assemblies by PISA under dispersion polymerization: Comparison between ATRP and RAFT. Polym. Chem. 2017, 8, 6407–6415. [Google Scholar] [CrossRef]
- Groison, E.; Brusseau, S.; D’Agosto, F.; Magnet, S.; Inoubli, R.; Couvreur, L.; Charleux, B. Well-defined amphiphilic block copolymer nanoobjects via nitroxide-mediated emulsion polymerization. ACS Macro Lett. 2012, 1, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Lansalot, M.; Bourgeat-Lami, E.; Charleux, B. Nitroxide-Mediated Polymerization-Induced Self-Assembly of Poly (poly (ethylene oxide) methyl ether methacrylate-co-styrene)-b-poly (n-butyl methacrylate-co-styrene) Amphiphilic Block Copolymers. Macromolecules 2013, 46, 4285–4295. [Google Scholar] [CrossRef]
- Qiao, X.; Dugas, P.Y.; Charleux, B.; Lansalot, M.; Bourgeat-Lami, E. Nitroxide-mediated polymerization-induced self-assembly of amphiphilic block copolymers with a pH/temperature dual sensitive stabilizer block. Polym. Chem. 2017, 8, 4014–4029. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Chertovich, A.V. Simulation of the RAFT polymerization in 3D: Steric restrictions and incompatibility between species. Polym. Chem. 2022, 13, 2143–2154. [Google Scholar] [CrossRef]
- Cai, D.; Li, J.; Ma, Z.; Gan, Z.; Shao, Y.; Xing, Q.; Tan, R.; Dong, X.H. Effect of Molecular Architecture and Symmetry on Self-Assembly: A Quantitative Revisit Using Discrete ABA Triblock Copolymers. ACS Macro Lett. 2022, 11, 555–561. [Google Scholar] [CrossRef]
- Huo, M.; Xu, Z.; Zeng, M.; Chen, P.; Liu, L.; Yan, L.T.; Wei, Y.; Yuan, J. Controlling vesicular size via topological engineering of amphiphilic polymer in polymerization-induced self-assembly. Macromolecules 2017, 50, 9750–9759. [Google Scholar] [CrossRef]
- Terreau, O.; Luo, L.; Eisenberg, A. Effect of poly (acrylic acid) block length distribution on polystyrene-b-poly (acrylic acid) aggregates in solution. 1. Vesicles. Langmuir 2003, 19, 5601–5607. [Google Scholar] [CrossRef]
- Terreau, O.; Bartels, C.; Eisenberg, A. Effect of poly (acrylic acid) block length distribution on polystyrene-b-poly (acrylic acid) block copolymer aggregates in solution. 2. A partial phase diagram. Langmuir 2004, 20, 637–645. [Google Scholar] [CrossRef]
- Gao, Z.; Eisenberg, A. A model of micellization for block copolymers in solutions. Macromolecules 1993, 26, 7353–7360. [Google Scholar] [CrossRef]
- Linse, P. Micellization of poly (ethylene oxide)-poly (propylene oxide) block copolymers in aqueous solution: Effect of polymer polydispersity. Macromolecules 1994, 27, 6404–6417. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, T.; Ye, F.; Liang, H.; Shi, A.C. Effect of polydispersity on the formation of vesicles from amphiphilic diblock copolymers. Macromolecules 2005, 38, 6710–6717. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Shupanov, R.M.; Chertovich, A.V. Phase Diagram for Ideal Diblock-Copolymer Micelles Compared to Polymerization-Induced Self Assembly. Polymers 2020, 12, 2599. [Google Scholar] [CrossRef]
- Huang, F.; Lv, Y.; Wang, L.; Xu, P.; Lin, J.; Lin, S. An insight into polymerization-induced self-assembly by dissipative particle dynamics simulation. Soft Matter 2016, 12, 6422–6429. [Google Scholar] [CrossRef]
- Yan, Y.D.; Xue, Y.H.; Zhao, H.Y.; Liu, H.; Lu, Z.Y.; Gu, F.L. Insight into the Polymerization-Induced Self-Assembly via a Realistic Computer Simulation Strategy. Macromolecules 2019, 52, 6169–6180. [Google Scholar] [CrossRef]
- Wang, J.; Fang, T.; Li, J.; Yan, Y.; Li, Z.; Zhang, J. Precise Mesoscopic Model Providing Insights into Polymerization-Induced Self-Assembly. Langmuir 2020, 36, 8009–8016. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Wang, L.; Liu, F.; Feng, W.; Wei, J.; Lin, S. Rod–coil block copolymer aggregates via polymerization-induced self-assembly. Soft Matter 2020, 16, 3466–3475. [Google Scholar] [CrossRef] [PubMed]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. Europhys. Lett. (EPL) 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Koelman, J.M.V.A.; Hoogerbrugge, P.J. Dynamic Simulations of Hard-Sphere Suspensions Under Steady Shear. Europhys. Lett. (EPL) 1993, 21, 363–368. [Google Scholar] [CrossRef]
- Schlijper, A.G.; Hoogerbrugge, P.J.; Manke, C.W. Computer simulation of dilute polymer solutions with the dissipative particle dynamics method. J. Rheol. 1995, 39, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Español, P.; Warren, P. Statistical Mechanics of Dissipative Particle Dynamics. Europhys. Lett. (EPL) 1995, 30, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef] [Green Version]
- Blanazs, A.; Ryan, A.; Armes, S. Predictive phase diagrams for RAFT aqueous dispersion polymerization: Effect of block copolymer composition, molecular weight, and copolymer concentration. Macromolecules 2012, 45, 5099–5107. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Chertovich, A.V. Copolymerization of partly incompatible monomers: An insight from computer simulations. Macromolecules 2017, 50, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Bai, Y.; Zhang, X.; Zhang, L. Room temperature synthesis of poly (poly (ethylene glycol) methyl ether methacrylate)-based diblock copolymer nano-objects via Photoinitiated Polymerization-Induced Self-Assembly (Photo-PISA). Polym. Chem. 2016, 7, 2372–2380. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Z.; Lee, B.; Yuan, R.; Lu, Z.; Yan, J.; Pan, X.; Song, Y.; Bockstaller, M.R.; Matyjaszewski, K. Polymerization-induced self-assembly of acrylonitrile via ICAR ATRP. Polymer 2017, 129, 57–67. [Google Scholar] [CrossRef]
- Zhao, D.; Ma, Y.; Wang, E.; Lodge, T.P. Micellization of Binary Diblock Co-polymer Mixtures in an Ionic Liquid. Macromolecules 2019, 52, 4729–4738. [Google Scholar] [CrossRef]
- Wang, Y.; Han, G.; Duan, W.; Zhang, W. ICAR ATRP in PEG with Low Concentration of Cu (II) Catalyst: A Versatile Method for Synthesis of Block Copolymer Nanoassemblies under Dispersion Polymerization. Macromol. Rapid Commun. 2019, 40, 1800140. [Google Scholar] [CrossRef]
- Genzer, J. In silico polymerization: Computer simulation of controlled radical polymerization in bulk and on flat surfaces. Macromolecules 2006, 39, 7157–7169. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrov, A.; Chertovich, A.V.; Gavrilov, A.A. Phase Diagrams of Polymerization-Induced Self-Assembly Are Largely Determined by Polymer Recombination. Polymers 2022, 14, 5331. https://doi.org/10.3390/polym14235331
Petrov A, Chertovich AV, Gavrilov AA. Phase Diagrams of Polymerization-Induced Self-Assembly Are Largely Determined by Polymer Recombination. Polymers. 2022; 14(23):5331. https://doi.org/10.3390/polym14235331
Chicago/Turabian StylePetrov, Artem, Alexander V. Chertovich, and Alexey A. Gavrilov. 2022. "Phase Diagrams of Polymerization-Induced Self-Assembly Are Largely Determined by Polymer Recombination" Polymers 14, no. 23: 5331. https://doi.org/10.3390/polym14235331