
Molecular Dynamics and Nuclear Magnetic Resonance Studies of Supercritical CO2 Sorption in Poly(Methyl Methacrylate)

 ,
,  ,
,  ,
,  and
and
Abstract
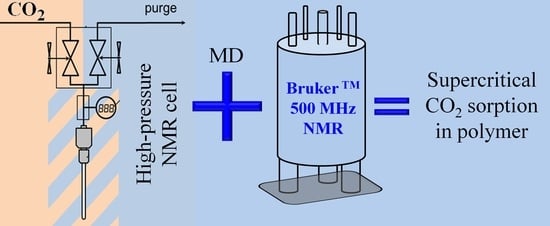
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
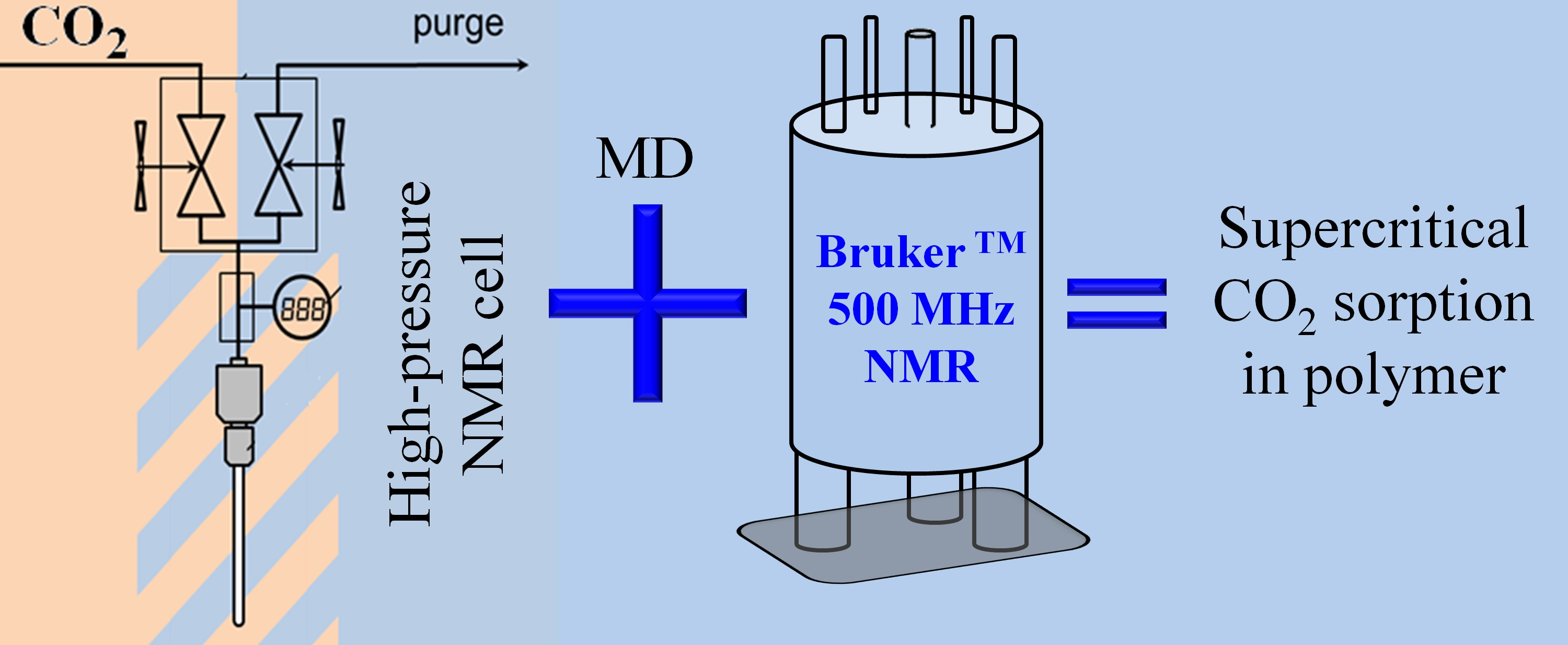
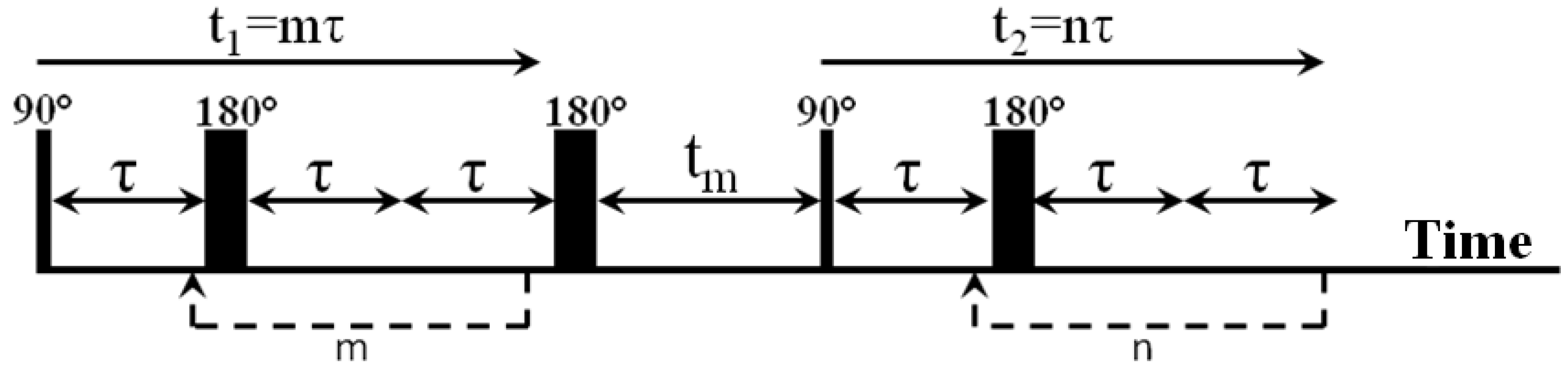
2.1. NMR Experiments and Methodology
2.2. NMR Experiment Parameters Selection
2.3. Computational Details/MD Method
2.4. RRCOSY Analysis
3. Results
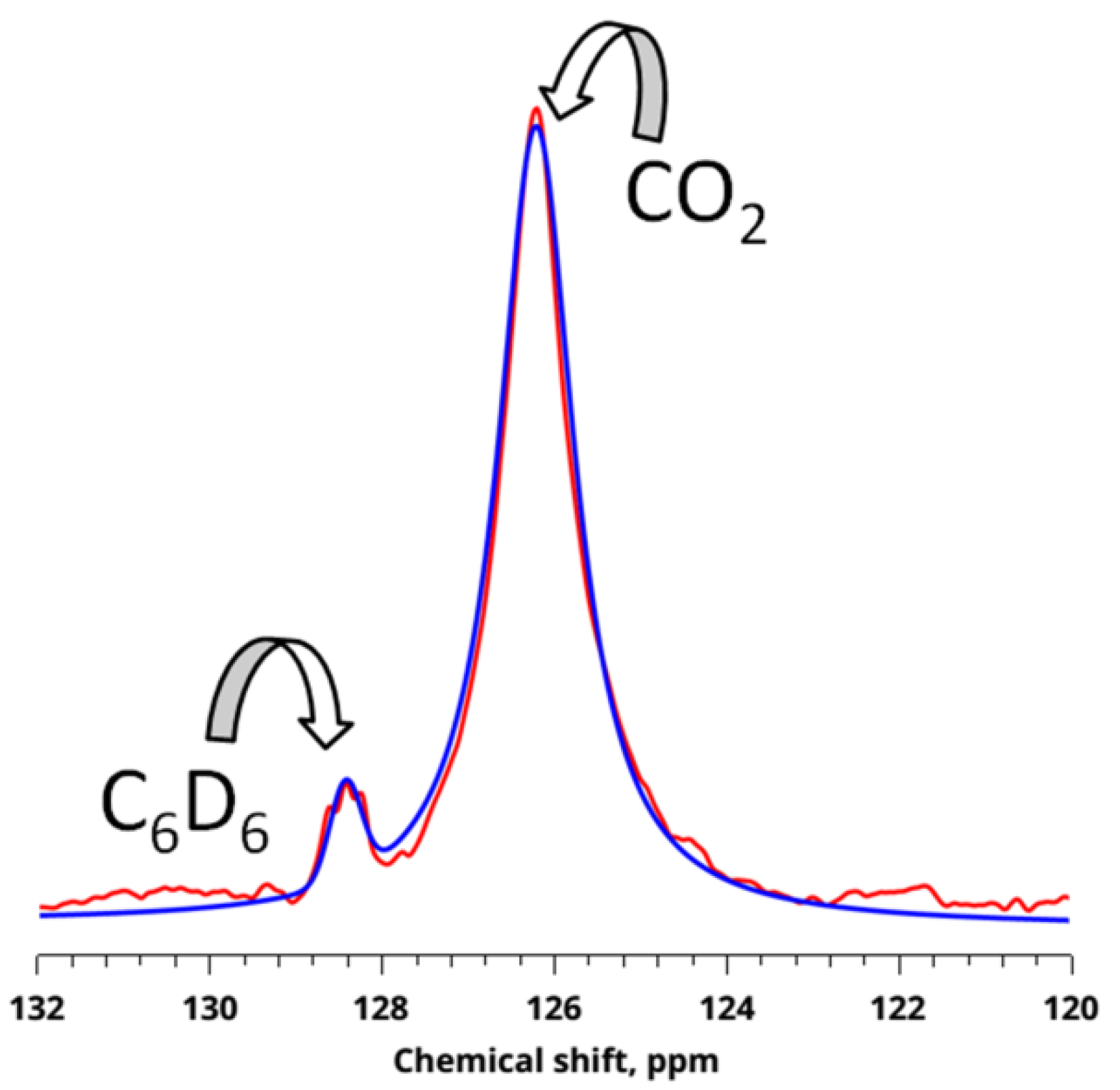
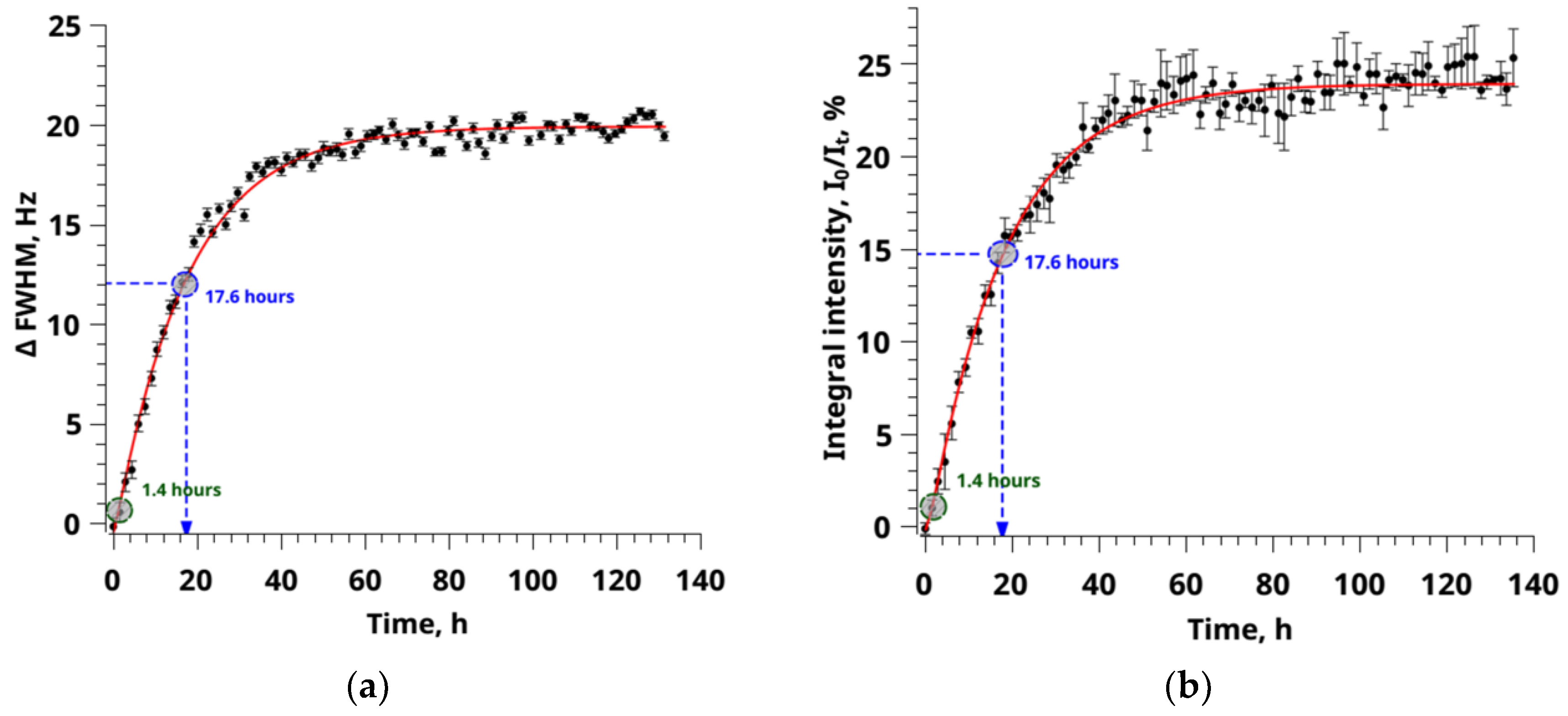
3.1. Sorption of CO2 from NMR Experiment
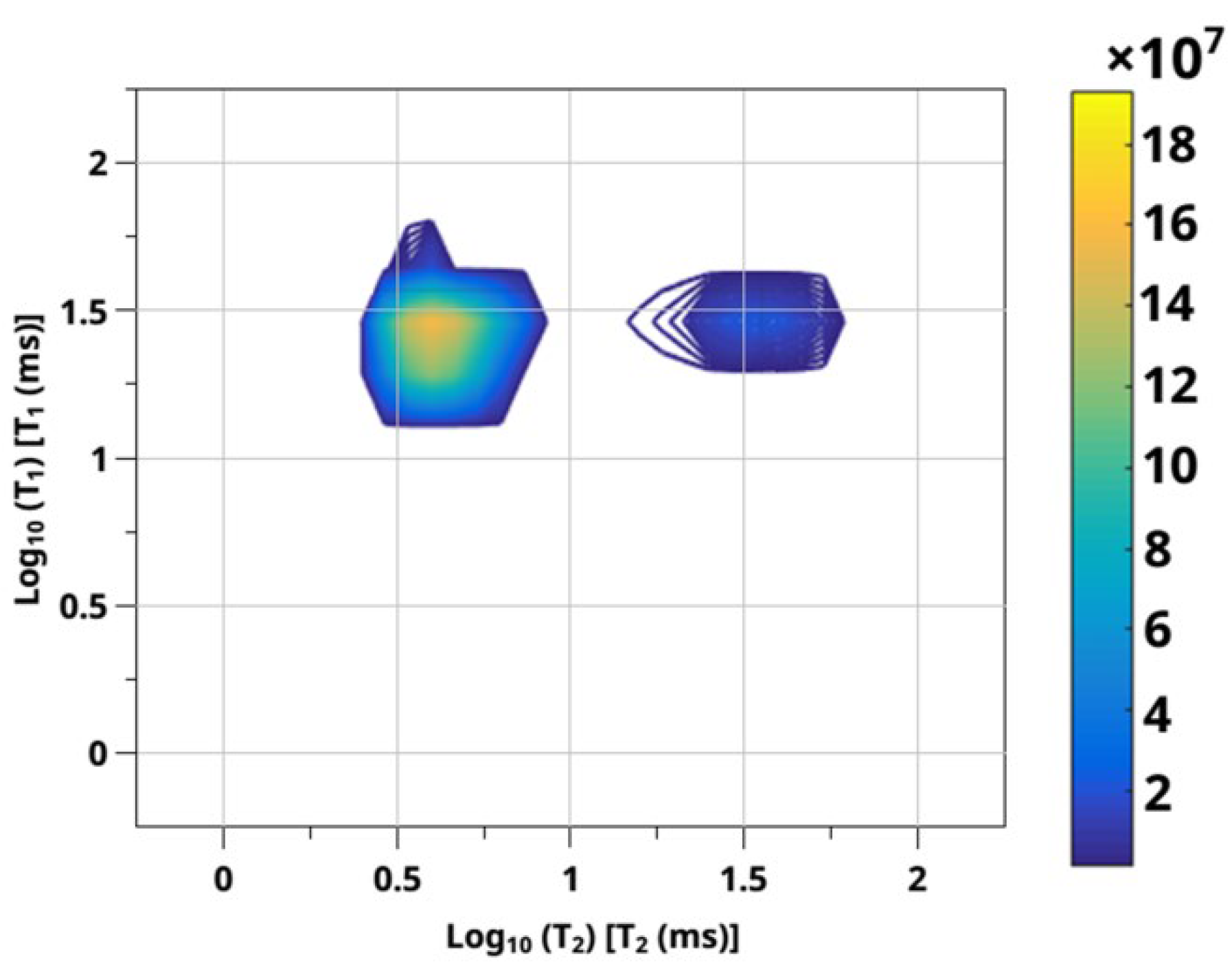
3.2. Sorption of CO2 from NMR Experiment—2D RRCOSY
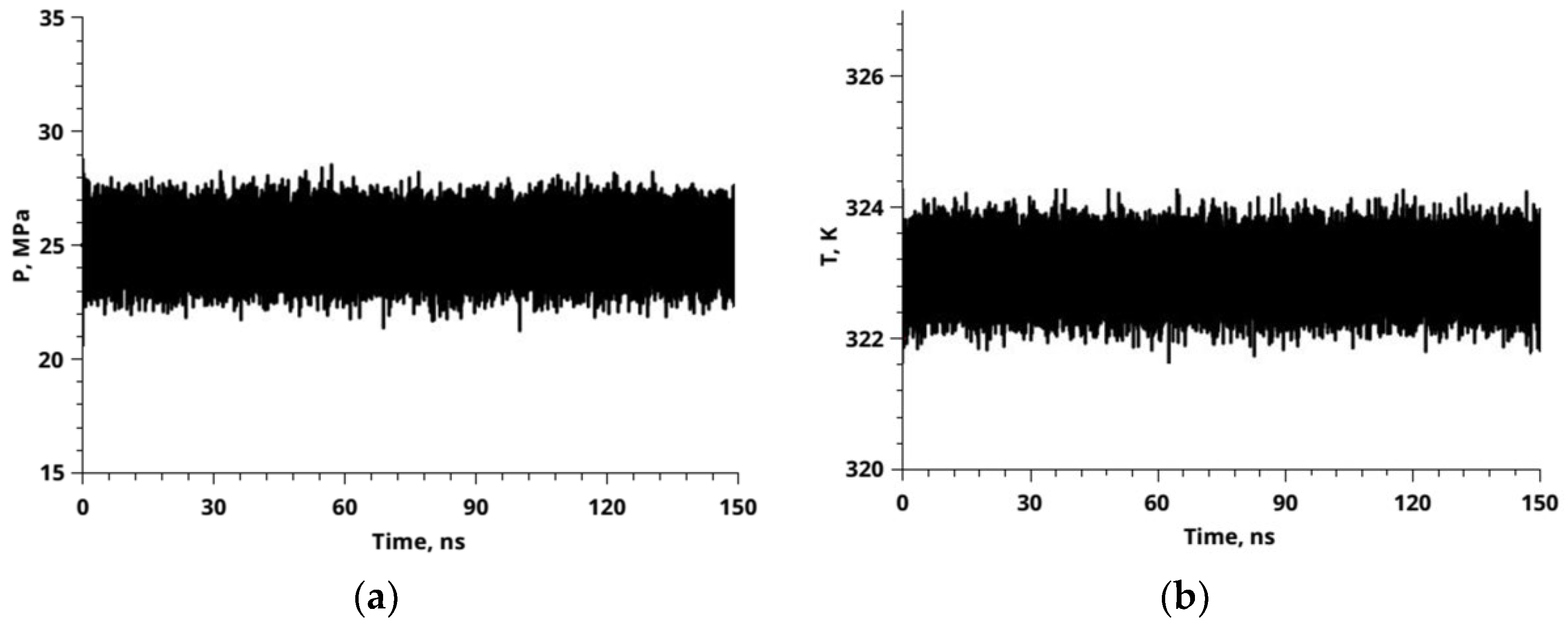
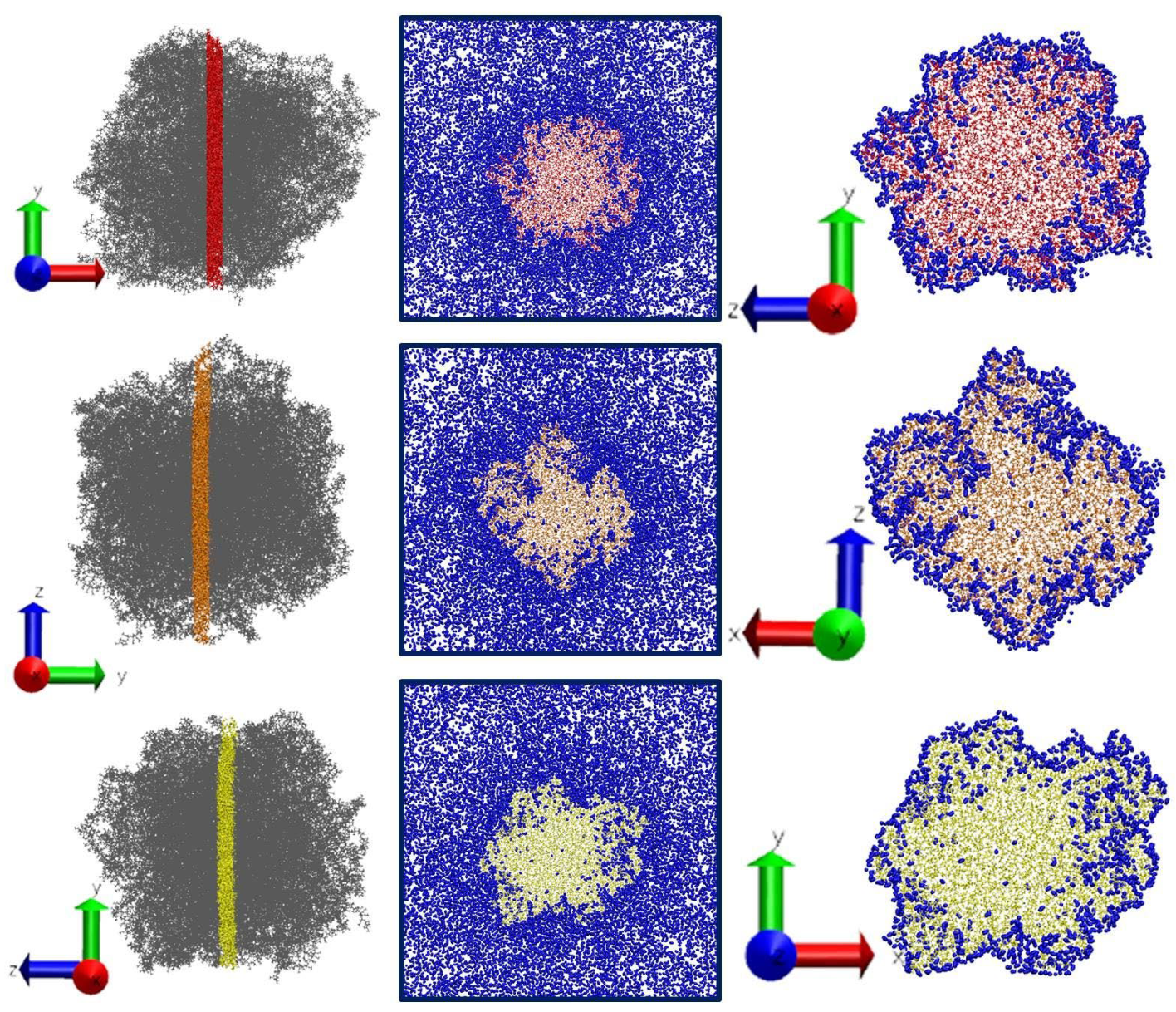
3.3. Sorption of CO2—MD Simulation Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Anagnostakos, K.; Fürst, O.; Kelm, J. Antibiotic-Impregnated PMMA Hip Spacers: Current Status. Acta Orthop. 2006, 77, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Pantoula, M.; Panayiotou, C. Sorption and Swelling in Glassy Polymer/carbon Dioxide Systems: Part I. Sorption. J. Supercrit. Fluids 2006, 37, 254–262. [Google Scholar] [CrossRef]
- Pantoula, M.; von Schnitzler, J.; Eggers, R.; Panayiotou, C. Sorption and Swelling in Glassy Polymer/carbon Dioxide Systems. Part II-Swelling. J. Supercrit. Fluids 2007, 39, 426–434. [Google Scholar] [CrossRef]
- Zhang, Y.; Gangwani, K.K.; Lemert, R.M. Sorption and Swelling of Block Copolymers in the Presence of Supercritical Fluid Carbon Dioxide. J. Supercrit. Fluids 1997, 11, 115–134. [Google Scholar] [CrossRef]
- Carla, V.; Wang, K.; Hussain, Y.; Efimenko, K.; Genzer, J.; Grant, C.; Sarti, G.C.; Carbonell, R.G.; Doghieri, F. Nonequilibrium Model for Sorption and Swelling of Bulk Glassy Polymer Films with Supercritical Carbon Dioxide. Macromolecules 2005, 38, 10299–10313. [Google Scholar] [CrossRef]
- Van der Lee, A.; Hamon, L.; Holl, Y.; Grohens, Y. Density Profiles in Thin PMMA Supported Films Investigated by X-Ray Reflectometry. Langmuir 2001, 17, 7664–7669. [Google Scholar] [CrossRef]
- Li, X.K.; Cao, G.P.; Chen, L.H.; Zhang, R.H.; Liu, H.L.; Shi, Y.H. Study of the Anomalous Sorption Behavior of CO2 into Poly(methyl Methacrylate) Films in the Vicinity of the Critical Pressure and Temperature Using a Quartz Crystal Microbalance (QCM). Langmuir 2013, 29, 14089–14100. [Google Scholar] [CrossRef]
- Aubert, J.H. Solubility of Carbon Dioxide in Polymers by the Quartz Crystal Microbalance Technique. J. Supercrit. Fluids 1998, 11, 163–172. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Chan, K.L.A. FTIR Imaging of Polymeric Materials under High-Pressure Carbon Dioxide. Macromolecules 2004, 37, 579–584. [Google Scholar] [CrossRef]
- Ichikawa, K.; Mori, T.; Kitano, H.; Fukuda, M.; Mochizuki, A.; Tanaka, M. Fourier Transform Infrared Study on the Sorption of Water to Various Kinds of Polymer Thin Films. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 2175–2182. [Google Scholar] [CrossRef]
- Naumann, D. FT-infrared and FT-Raman spectroscopy in biomedical research. Appl. Spectrosc. Rev. 2001, 36, 239–298. [Google Scholar] [CrossRef]
- Dubois, J.; Grau, E.; Tassaing, T.; Dumon, M. On the CO2 Sorption and Swelling of Elastomers by Supercritical CO2 as Studied by in Situ High Pressure FTIR Microscopy. J. Supercrit. Fluids 2018, 131, 150–156. [Google Scholar] [CrossRef]
- Meunier, F.C. Pitfalls and Benefits of: In Situ and Operando Diffuse Reflectance FT-IR Spectroscopy (DRIFTS) Applied to Catalytic Reactions. React. Chem. Eng. 2016, 1, 134–141. [Google Scholar] [CrossRef]
- Fried, J.R.; Li, W. High-Pressure FTIR Studies of Gas–polymer Interactions. J. Appl. Polym. Sci. 1990, 41, 1123–1131. [Google Scholar] [CrossRef]
- Di Noto, V.; Vezzù, K.; Giffin, G.A.; Conti, F.; Bertucco, A. Effect of High Pressure CO2 on the Structure of PMMA: A FT-IR Study. J. Phys. Chem. B 2011, 115, 13519–13525. [Google Scholar] [CrossRef]
- Briscoe, B.J.; Lorge, O.; Wajs, A.; Dang, P. Carbon Dioxide-Poly(vinylidene Fluoride) Interactions at High Pressure. J. Polym. Sci. Part B Polym. Phys. 1998, 36, 2435–2447. [Google Scholar] [CrossRef]
- Wissinger, R.G.; Paulaitis, M.E. Swelling and Sorption in polymer–CO2 Mixtures at Elevated Pressures. J. Polym. Sci. Part B Polym. Phys. 1987, 25, 2497–2510. [Google Scholar] [CrossRef]
- Keller, J.U.; Rave, H.; Staudt, R. Measurement of Gas Absorption in a Swelling Polymeric Material by a Combined Gravimetric-Dynamic Method. Macromol. Chem. Phys. 1999, 200, 2269–2275. [Google Scholar] [CrossRef]
- Royer, J.R.; DeSimone, J.M.; Khan, S.A. Carbon Dioxide-Induced Swelling of Poly(dimethylsiloxane). Macromolecules 1999, 32, 8965–8973. [Google Scholar] [CrossRef]
- Nikitin, L.N.; Said-Galiyev, E.E.; Vinokur, R.A.; Khokhlov, A.R.; Gallyamov, M.O.; Schaumburg, K. Poly(methyl Methacrylate) and Poly(butyl Methacrylate) Swelling in Supercritical Carbon Dioxide. Macromolecules 2002, 35, 934–940. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Schaefer, J.; Henis, J.M.S.; Tripodi, M.K. Magic-Angle Carbon-13 NMR Study of CO2 Adsorbed on Some Molecular Sieves. J. Chem. Phys. 1974, 61, 2351–2355. [Google Scholar] [CrossRef]
- Michael, A.; Meiler, W.; Michel, D.; Pfeifer, H. 13C NMR Investigations of Carbon Monoxide Adsorbed in Zeolites. Chem. Phys. Lett. 1981, 84, 30–32. [Google Scholar] [CrossRef]
- Dardin, A.; Cain, J.B.; Desimone, J.M.; Johnson, C.S.; Samulski, E.T. High-Pressure NMR of Polymers Dissolved in Supercritical Carbon Dioxide. Macromolecules 1997, 30, 3593–3599. [Google Scholar] [CrossRef]
- Iijima, M.; Nagasaki, Y.; Okada, T.; Kato, M.; Kataoka, K. Core-Polymerized Reactive Micelles from Heterotelechelic Amphiphilic Block Copolymers. Macromolecules 1999, 32, 1140–1146. [Google Scholar] [CrossRef]
- Niessen, H.G.; Trautner, P.; Wiemann, S.; Bargon, J.; Woelk, K. The Toroid Cavity Autoclave for High-Pressure and Variable-Temperature in Situ Nuclear Magnetic Resonance Studies. Rev. Sci. Instrum. 2002, 73, 1259. [Google Scholar] [CrossRef]
- Ballard, L.; Reiner, C.; Jonas, J. High-Resolution NMR Probe for Experiments at High Pressures. J. Magn. Reson. Ser. A 1996, 123, 81–86. [Google Scholar] [CrossRef]
- Wissinger, R.G.; Paulaitis, M.E. Molecular Thermodynamic Model for Sorption and Swelling in Glassy Polymer-CO2 Systems at Elevated Pressures. Ind. Eng. Chem. Res. 1991, 30, 842–851. [Google Scholar] [CrossRef]
- Kamiya, Y.; Mizoguchi, K.; Terada, K.; Fujiwara, Y.; Wang, J.-S. CO2 Sorption and Dilation of Poly(methyl Methacrylate). Macromolecules 1998, 31, 472–478. [Google Scholar] [CrossRef]
- Rajendran, A.; Bonavoglia, B.; Forrer, N.; Storti, G.; Mazzotti, M.; Morbidelli, M. Simultaneous Measurement of Swelling and Sorption in a Supercritical CO2-Poly(methyl Methacrylate) System. Ind. Eng. Chem. Res. 2005, 44, 2549–2560. [Google Scholar] [CrossRef]
- Vogt, B.D.; RamachandraRao, V.S.; Gupta, R.R.; Lavery, K.A.; Francis, T.J.; Russell, T.P.; Watkins, J.J. Phase Behavior of Polystyrene-Block-Poly(n-Alkyl Methacrylate)s Dilated with Carbon Dioxide. Macromolecules 2003, 36, 4029–4036. [Google Scholar] [CrossRef]
- Shieh, Y.-T.; Liu, K.-H. The Effect of Carbonyl Group on Sorption of CO2 in Glassy Polymers. J. Supercrit. Fluids 2003, 25, 261–268. [Google Scholar] [CrossRef]
- Gurina, D.L.; Budkov, Y.A.; Kiselev, M.G. Impregnation of Poly(methyl Methacrylate) with Carbamazepine in Supercritical Carbon Dioxide: Molecular Dynamics Simulation. J. Phys. Chem. B 2020, 124, 8410–8417. [Google Scholar] [CrossRef]
- Gurina, D.; Budkov, Y.; Kiselev, M. Molecular Dynamics Study of the Swelling of Poly(methyl Methacrylate) in Supercritical Carbon Dioxide. Materials 2019, 12, 3315. [Google Scholar] [CrossRef] [Green Version]
- Gurina, D.L.; Budkov, Y.A.; Kiselev, M.G. A Molecular Insight into Poly(methyl Methacrylate) Impregnation with Mefenamic Acid in Supercritical Carbon Dioxide: A Computational Simulation. J. Mol. Liq. 2021, 337, 116424. [Google Scholar] [CrossRef]
- Kikic, I. Polymer–supercritical Fluid Interactions. J. Supercrit. Fluids 2009, 47, 458–465. [Google Scholar] [CrossRef]
- Belov, K.V.; Dyshin, A.A.; Krestyaninov, M.A.; Efimov, S.V.; Khodov, I.A.; Kiselev, M.G. Conformational Preferences of Tolfenamic Acid in DMSO-CO2 Solvent System by 2D NOESY. J. Mol. Liq. 2022, 367, 120481. [Google Scholar] [CrossRef]
- Belov, K.V.; Batista de Carvalho, L.A.E.; Dyshin, A.A.; Efimov, S.V.; Khodov, I.A. The Role of Hidden Conformers in Determination of Conformational Preferences of Mefenamic Acid by NOESY Spectroscopy. Pharmaceutics 2022, 14, 2276. [Google Scholar] [CrossRef]
- Khodov, I.A.; Belov, K.V.; Dyshin, A.A.; Krestyaninov, M.A.; Kiselev, M.G. Pressure Effect on Lidocaine Conformational Equilibria in scCO2: A Study by 2D NOESY. J. Mol. Liq. 2022, 367, 120525. [Google Scholar] [CrossRef]
- Khodov, I.; Dyshin, A.; Efimov, S.; Ivlev, D.; Kiselev, M. High-Pressure NMR Spectroscopy in Studies of the Conformational Composition of Small Molecules in Supercritical Carbon Dioxide. J. Mol. Liq. 2020, 309, 113113. [Google Scholar] [CrossRef] [Green Version]
- Raiford, D.S.; Fisk, C.L.; Becker, E.D. Calibration of Methanol and Ethylene Glycol Nuclear Magnetic Resonance Thermometers. Anal. Chem. 1979, 51, 2050–2051. [Google Scholar] [CrossRef]
- Shieh, Y.-T.; Liu, K.-H. Solubility of CO2 in Glassy PMMA and PS over a Wide Pressure Range: The Effect of Carbonyl Groups. J. Polym. Res. 2002, 9, 107–113. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Zhang, Z.; Duan, Z. An Optimized Molecular Potential for Carbon Dioxide. J. Chem. Phys. 2005, 122, 214507. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Provencher, S.W. CONTIN: A General Purpose Constrained Regularization Program for Inverting Noisy Linear Algebraic and Integral Equations. Comput. Phys. Commun. 1982, 27, 229–242. [Google Scholar] [CrossRef]
- Lee, J.H.; Labadie, C.; Springer, C.S.; Harbison, G.S.; Lee, J.H.; Labadie, C.; Springer, C.S.; Harbison, G.S. Two-Dimensional Inverse Laplace Transform NMR: Altered Relaxation Times Allow Detection of Exchange Correlation. J. Am. Chem. Soc. 1993, 115, 7761–7764. [Google Scholar] [CrossRef]
- Caputo, P.; Loise, V.; Ashimova, S.; Teltayev, B.; Vaiana, R.; Oliviero Rossi, C. Inverse Laplace Transform (ILT) NMR: A Powerful Tool to Differentiate a Real Rejuvenator and a Softener of Aged Bitumen. Colloids Surfaces A Physicochem. Eng. Asp. 2019, 574, 154–161. [Google Scholar] [CrossRef]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Uniform-Penalty Inversion of Multiexponential Decay Data. J. Magn. Reson. 1998, 132, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.Q.; Venkataramanan, L.; Hürlimann, M.D.; Flaum, M.; Frulla, P.; Straley, C. T1-T2 Correlation Spectra Obtained Using a Fast Two-Dimensional Laplace Inversion. J. Magn. Reson. 2002, 154, 261–268. [Google Scholar] [CrossRef]
- Venkataramanan, L.; Song, Y.Q.; Hürlimann, M.D. Solving Fredholm Integrals of the First Kind with Tensor Product Structure in 2 and 2.5 Dimensions. IEEE Trans. Signal Process. 2002, 50, 1017–1026. [Google Scholar] [CrossRef]
- Van Landeghem, M.; Haber, A.; D’espinose De Lacaillerie, J.-B.; Blümich, B. Analysis of Multisite 2D Relaxation Exchange NMR. Concepts Magn. Reson. Part A 2010, 36A, 153–169. [Google Scholar] [CrossRef]
- Dortch, R.D.; Horch, R.A.; Does, M.D. Development, Simulation, and Validation of NMR Relaxation-Based Exchange Measurements. J. Chem. Phys. 2009, 131, 164502. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, L.M.; Johnson, D.L.; Mitchell, J.; Chandrasekera, T.C.; Fordham, E.J. Modeling Two-Dimensional Magnetic Resonance Measurements in Coupled Pore Systems. Phys. Rev. E—Stat. Nonlinear Soft Matter Phys. 2013, 88, 032813. [Google Scholar] [CrossRef]
- Monteilhet, L.; Korb, J.P.; Mitchell, J.; McDonald, P.J. Observation of Exchange of Micropore Water in Cement Pastes by Two-Dimensional T2—T2 Nuclear Magnetic Resonance Relaxometry. Phys. Rev. E—Stat. Nonlinear Soft Matter Phys. 2006, 74, 061404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, P.J.; Korb, J.P.; Mitchell, J.; Monteilhet, L. Surface Relaxation and Chemical Exchange in Hydrating Cement Pastes: A Two-Dimensional NMR Relaxation Study. Phys. Rev. E—Stat. Nonlinear Soft Matter Phys. 2005, 72, 011409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, H.; Harwood, J.S.; Yang, D.; Post, C.B. A Simple Method for NMR t1 Noise Suppression. J. Magn. Reson. 2017, 276, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, H.M. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958, 28, 430–431. [Google Scholar] [CrossRef] [Green Version]
- Tracy, D.S.; Singh, R.P. A New Matrix Product and Its Applications in Partitioned Matrix Differentiation. Stat. Neerl. 1972, 26, 143–157. [Google Scholar] [CrossRef]
- Liu, S. Matrix Results on the Khatri-Rao and Tracy-Singh Products. Linear Algebra Appl. 1999, 289, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Omi, H.; Ueda, T.; Miyakubo, K.; Eguchi, T. Dynamics of CO2 Molecules Confined in the Micropores of Solids as Studied by 13C NMR. Appl. Surf. Sci. 2005, 252, 660–667. [Google Scholar] [CrossRef]
- Nakayama, A.; Ishii, C.; Takayama, T.; Watanabe, M.; Zanma, A.; Kaneko, K.; Sugihara, K. Magnetic and Electronic Properties of Ferrocene-Doped Activated Carbon Fibers. Synth. Met. 1997, 86, 2335–2336. [Google Scholar] [CrossRef]
- Petrakis, L. Spectral Line Shapes: Gaussian and Lorentzian Functions in Magnetic Resonance. J. Chem. Educ. 1967, 44, 432–436. [Google Scholar] [CrossRef]
- Bleaney, B. John Hasbrouck Van Vleck, 13 March 1899—27 October 1980. Biogr. Mem. Fellows R. Soc. 1982, 28, 627–665. [Google Scholar] [CrossRef]
- Seshadri, K.S.; Jones, R.N. The Shapes and Intensities of Infrared Absorption Bands—A Review. Spectrochim. Acta 1963, 19, 1013–1085. [Google Scholar] [CrossRef]
- Earl, W.L.; Kim, Y.W.; Smith, D.M. NMR Measurement of Pore Structure. Stud. Surf. Sci. Catal. 1994, 87, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, M.; Moradi, O.; Zare, K. Kinetics Adsorption Study of the Ethidium Bromide by Graphene Oxide as Adsorbent from Aqueous Matrices. Int. Nano Lett. 2017, 7, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Üzer, S.; Akman, U.; Hortaçsu, O. Polymer Swelling and Impregnation Using Supercritical CO2: A Model-Component Study towards Producing Controlled-Release Drugs. J. Supercrit. Fluids 2006, 38, 119–128. [Google Scholar] [CrossRef]
- D’Agostino, C.; Mitchell, J.; Mantle, M.D.; Gladden, L.F. Interpretation of NMR Relaxation as a Tool for Characterising the Adsorption Strength of Liquids inside Porous Materials. Chem.—A Eur. J. 2014, 20, 13009–13015. [Google Scholar] [CrossRef] [Green Version]
- Mailhiot, S.E.; Zong, F.; Maneval, J.E.; June, R.K.; Galvosas, P.; Seymour, J.D. Quantifying NMR Relaxation Correlation and Exchange in Articular Cartilage with Time Domain Analysis. J. Magn. Reson. 2018, 287, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Rodts, S.; Bytchenkoff, D. Structural Properties of 2D NMR Relaxation Spectra of Diffusive Systems. J. Magn. Reson. 2010, 205, 315–318. [Google Scholar] [CrossRef]
- Bytchenkoff, D.; Rodts, S. Structure of the Two-Dimensional Relaxation Spectra Seen within the Eigenmode Perturbation Theory and the Two-Site Exchange Model. J. Magn. Reson. 2011, 208, 4–19. [Google Scholar] [CrossRef]
- Song, Y.Q.; Zielinski, L.; Ryu, S. Two-Dimensional NMR of Diffusion Systems. Phys. Rev. Lett. 2008, 100, 248002. [Google Scholar] [CrossRef]
- Song, Y.Q.; Carneiro, G.; Schwartz, L.M.; Johnson, D.L. Experimental Identification of Diffusive Coupling Using 2D NMR. Phys. Rev. Lett. 2014, 113, 235503. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobornova, V.V.; Belov, K.V.; Dyshin, A.A.; Gurina, D.L.; Khodov, I.A.; Kiselev, M.G. Molecular Dynamics and Nuclear Magnetic Resonance Studies of Supercritical CO2 Sorption in Poly(Methyl Methacrylate). Polymers 2022, 14, 5332. https://doi.org/10.3390/polym14235332
Sobornova VV, Belov KV, Dyshin AA, Gurina DL, Khodov IA, Kiselev MG. Molecular Dynamics and Nuclear Magnetic Resonance Studies of Supercritical CO2 Sorption in Poly(Methyl Methacrylate). Polymers. 2022; 14(23):5332. https://doi.org/10.3390/polym14235332
Chicago/Turabian StyleSobornova, Valentina V., Konstantin V. Belov, Alexey A. Dyshin, Darya L. Gurina, Ilya A. Khodov, and Michael G. Kiselev. 2022. "Molecular Dynamics and Nuclear Magnetic Resonance Studies of Supercritical CO2 Sorption in Poly(Methyl Methacrylate)" Polymers 14, no. 23: 5332. https://doi.org/10.3390/polym14235332