
Novel Metallo-Supramolecular Polymers with 1-Thioxophosphole Main-Chain Units and Remarkable Photoinduced Changes in Their Resonance Raman Spectra

Abstract
:
1. Introduction
2. Materials and Methods
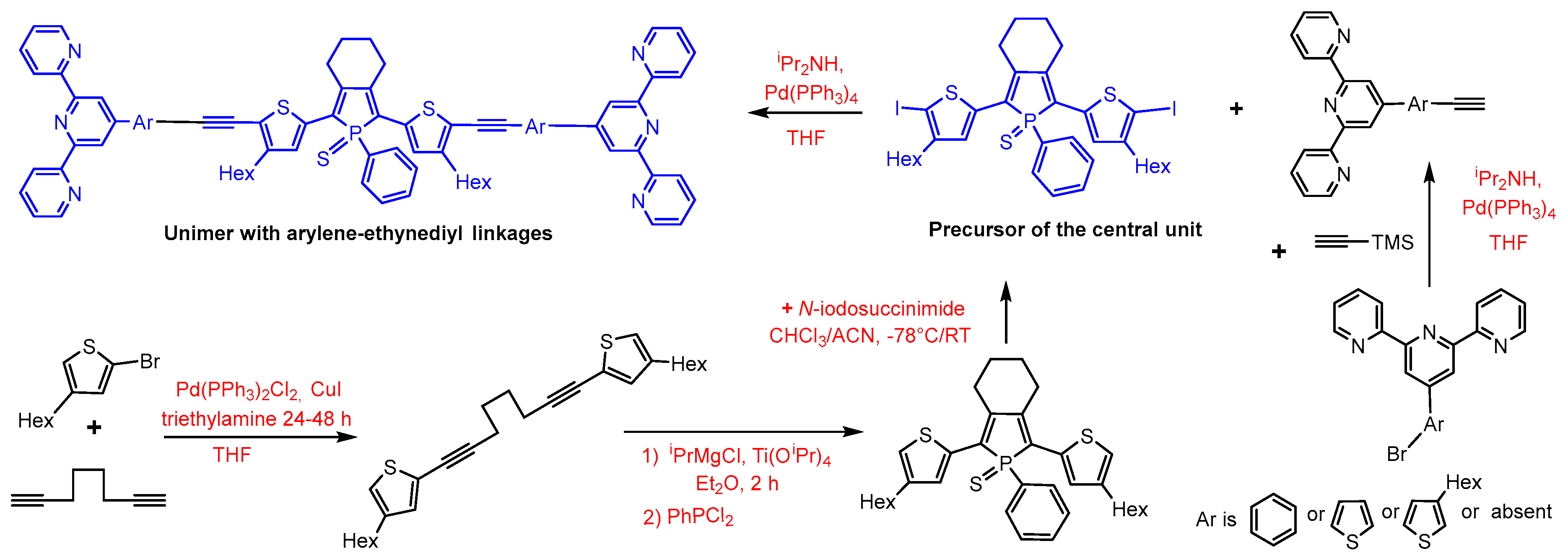
2.1. Synthesis
2.2. Precursors of Linkers
2.3. Precursor of the Central Unit
2.4. Coupling of Precursors to Unimers
2.5. Unimer E
2.6. Unimer EPh
2.7. Unimer ET
2.8. Unimer ET6
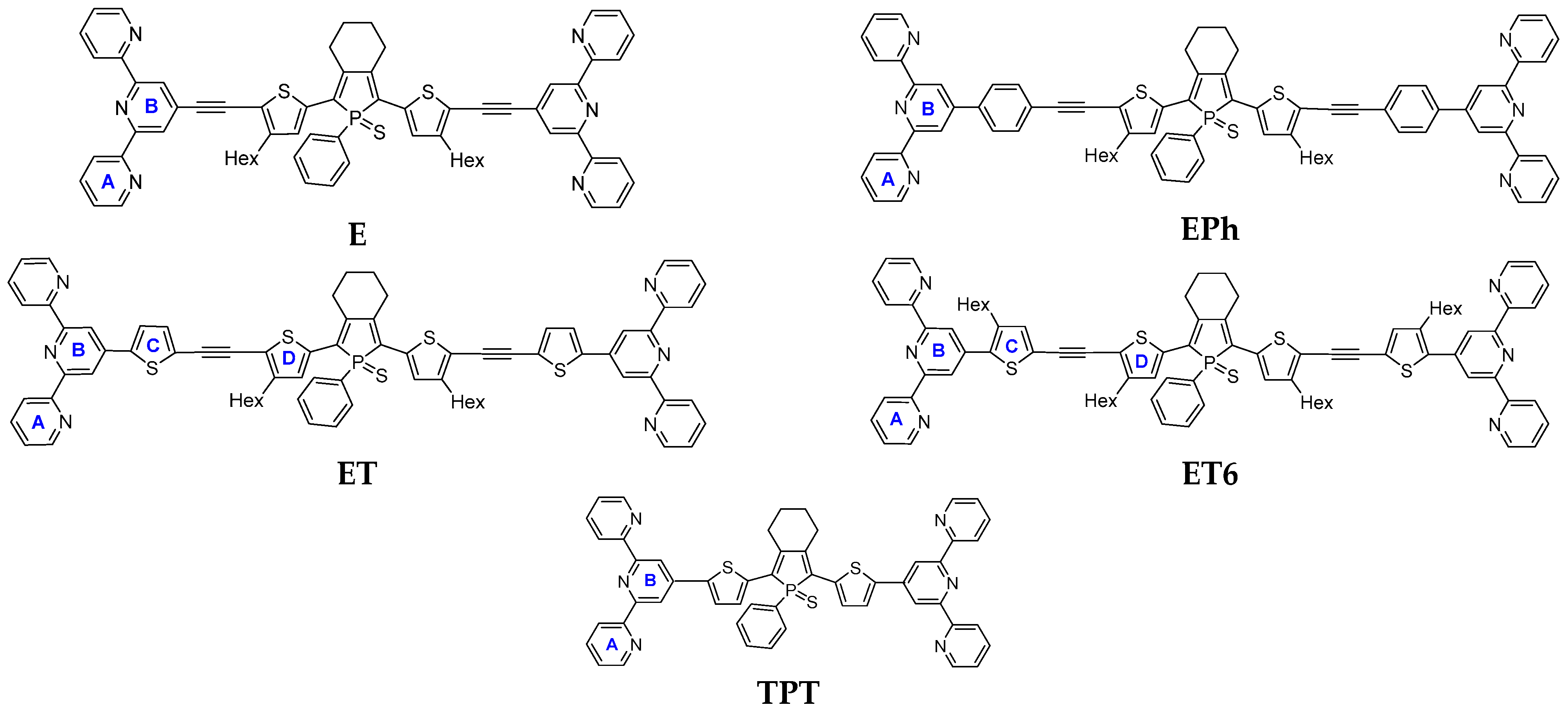
3. Results
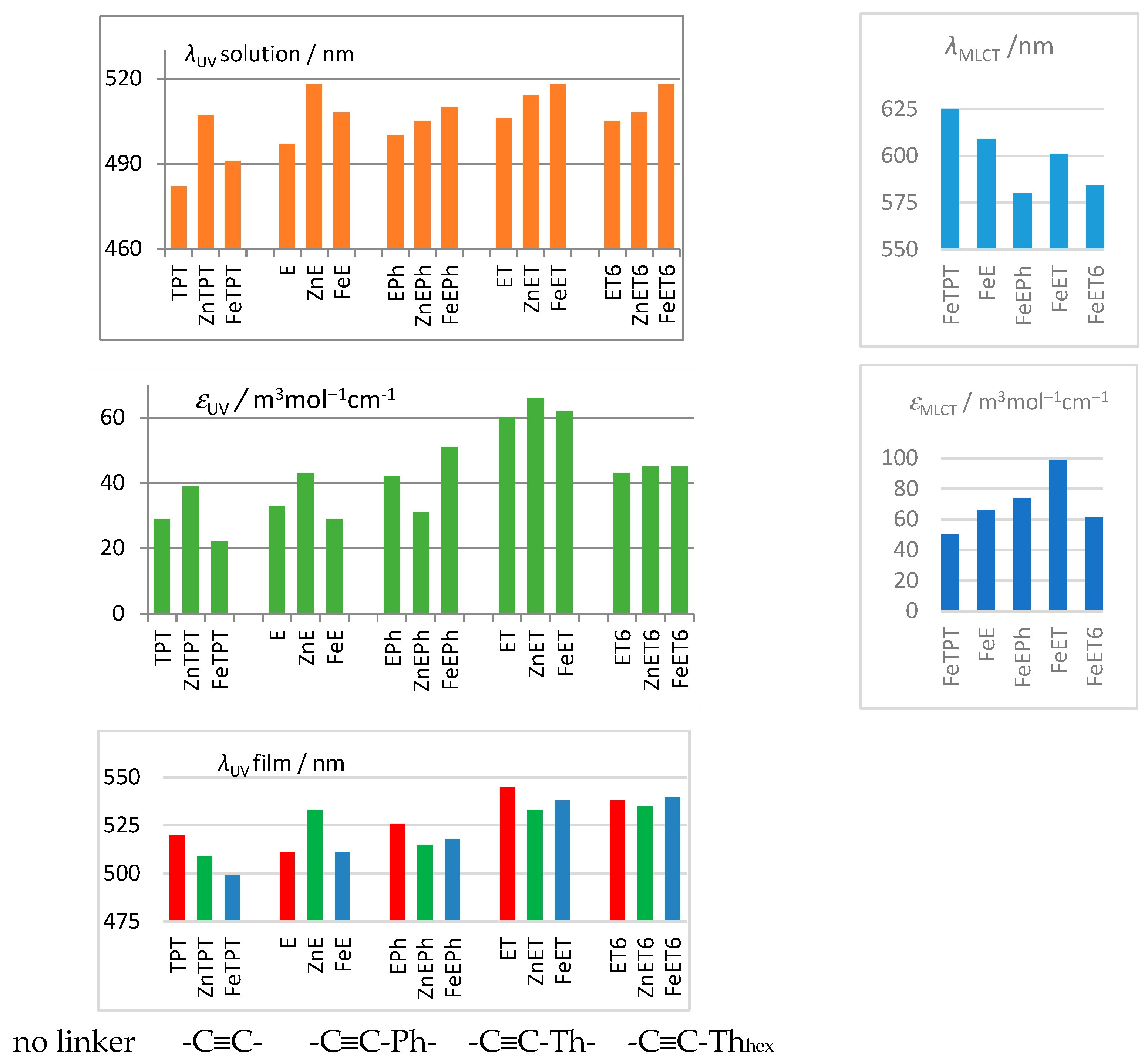
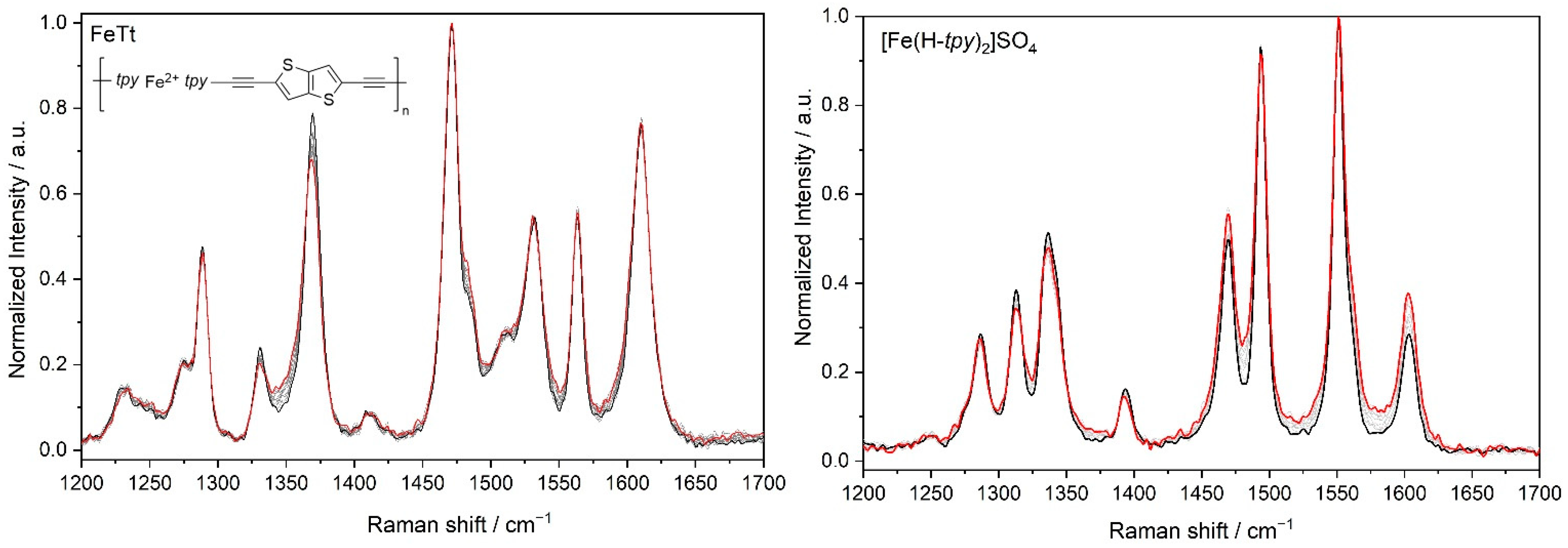
3.1. Characteristics of Phosphole Unimers and Their Polymers
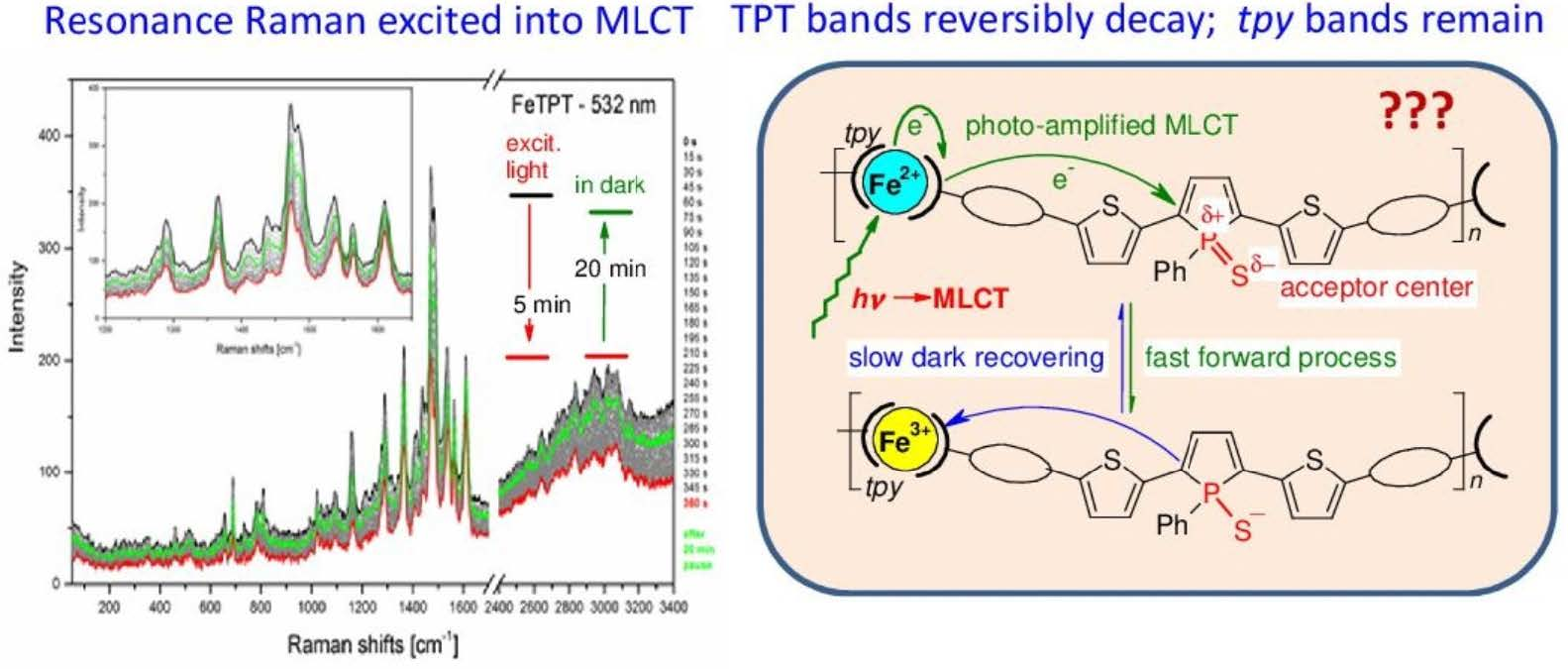
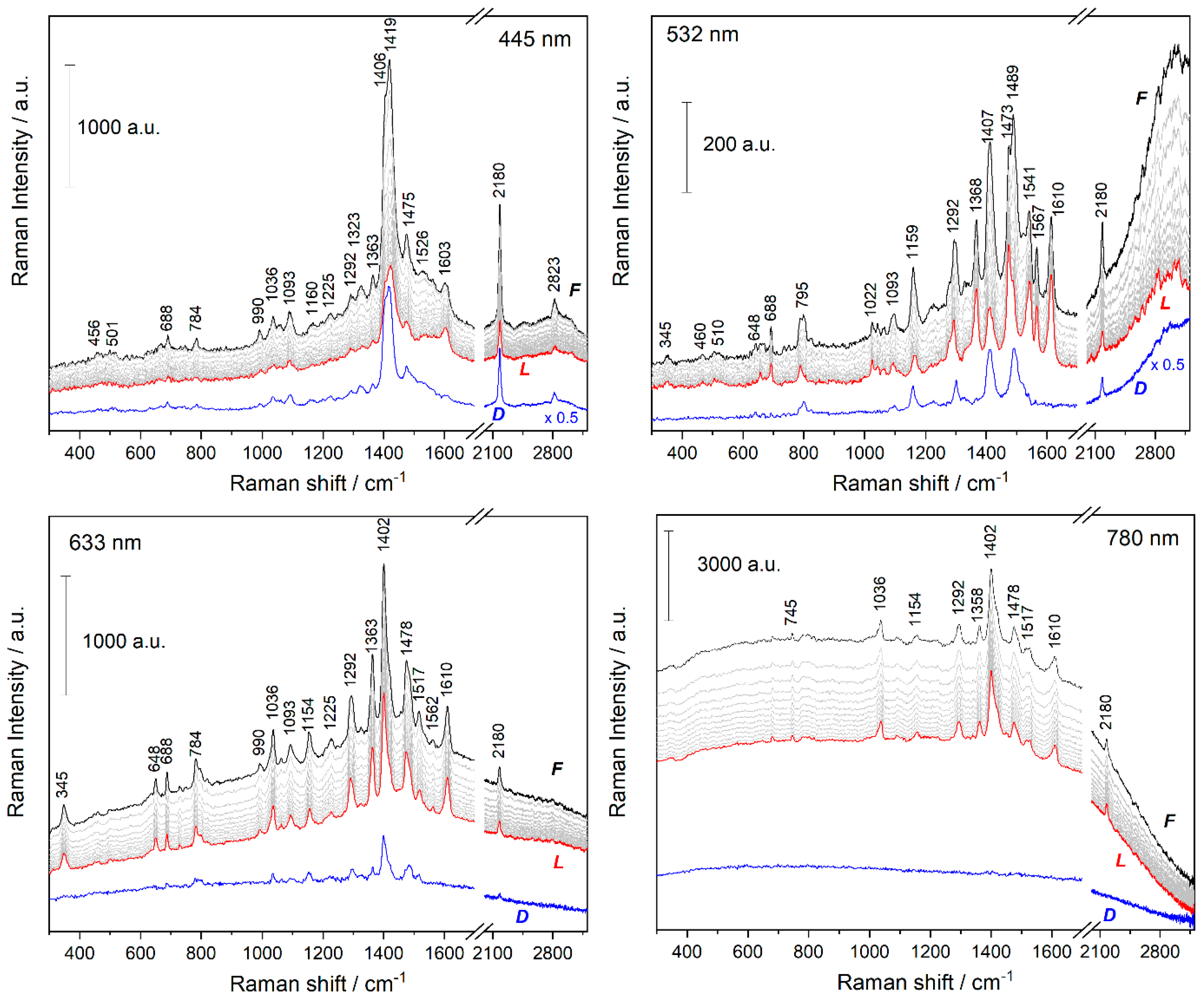
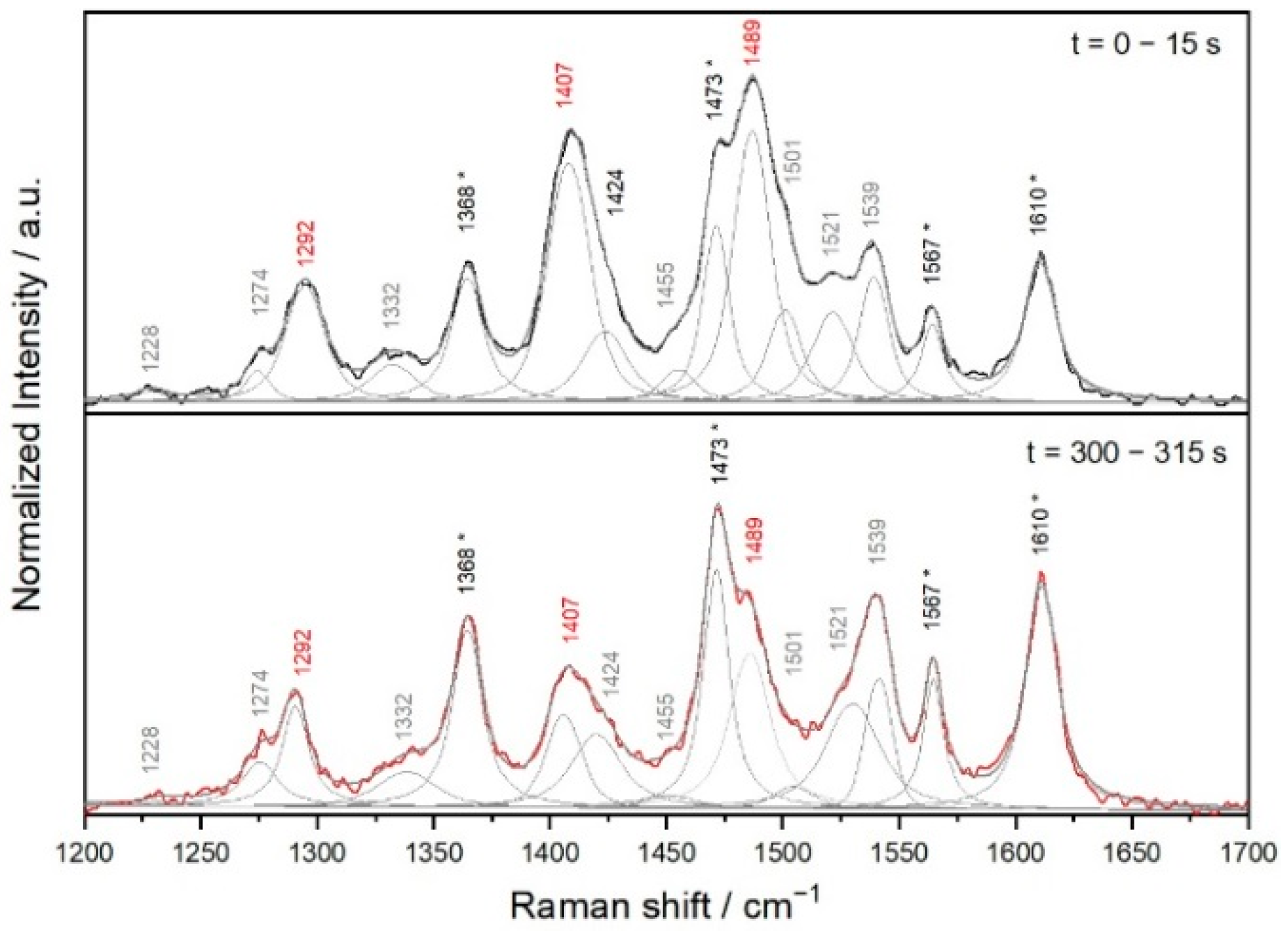
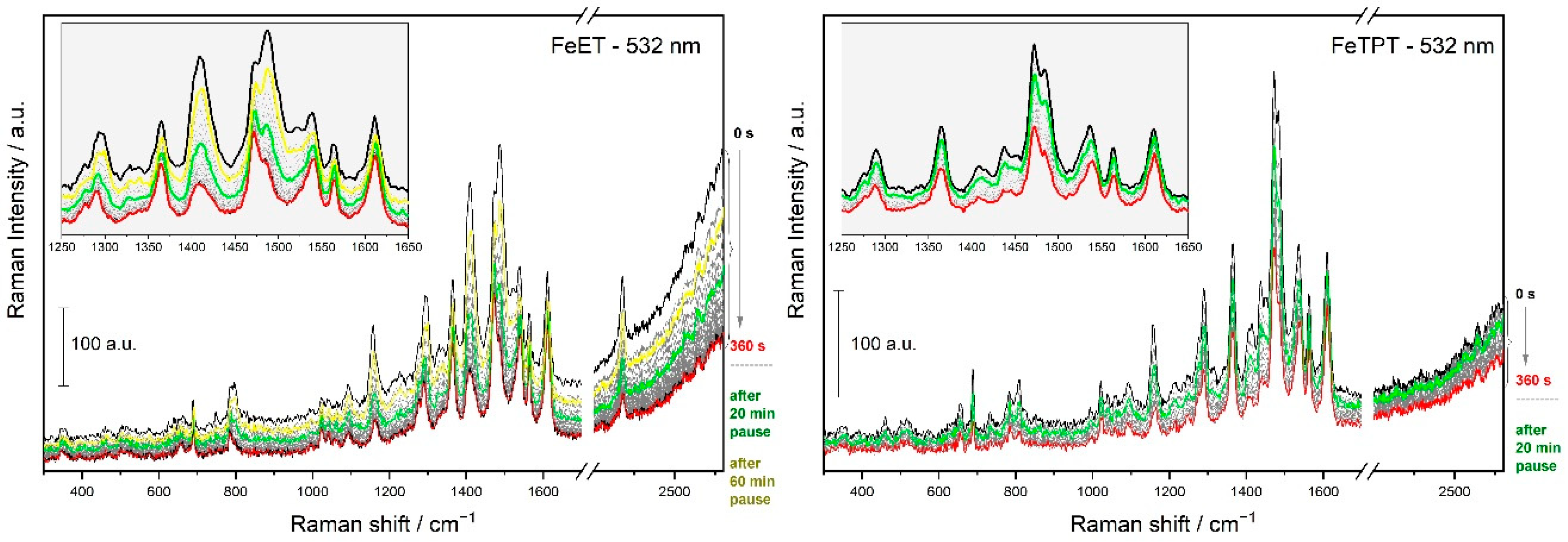
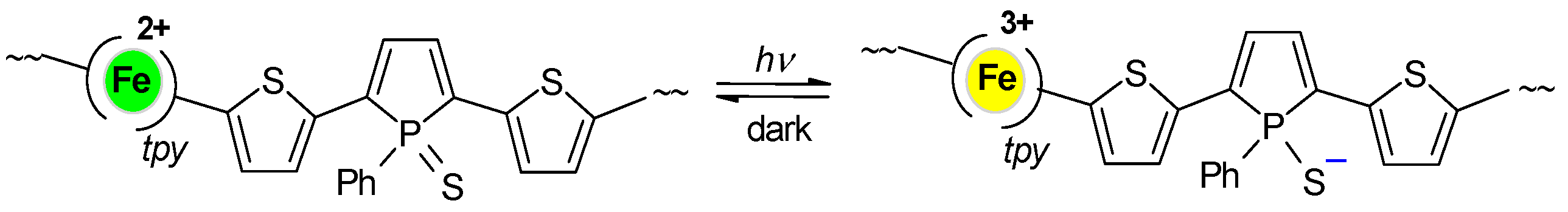
3.2. Time Changes in Raman Spectra of Films of Fe-MSPs with Phosphole Units
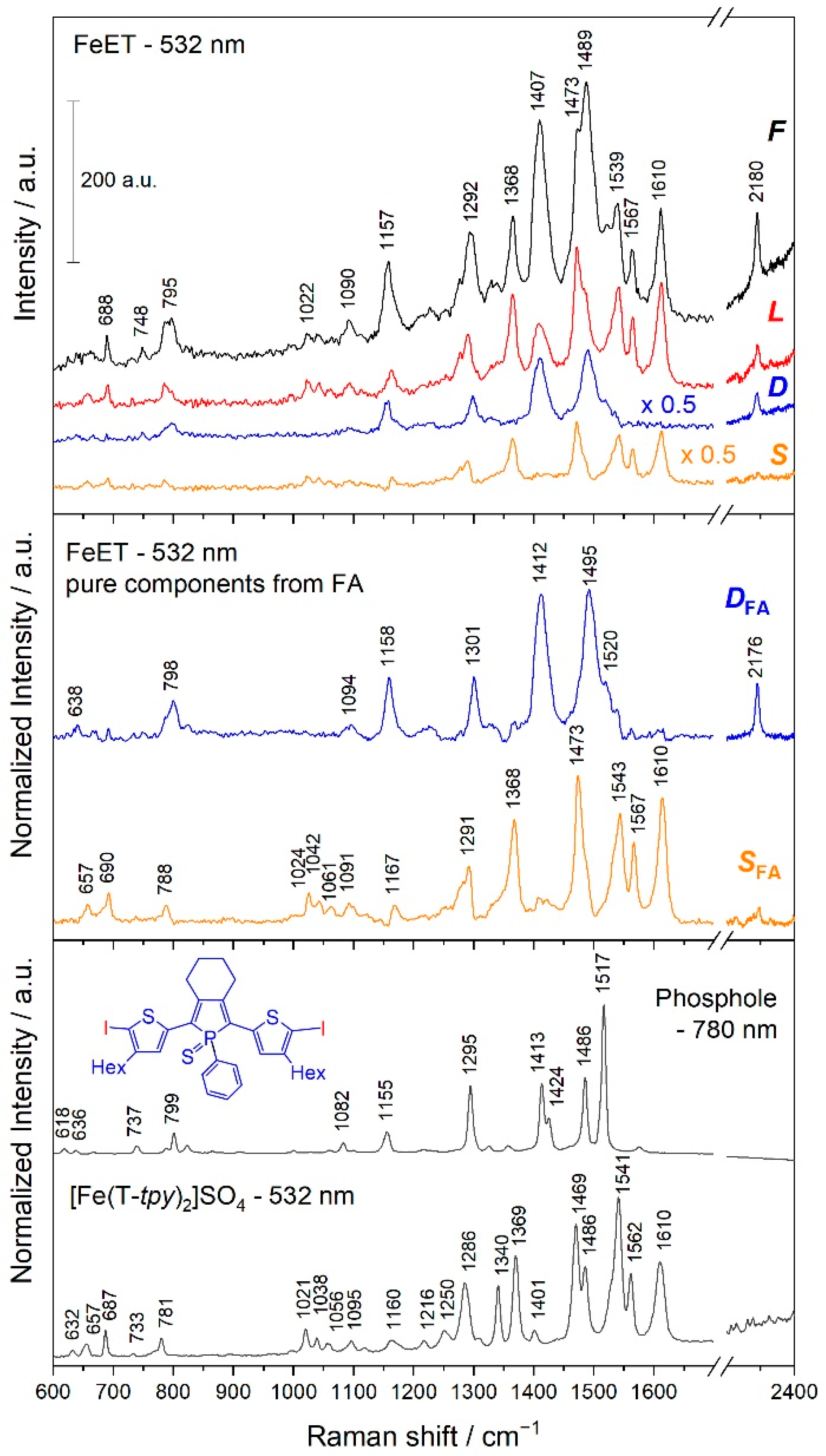
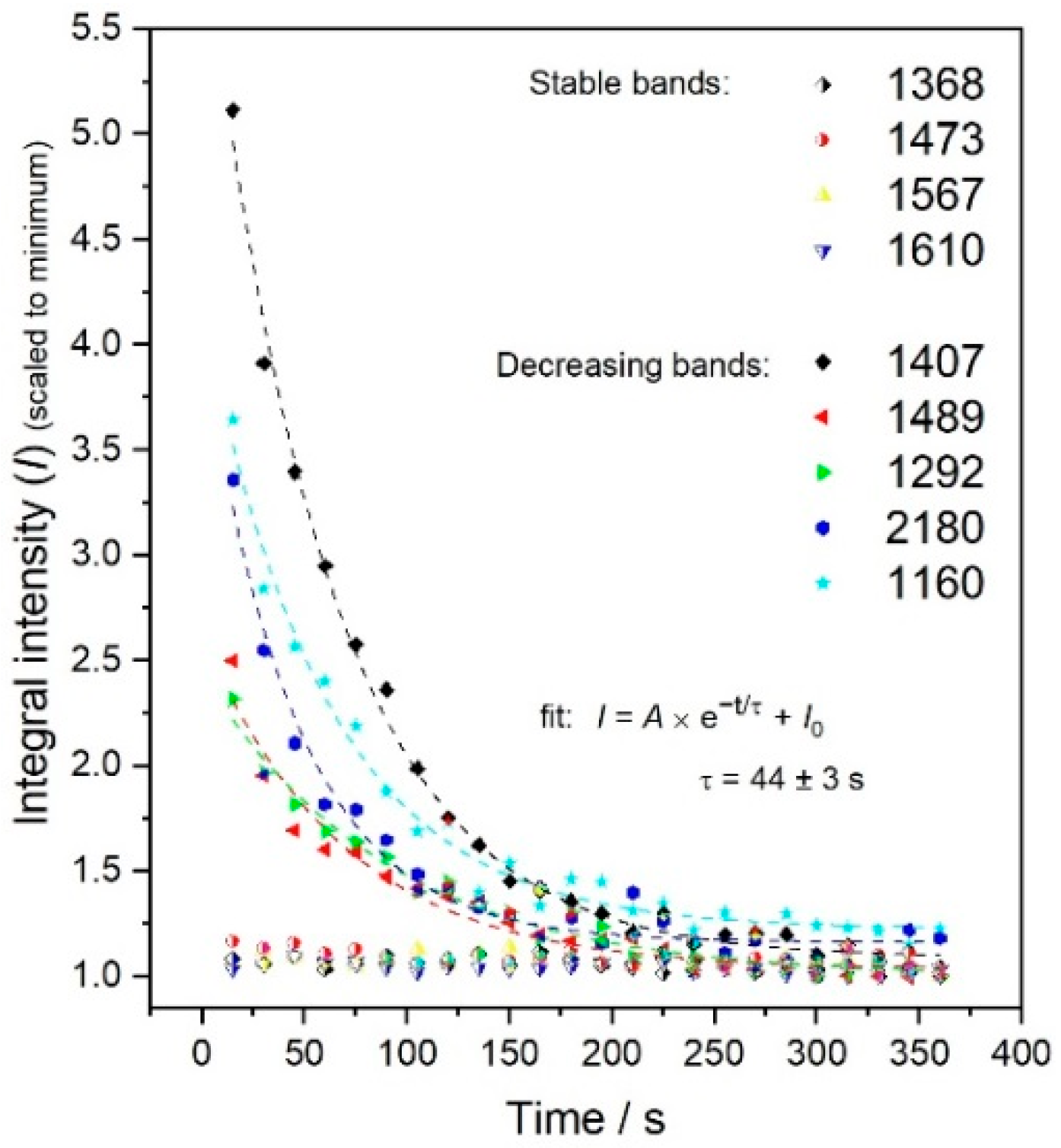
3.3. Analyses of the Spectral Changes
4. Discussion
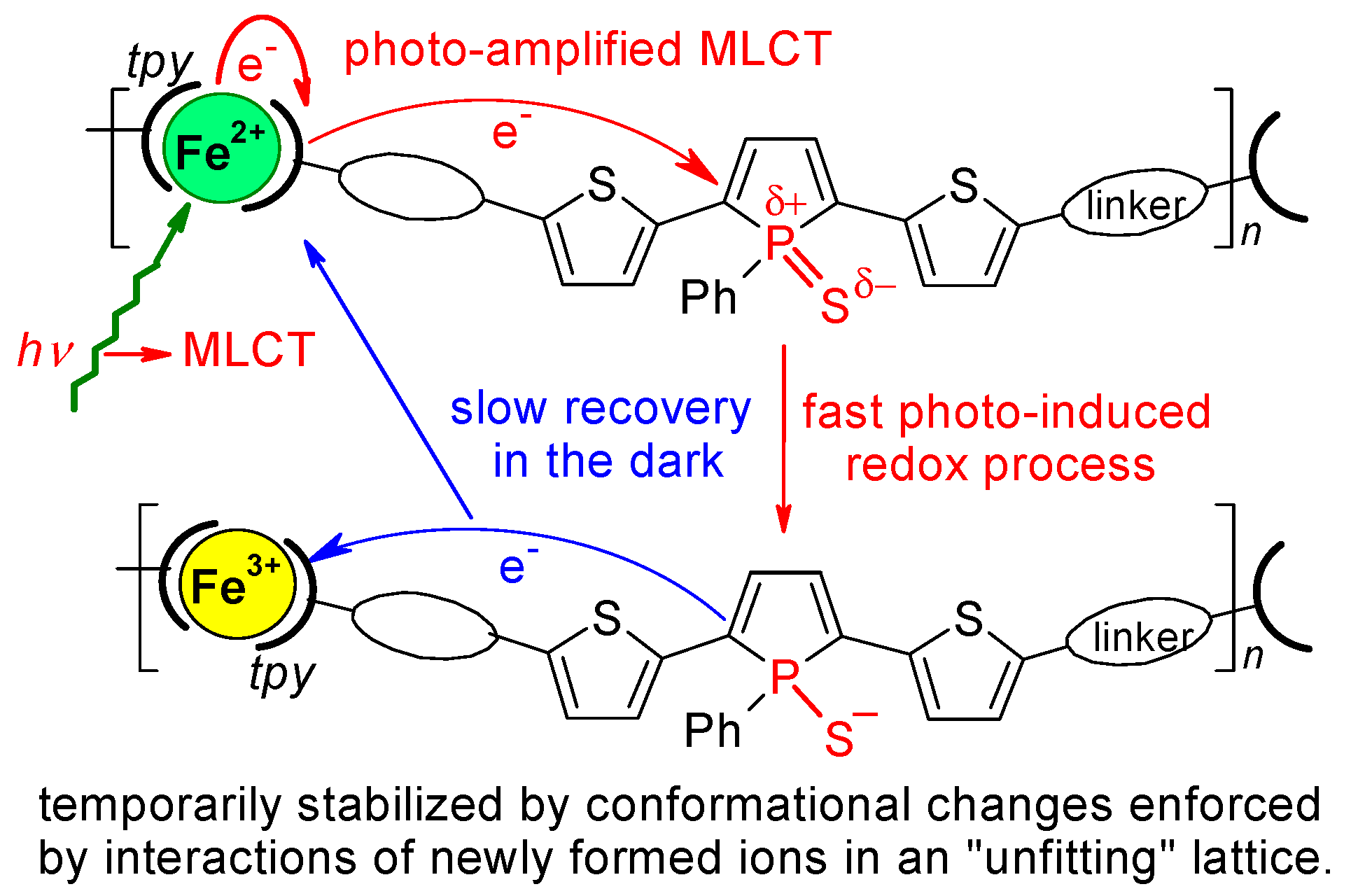
4.1. Structures Responsible for the Spectral Changes
4.2. Mechanistic Considerations
- (a)
- (b)
- Repeating units with newly formed ions Fe3+ and S− represent “structure islands”, with the distribution of localized charges significantly differing from the original distribution in the surrounding intact domains, which introduces instability into the lattice. The greater the number of new ions in the original lattice, the less stable this lattice is.
- (c)
- Stabilization of the disrupted lattice requires either restoring its original structure or creating a new structure by conformational changes and displacements of mobile counterions, which is not easy in a solid.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar] [CrossRef]
- Winter, A.; Schubert, U.S. Synthesis and Characterization of Metallo-Supramolecular Polymers. Chem. Soc. Rev. 2016, 45, 5311–5357. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, A. (Ed.) Supramolecular Polymers, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar] [CrossRef]
- Vohlidal, J.; Hissler, M. Metallo-Supramolecular Polymers. In Smart Inorganic Polymers: Synthesis, Properties, and Emerging Applications in Materials and Life Sciences; Evamarie, H.-H., Murriel, H., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp. 141–162. [Google Scholar] [CrossRef]
- Higuchi, M. Metallo-Supramolecular Polymers: Synthesis, Properties, and Device Applications; Springer: Tokyo, Japan, 2019. [Google Scholar] [CrossRef]
- Zhu, Y.; Zheng, W.; Wang, W.; Yang, H.-B. When Polymerization Meets Coordination-Driven Self-Assembly: Metallo-Supramolecular Polymers Based on Supramolecular Coordination Complexes. Chem. Soc. Rev. 2021, 50, 7395–7417. [Google Scholar] [CrossRef]
- Ciferri, A. Supramolecular Polymerizations. Macromol. Rapid Commun. 2002, 23, 511–529. [Google Scholar] [CrossRef]
- Lehn, J.-M. From Supramolecular Chemistry towards Constitutional Dynamic Chemistry and Adaptive Chemistry. Chem. Soc. Rev. 2007, 36, 151–160. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, F.; Lu, C.; Zhu, J.; Ke, C.; Han, S.; Zhuang, X. A Terpyridine-Fe2+-Based Coordination Polymer Film for On-Chip Micro-Supercapacitor with AC Line-Filtering Performance. Polymers 2021, 13, 1002. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Chakraborty, C. Interfacial Coordination Nanosheet Based on Nonconjugated Three-Arm Terpyridine: A Highly Color-Efficient Electrochromic Material to Converge Fast Switching with Long Optical Memory. ACS Appl. Mater. Interfaces 2020, 12, 35181–35192. [Google Scholar] [CrossRef]
- Liu, Y.; Sakamoto, R.; Ho, C.-L.; Nishihara, H.; Wong, W.-Y. Electrochromic Triphenylamine-Based Cobalt(II) Complex Nanosheets. J. Mater. Chem. C 2019, 7, 9159–9166. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Takada, K.; Sakamoto, R.; Matsuoka, R.; Toyoda, R.; Maeda, H.; Yagi, T.; Nishikawa, M.; Shinjo, N.; Amano, S.; et al. Coordination Nanosheets Based on Terpyridine-Zinc(II) Complexes: As Photoactive Host Materials. J. Am. Chem. Soc. 2017, 139, 5359–5366. [Google Scholar] [CrossRef]
- Sakamoto, R.; Hoshiko, K.; Liu, Q.; Yagi, T.; Nagayama, T.; Kusaka, S.; Tsuchiya, M.; Kitagawa, Y.; Wong, W.-Y.; Nishihara, H. A Photofunctional Bottom-up Bis(Dipyrrinato)Zinc(II) Complex Nanosheet. Nat. Commun. 2015, 6, 6713. [Google Scholar] [CrossRef] [Green Version]
- Whittell, G.R.; Hager, M.D.; Schubert, U.S.; Manners, I. Functional Soft Materials from Metallopolymers and Metallosupramolecular Polymers. Nat. Mater. 2011, 10, 176–188. [Google Scholar] [CrossRef]
- Chernyshev, A.; Acharya, U.; Pfleger, J.; Trhlíková, O.; Zedník, J.; Vohlídal, J. Iron (II) Metallo-Supramolecular Polymers Based on Thieno[3,2-b]Thiophene for Electrochromic Applications. Polymers 2021, 13, 362. [Google Scholar] [CrossRef]
- Schubert, U.S.; Hofmeier, H.; Newkome, G.R. Modern Terpyridine Chemistry; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar] [CrossRef]
- Hogg, R.; Wilkins, R.G. Exchange Studies of Certain Chelate Compounds of Transitional Metals. 8. 2,2′,2″-Terpyridine Complexes. J. Chem. Soc. 1962, 57, 341–350. [Google Scholar] [CrossRef]
- Bláhová, P.; Zedník, J.; Šloufová, I.; Vohlídal, J.; Svoboda, J. Synthesis and Photophysical Properties of New α,ω-Bis(Tpy)Oligothiophenes and Their Metallo-Supramolecular Polymers with Zn2+ Ion Couplers. Soft Mater. 2014, 12, 214–229. [Google Scholar] [CrossRef]
- Vitvarova, T.; Svoboda, J.; Hissler, M.; Vohlidal, J. Conjugated Metallo-Supramolecular Polymers Containing a Phosphole Unit. Organometallics 2017, 36, 777–786. [Google Scholar] [CrossRef]
- Han, F.S.; Higuchi, M.; Kurth, D.G. Metallosupramolecular Polyelectrolytes Self-Assembled from Various Pyridine Ring-Substituted Bisterpyridines and Metal Ions: Photophysical, Electrochemical, and Electrochromic Properties. J. Am. Chem. Soc. 2008, 130, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-C.; Chen, H.-B.; Zhang, Y.; Pei, J. Rigid Linear and Star-Shaped Pi-Conjugated 2,2′:6′,2″-Terpyridine Ligands with Blue Emission. Org. Lett. 2006, 8, 5700–5704. [Google Scholar] [CrossRef]
- Wild, A.; Schluetter, F.; Pavlov, G.M.; Friebe, C.; Festag, G.; Winter, A.; Hager, M.D.; Cimrova, V.; Schubert, U.S. Pi-Conjugated Donor and Donor Acceptor Metallo-Polymers. Macromol. Rapid Commun. 2010, 31, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Schmelz, O.; Rehahn, M. Synthesis of Rodlike Ruthenium(II) Coordination Polymers Having Metal-Nitrogen and Metal-Carbon Bonds in Their Main Chains. E-Polym. 2002, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Kelch, S.; Rehahn, M. Synthesis and Properties in Solution of Rodlike, 2,2′:6′,2″-Terpyridine-Based Ruthenium(II) Coordination Polymers. Macromolecules 1999, 32, 5818–5828. [Google Scholar] [CrossRef]
- Hrma, M.; Šichová, K.; Svoboda, J.; Vohlídal, J. Assembling of Bis(Tpy)Fluorenes with Zn2+ and Fe2+ Ions into Metallo-Supramolecular Polymers with Highly Efficient White-Light Emission. Polymer 2017, 122, 22–23. [Google Scholar] [CrossRef]
- Schluetter, F.; Wild, A.; Winter, A.; Hager, M.D.; Baumgaertel, A.; Friebe, C.; Schubert, U.S. Synthesis and Characterization of New Self-Assembled Metallo-Polymers Containing Electron-Withdrawing and Electron-Donating Bis(Terpyridine) Zinc(II) Moieties. Macromolecules 2010, 43, 2759–2771. [Google Scholar] [CrossRef]
- Han, F.-S.; Higuchi, M.; Kurth, D.G. Synthesis of Pi-Conjugated, Pyridine Ring Functionalized Bis-Terpyridines with Efficient Green, Blue, and Purple Emission. Tetrahedron 2008, 64, 9108–9116. [Google Scholar] [CrossRef]
- Stenclova, P.; Sichova, K.; Sloufova, I.; Zednik, J.; Vohlidal, J.; Svoboda, J. Alcohol- and Water-Soluble Bis(Tpy)Quaterthiophenes with Phosphonium Side Groups: New Conjugated Units for Metallo-Supramolecular Polymers. Dalt. Trans. 2016, 45, 1208–1224. [Google Scholar] [CrossRef] [Green Version]
- Stenclova-Blahova, P.; Svoboda, J.; Sloufova, I.; Vohlidal, J. Alcohol-Soluble Bis(Tpy)Thiophenes: New Building Units for Constitutional Dynamic Conjugated Polyelectrolytes. Phys. Chem. Chem. Phys. 2015, 17, 13743–13756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svoboda, J.; Stenclova, P.; Uhlik, F.; Zednik, J.; Vohlidal, J. Synthesis and Photophysical Properties of α,ω-Bis(Terpyridine)Oligothiophenes. Tetrahedron 2011, 67, 75–79. [Google Scholar] [CrossRef]
- Liang, Y.; Strohecker, D.; Lynch, V.; Holliday, B.J.; Jones, R.A. A Thiophene-Containing Conductive Metallopolymer Using an Fe(II) Bis(Terpyridine) Core for Electrochromic Materials. ACS Appl. Mater. Interfaces 2016, 8, 34568–34580. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.; Schott, M.; Niklaus, L.; Posset, U.; Kurth, D.G. A Study of the Effect of Pyridine Linkers on the Viscosity and Electrochromic Properties of Metallo-Supramolecular Coordination Polymers. J. Mater. Chem. C 2018, 6, 3310–3321. [Google Scholar] [CrossRef]
- Bai, D.R.; Romero-Nieto, C.; Baumgartner, T. Highly Luminescent Terpyridinyl-Ethynyl Functionalized Dithieno [3,2-b:2’,3’-d]Phospholes: Synthesis, Properties and Complexation Behavior. Dalt. Trans. 2010, 39, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, M.G.; Baumgartner, T. Recent Developments in Phosphole-Containing Oligo- and Polythiophene Materials. Eur. J. Inorg. Chem. 2007, 23, 3611–3628. [Google Scholar] [CrossRef]
- Matano, Y.; Imahoria, H. Design and Synthesis of Phosphole-Based Pi Systems for Novel Organic Materials. Org. Biomol. Chem. 2009, 7, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Matano, Y.; Miyajima, T.; Ochi, N.; Nakabuchi, T.; Shiro, M.; Nakao, Y.; Sakaki, S.; Imahori, H. Syntheses, Structures, and Coordination Chemistry of Phosphole-Containing Hybrid Calixphyrins: Promising Macrocyclic P,N-2,X-Mixed Donor Ligands for Designing Reactive Transition-Metal Complexes. J. Am. Chem. Soc. 2008, 130, 990–1002. [Google Scholar] [CrossRef]
- Nyulaszi, L. Aromaticity of Phosphorus Heterocycles. Chem. Rev. 2001, 101, 1229–1246. [Google Scholar] [CrossRef] [PubMed]
- Nyulaszi, L.; Holloczki, O.; Lescop, C.; Hissler, M.; Reau, R. An Aromatic-Antiaromatic Switch in P-Heteroles. A Small Change in Delocalisation Makes a Big Reactivity Difference. Org. Biomol. Chem. 2006, 4, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Fave, C.; Hissler, M.; Karpati, T.; Rault-Berthelot, J.; Deborde, V.; Toupet, L.; Nyulaszi, L.; Reau, R. Connecting Pi-Chromophores by Sigma-P-P Bonds: New Type of Assemblies Exhibiting Sigma-Pi-Conjugation. J. Am. Chem. Soc. 2004, 126, 6058–6063. [Google Scholar] [CrossRef] [PubMed]
- Salzner, U.; Lagowski, J.B.; Pickup, P.G.; Poirier, R.A. Comparison of Geometries and Electronic Structures of Polyacetylene, Polyborole, Polycyclopentadiene, Polypyrrole, Polyfuran, Polysilole, Polyphosphole, Polythiophene, Polyselenophene and Polytellurophene. Synth. Met. 1998, 96, 177–189. [Google Scholar] [CrossRef]
- Sebastian, M.; Hissler, M.; Fave, C.; Rault-Berthelot, J.; Odin, C.; Reau, R. Phosphole-Modified Poly(Thiophene)s: Unique Postfunctionalizable Conjugated Polymers That Sense Elemental Chalcogenides. Angew. Chem. Int. Ed. 2006, 45, 6152–6155. [Google Scholar] [CrossRef] [PubMed]
- Fadhel, O.; Gras, M.; Lemaitre, N.; Deborde, V.; Hissler, M.; Geffroy, B.; Reau, R. Tunable Organophosphorus Dopants for Bright White Organic Light-Emitting Diodes with Simple Structures. Adv. Mater. 2009, 21, 1261–1265. [Google Scholar] [CrossRef]
- Casado, J.; Réau, R.; López Navarrete, J.T. Tuning of Electronic Properties in Thienyl-Phosphole $π$-Conjugated Systems through P-Functionalization Monitored by Raman Spectroscopy. Chem. A Eur. J. 2006, 12, 3759–3767. [Google Scholar] [CrossRef]
- Casado, J.; Réau, R.; Hernández, V.; Navarrete, J.T.L. Combined Raman, Electrochemical and DFT Studies on a Series of $α$, $α$′-Thiophene-Phosphole Oligomers and Their Corresponding Polymers. Synth. Met. 2005, 153, 249–252. [Google Scholar] [CrossRef]
- Harada, K.; Urabe, H.; Sato, F. Generation and Reactions of Low-Valent Titanium Alkoxide-Acetylene Complexes—A Practical Preparation of Allyl Alcohols. Tetrahedron Lett. 1995, 36, 3203–3206. [Google Scholar] [CrossRef]
- Vitvarova, T.; Zednik, J.; Blaha, M.; Vohlidal, J.; Svoboda, J. Effect of Ethynyl and 2-Thienyl Substituents on the Complexation of 4’-Substituted 2,2′:6′,2″-Terpyridines with Zn2+ and Fe2+ Ions, and the Spectroscopic Properties of the Ligands and Formed Complex Species. Eur. J. Inorg. Chem. 2012, 24, 3866–3874. [Google Scholar] [CrossRef]
- Grosshenny, V.; Romero, F.M.; Ziessel, R. Construction of Preorganized Polytopic Ligands via Palladium-Promoted Cross-Coupling Reactions. J. Org. Chem. 1997, 62, 1491–1500. [Google Scholar] [CrossRef]
- Constable, E.C.; Figgemeier, E.; Housecroft, C.E.; Kokatam, S.L.; Medlycott, E.A.; Neuburger, M.; Schaffner, S.; Zampese, J.A. Wiring Terpyridine: Approaches to Alkynylthienyl 2,2′:6′,2″-Terpyridines. Dalt. Trans. 2008, 47, 6752–6762. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.; Winter, A.; Schluetter, F.; Schubert, U.S. Advances in the Field of Pi-Conjugated 2,2′:6′,2′′-Terpyridines. Chem. Soc. Rev. 2011, 40, 1459–1511. [Google Scholar] [CrossRef]
- Sloufova, I.; Vlckova, B.; Prochazka, M.; Svoboda, J.; Vohlidal, J. Comparison of SERRS and RRS Excitation Profiles of [Fe(Tpy)₂]2+ (Tpy = 2,2′:6′,2″--Terpyridine) Supported by DFT Calculations: Effect of the Electrostatic Bonding to Chloride-Modified Ag Nanoparticles on Its Vibrational and Electronic Structure. J. Raman Spectrosc. 2014, 45, 338–348. [Google Scholar] [CrossRef]
- Dobrawa, R.; Wurthner, F. Metallosupramolecular Approach toward Functional Coordination Polymers. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 4981–4995. [Google Scholar] [CrossRef]
- Hofmeier, H.; Schubert, U.S. Recent Developments in the Supramolecular Chemistry of Terpyridine-Metal Complexes. Chem. Soc. Rev. 2004, 33, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Vohlidal, J.; Graeff, C.F.O.; Hiorns, R.C.; Jones, R.G.; Luscombe, C.; Schue, F.; Stingelin, N.; Walter, M.G. Glossary of Terms Relating to Electronic, Photonic and Magnetic Properties of Polymers (IUPAC Recommendations 2021). PURE Appl. Chem. 2022, 94, 15–69. [Google Scholar] [CrossRef]
- Palacký, J.; Mojzeš, P.; Bok, J. SVD-Based Method for Intensity Normalization, Background Correction and Solvent Subtraction in Raman Spectroscopy Exploiting the Properties of Water Stretching Vibrations. J. Raman Spectrosc. 2011, 42, 1528–1539. [Google Scholar] [CrossRef]
- Rais, D.; Menšík, M.; Štenclová-Bláhová, P.; Svoboda, J.; Vohlídal, J.; Pfleger, J. Time-Resolved Transient Optical Absorption Study of Bis(Terpyridyl)Oligothiophenes and Their Metallo-Supramolecular Polymers with Zn(II) Ion Couplers. J. Phys. Chem. A 2015, 119, 6203–6214. [Google Scholar] [CrossRef] [PubMed]
- Rais, D.; Pfleger, J.; Menšík, M.; Zhigunov, A.; Štenclová, P.; Svoboda, J.; Vohlídal, J. Singlet Fission in Thin Films of Metallo-Supramolecular Polymers with Ditopic Thiophene-Bridged Terpyridine Ligands. J. Mater. Chem. C 2017, 5, 8041–8051. [Google Scholar] [CrossRef] [Green Version]
- Hladysh, S.; Václavková, D.; Vrbata, D.; Bondarev, D.; Havlíček, D.; Svoboda, J.; Zedník, J.; Vohlídal, J. Synthesis and Characterization of Metallo-Supramolecular Polymers from Thiophene-Based Unimers Bearing Pybox Ligands. RSC Adv. 2017, 7, 10718–10728. [Google Scholar] [CrossRef] [Green Version]
- Kupfer, S.; Zedler, L.; Guthmuller, J.; Bode, S.; Hager, M.D.; Schubert, U.S.; Popp, J.; Graefe, S.; Dietzek, B. Self-Healing Mechanism of Metallopolymers Investigated by QM/MM Simulations and Raman Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 12422–12432. [Google Scholar] [CrossRef]
- Bode, S.; Zedler, L.; Schacher, F.H.; Dietzek, B.; Schmitt, M.; Popp, J.; Hager, M.D.; Schubert, U.S. Self-Healing Polymer Coatings Based on Crosslinked Metallosupramolecular Copolymers. Adv. Mater. 2013, 25, 1634–1638. [Google Scholar] [CrossRef]
- Bousseksou, A.; McGarvey, J.J.; Varret, F.; Real, J.A.; Tuchagues, J.P.; Dennis, A.C.; Boillot, M.L. Raman Spectroscopy of the High- and Low-Spin States of the Spin Crossover Complex Fe(Phen)₂(NCS)₂: An Initial Approach to Estimation of Vibrational Contributions to the Associated Entropy Change. Chem. Phys. Lett. 2000, 318, 409–416. [Google Scholar] [CrossRef]
- Guetlich, P.; Gaspar, A.B.; Garcia, Y. Spin State Switching in Iron Coordination Compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef] [Green Version]
- Khusniyarov, M.M. How to Switch Spin-Crossover Metal Complexes at Constant Room Temperature. Chem. A Eur. J. 2016, 22, 15178–15191. [Google Scholar] [CrossRef] [PubMed]
- Devid, E.J.; Martinho, P.N.; Kamalakar, M.V.; Šalitroš, I.; Prendergast, Ú.; Dayen, J.F.; Meded, V.; Lemma, T.; González-Prieto, R.; Evers, F.; et al. Spin Transition in Arrays of Gold Nanoparticles and Spin Crossover Molecules. ACS Nano 2015, 9, 4496–4507. [Google Scholar] [CrossRef]
- Attwood, M.; Turner, S.S. Back to Back 2,6-Bis(Pyrazol-1-Yl)Pyridine and 2,2′:6′,2″-Terpyridine Ligands: Untapped Potential for Spin Crossover Research and Beyond. Coord. Chem. Rev. 2017, 353, 247–277. [Google Scholar] [CrossRef]
- Kittel, C. Kittel’s Introduction to Solid State Physics, 8th ed.; Global Edition; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unimer | λUV, nm | λMLCT, nm | λF, nm (Φ, %) | ∆νS, cm–1 | ||||
|---|---|---|---|---|---|---|---|---|
| Polymer | Solution | Film | Solution | Film | Solution | Film | Solution | Film |
| TPT [19] | 482 (ε = 29) | 520 | - | - | 603 (19%) | 685 (0.7%) | 4160 | 4630 |
| ZnTPT | 507 (ε = 39) | 509 | - | - | 634 | 641 (0.5%) | 3950 | 4100 |
| FeTPT | 491 (ε = 22) | 499 | 625 (ε = 50) | 626 | - | - | - | - |
| E | 497 (ε = 33) | 511 | - | - | 623 (9%) | 705 (0.2%) | 4070 | 5390 |
| ZnE | 518 (ε = 43) | 533 | - | - | 642 | 702 (7%) | 3730 | 4520 |
| FeE | 508 (ε = 29) | 511 | 609 (ε = 66) | 616 | - | - | - | - |
| EPh | 500 (ε = 42) | 526 | - | - | 635(15%) | 669 (0.3%) | 4250 | 4060 |
| ZnEPh | 505 (ε = 51) | 515 | - | - | 649 | 673 (4%) | 4350 | 4560 |
| FeEPh | 510 (ε = 51) | 518 | 580 (ε = 74) | 590 | - | - | - | - |
| ET | 506 (ε = 60) | 545 | - | - | 641 (22%) | 730 (0.3%) | 4160 | 4650 |
| ZnET | 513 (ε = 66) | 533 | - | - | 680 | 723 (3%) | 4710 | 4950 |
| FeET | 518 (ε = 62) | 538 | 601 (ε = 99) | 615 | - | - | - | - |
| ET6 | 505 (ε = 43) | 538 | - | - | 642 (8%) | 700 (0.2%) | 4230 | 4300 |
| ZnET6 | 508 (ε = 45) | 535 | - | - | 674 | 692 (5%) | 4850 | 4490 |
| FeET6 | 518 (ε = 45) | 540 | 584 (ε = 61) | 592 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šloufová, I.; Urválková, T.; Hissler, M.; Vohlídal, J. Novel Metallo-Supramolecular Polymers with 1-Thioxophosphole Main-Chain Units and Remarkable Photoinduced Changes in Their Resonance Raman Spectra. Polymers 2022, 14, 5207. https://doi.org/10.3390/polym14235207
Šloufová I, Urválková T, Hissler M, Vohlídal J. Novel Metallo-Supramolecular Polymers with 1-Thioxophosphole Main-Chain Units and Remarkable Photoinduced Changes in Their Resonance Raman Spectra. Polymers. 2022; 14(23):5207. https://doi.org/10.3390/polym14235207
Chicago/Turabian StyleŠloufová, Ivana, Tereza Urválková, Muriel Hissler, and Jiří Vohlídal. 2022. "Novel Metallo-Supramolecular Polymers with 1-Thioxophosphole Main-Chain Units and Remarkable Photoinduced Changes in Their Resonance Raman Spectra" Polymers 14, no. 23: 5207. https://doi.org/10.3390/polym14235207