

Tailored Supersaturable Immediate Release Behaviors of Hypotensive Supersaturating Drug-Delivery Systems Combined with Hot-Melt Extrusion Technique and Self-Micellizing Polymer
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Development of Various faSDDS Systems Based on Hot-Melt Extrusion Technique and Other Discontinuous Approaches
2.2.1. Fabrication of faSDDSHME Based on Hot-Melt Extrusion Technique
2.2.2. Fabrication of faSDDSSE Based on Solvent Evaporation Technique
2.2.3. Fabrication of faSDDSQC Based on Microwave Irradiation-Quench Cooling Technique
2.2.4. Preparation of Physical Mixtures and Pure Amorphous FEL
2.3. Solid-State Characterization
2.3.1. Scanning Electron Microscopy (SEM) Experimentation
2.3.2. Powder X-ray Diffraction (PXRD)
2.4. Initial Release in Various Media Environments Measured by Dissolution
2.5. Evolved Release during Longer Dissolution Period Monitored by “Spring-Parachute” Process
2.5.1. Semi-Continuous Determinations
2.5.2. Process Quantification
2.6. Terminal Release in Various Media Environments Measured by Solubility
2.7. Stability of faSDDS and Possible Mechanisms for Supersaturable Immediate Release
2.7.1. Stability of the Amorphous State
2.7.2. Fourier Transform Infrared Spectroscopy (FT-IR)
2.7.3. Simulated Crystallization Assay
2.7.4. Phase Solubility
2.8. Data Analysis
3. Results and Discussion
3.1. Fabrication and Characterization of Various faSDDS Systems
3.2. Tailored Supersaturable Immediate-Release Behaviors
3.2.1. Initial Release Behavior via Dissolution Measurement
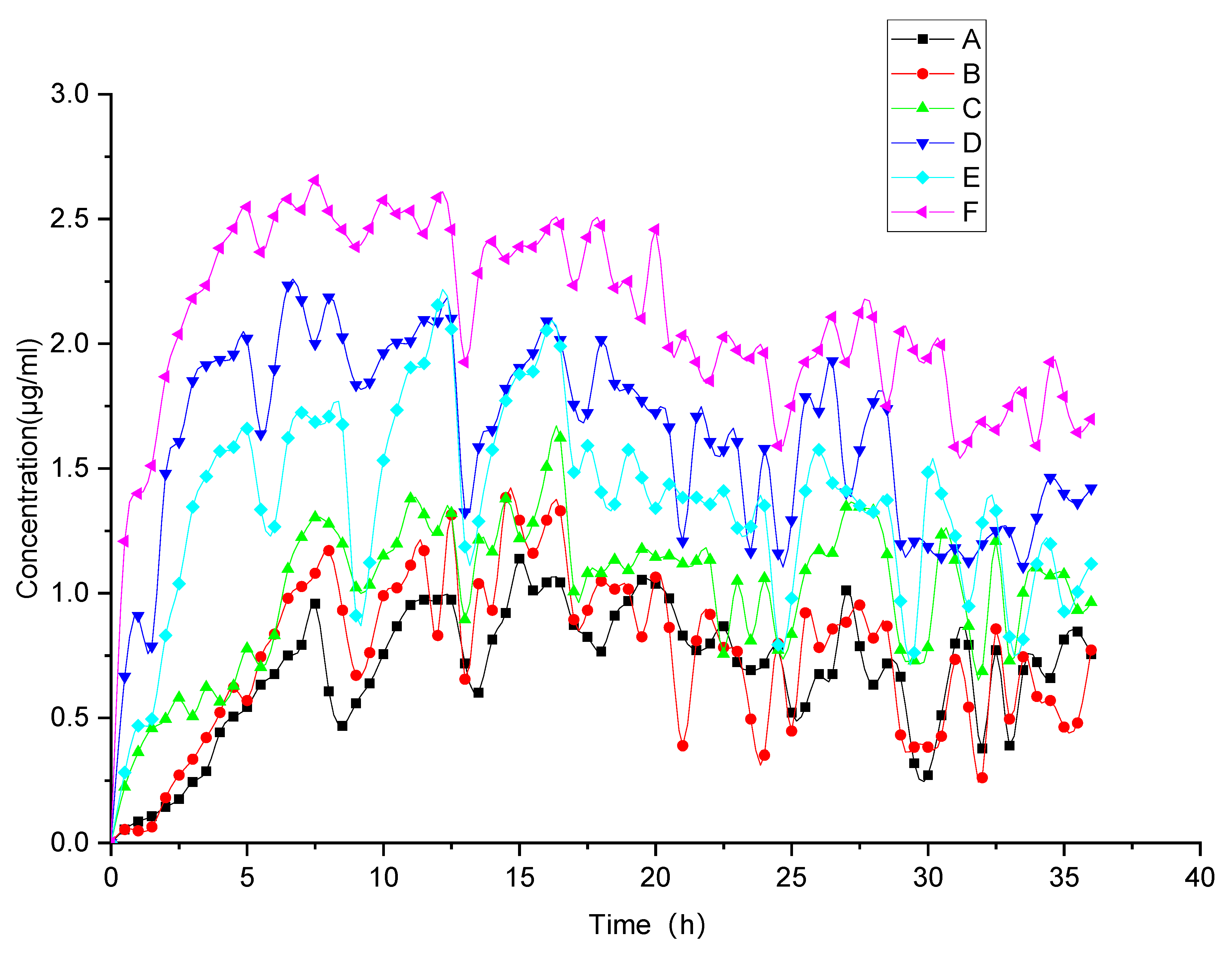
3.2.2. Evolved Release Behavior via Monitoring of the “Spring-Parachute” Process
3.2.3. Terminal Release Behavior via Solubility Measurement
3.3. Stability of faSDDSs and Possible Mechanisms Controlling Supersaturable Immediate Release
3.3.1. Stability of Various faSDDS Systems
3.3.2. Underlying Molecular Interaction Facilitates the Immediate Release of faSDDS Systems
3.3.3. Soluplus® in faSDDS Can Inhibit FEL Crystallization from a Supersaturated State and Improve Immediate Release
3.3.4. Self-Micellizing Soluplus® Itself Could Solubilize FEL in a Favorable/Spontaneous Manner and Enhance Immediate Release in faSDDS Systems
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thakore, S.D.; Sirvi, A.; Joshi, V.C.; Panigrahi, S.S.; Manna, A.; Singh, R.; Sangamwar, A.T.; Bansal, A.K. Biorelevant dissolution testing and physiologically based absorption modeling to predict in vivo performance of supersaturating drug delivery systems. Int. J. Pharm. 2021, 607, 120958. [Google Scholar] [CrossRef]
- Charalabidis, A.; Sfouni, M.; Bergstrom, C.; Macheras, P. The Biopharmaceutics Classification System (BCS) and the Biopharmaceutics Drug Disposition Classification System (BDDCS): Beyond guidelines. Int. J. Pharm. 2019, 566, 264–281. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, H.; Fathalla, Z. Investigation into the Emerging Role of the Basic Amino Acid L-Lysine in Enhancing Solubility and Permeability of BCS Class II and BCS Class IV Drugs. Pharm. Res. 2018, 35, 160. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.A.; Williams, R.O., 3rd. Specific mechanical energy—An essential parameter in the processing of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2021, 173, 374–393. [Google Scholar] [CrossRef]
- Yang, L.; Wu, P.; Xu, J.; Xie, D.; Wang, Z.; Wang, Q.; Chen, Y.; Li, C.H.; Zhang, J.; Chen, H.; et al. Development of Apremilast Solid Dispersion Using TPGS and PVPVA with Enhanced Solubility and Bioavailability. AAPS PharmSciTech 2021, 22, 142. [Google Scholar] [CrossRef]
- Xu, S.; Dai, W.G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef]
- Kawakami, K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv. Drug Deliv. Rev. 2012, 64, 480–495. [Google Scholar] [CrossRef]
- Kuentz, M. Drug supersaturation during formulation digestion, including real-time analytical approaches. Adv. Drug Deliv. Rev. 2019, 142, 50–61. [Google Scholar] [CrossRef]
- Shi, N.Q.; Zhou, J.; Walker, J.; Li, L.; Hong, J.K.Y.; Olsen, K.F.; Tang, J.; Ackermann, R.; Wang, Y.; Qin, B.; et al. Microencapsulation of luteinizing hormone-releasing hormone agonist in poly (lactic-co-glycolic acid) microspheres by spray-drying. J. Control Release 2020, 321, 756–772. [Google Scholar] [CrossRef]
- Tran, P.H.L.; Tran, T.T.D. Dosage form designs for the controlled drug release of solid dispersions. Int. J. Pharm. 2020, 581, 119274. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Drug release mechanisms from amorphous solid dispersions. J. Control Release 2015, 211, 171. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.R.; Ma, Y.; Lee, P.I. Impact of phase separation morphology on release mechanism of amorphous solid dispersions. Eur. J. Pharm. Sci. 2019, 136, 104955. [Google Scholar] [CrossRef]
- Fong, S.Y.; Bauer-Brandl, A.; Brandl, M. Oral bioavailability enhancement through supersaturation: An update and meta-analysis. Expert Opin. Drug Deliv. 2017, 14, 403–426. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Haste Makes Waste: The Interplay Between Dissolution and Precipitation of Supersaturating Formulations. AAPS J. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Yang, Y.; Covington, R.A.; Bi, Y.V.; Durig, T.; Ilies, M.A.; Fassihi, R. Supersaturated controlled release matrix using amorphous dispersions of glipizide. Int. J. Pharm. 2016, 511, 957–968. [Google Scholar] [PubMed]
- Suzuki, H.; Ueno, K.; Mizumoto, T.; Seto, Y.; Sato, H.; Onoue, S. Self-micellizing solid dispersion of cyclosporine A for pulmonary delivery: Physicochemical, pharmacokinetic and safety assessments. Eur. J. Pharm. Sci. 2017, 96, 107–114. [Google Scholar] [CrossRef]
- Torrado-Salmeron, C.; Guarnizo-Herrero, V.; Cerezo-Garreta, J.; Torrado Duran, G.; Torrado-Santiago, S. Self-Micellizing Technology Improves the Properties of Ezetimibe and Increases Its Effect on Hyperlipidemic Rats. Pharmaceutics 2019, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Kojo, Y.; Matsunaga, S.; Suzuki, H.; Sato, H.; Seto, Y.; Onoue, S. Improved oral absorption profile of itraconazole in hypochlorhydria by self-micellizing solid dispersion approach. Eur. J. Pharm. Sci. 2017, 97, 55–61. [Google Scholar] [CrossRef]
- Sundaramoorthy, P.; Baskaran, R.; Mishra, S.K.; Jeong, K.Y.; Oh, S.H.; Kyu Yoo, B.; Kim, H.M. Novel self-micellizing anticancer lipid nanoparticles induce cell death of colorectal cancer cells. Colloids Surf. B Biointerfaces 2015, 135, 793–801. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Lobmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef]
- Alleso, M.; Chieng, N.; Rehder, S.; Rantanen, J.; Rades, T.; Aaltonen, J. Enhanced dissolution rate and synchronized release of drugs in binary systems through formulation: Amorphous naproxen-cimetidine mixtures prepared by mechanical activation. J. Control Release 2009, 136, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Butreddy, A.; Bandari, S.; Repka, M.A. Quality-by-design in hot melt extrusion based amorphous solid dispersions: An industrial perspective on product development. Eur. J. Pharm. Sci. 2021, 158, 105655. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, part II: New strategies in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1663–1680. [Google Scholar] [CrossRef]
- Maurya, P.; Pandey, P.; Singh, S.; Sonkar, A.; Singh, S.; Saraf, S.A. Appraisal of Felodipine Nanocrystals for Solubility Enhancement and Pharmacodynamic Parameters on Cadmium Chloride Induced Hypertension in Rats. Curr. Drug Deliv. 2022, 19, 625–634. [Google Scholar]
- van der Lee, R.; Pfaffendorf, M.; Koopmans, R.P.; van Lieshout, J.J.; van Montfrans, G.A.; van Zwieten, P.A. Comparison of the time courses and potencies of the vasodilator effects of nifedipine and felodipine in the human forearm. Blood Press. 2001, 10, 217–222. [Google Scholar] [CrossRef]
- Prasad, R.; Radhakrishnan, P.; Singh, S.K.; Verma, P.R.P. Furosemide—Soluplus(R) Solid Dispersion: Development and Characterization. Recent Pat. Drug Deliv. Formul. 2017, 11, 211–220. [Google Scholar] [CrossRef]
- Hitzer, P.; Bauerle, T.; Drieschner, T.; Ostertag, E.; Paulsen, K.; van Lishaut, H.; Lorenz, G.; Rebner, K. Process analytical techniques for hot-melt extrusion and their application to amorphous solid dispersions. Anal. Bioanal. Chem. 2017, 409, 4321–4333. [Google Scholar] [CrossRef]
- Song, B.; Wang, J.; Lu, S.; Shan, L. Andrographolide solid dispersions formulated by Soluplus to enhance interface wetting, dissolution, and absorption. J. Appl. Polym. Sci. 2020, 137, 48354. [Google Scholar] [CrossRef]
- Zi, P.; Zhang, C.; Ju, C.; Su, Z.; Bao, Y.; Gao, J.; Sun, J.; Lu, J.; Zhang, C. Solubility and bioavailability enhancement study of lopinavir solid dispersion matrixed with a polymeric surfactant—Soluplus. Eur. J. Pharm. Sci. 2019, 134, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Almotairy, A.; Almutairi, M.; Althobaiti, A.; Alyahya, M.; Sarabu, S.; Alzahrani, A.; Zhang, F.; Bandari, S.; Repka, M.A. Effect of pH Modifiers on the Solubility, Dissolution Rate, and Stability of Telmisartan Solid Dispersions Produced by Hot-melt Extrusion Technology. J. Drug Deliv. Sci. Technol. 2021, 65. [Google Scholar] [CrossRef] [PubMed]
- Fael, H.; Demirel, A.L. Tannic acid as a co-former in co-amorphous systems: Enhancing their physical stability, solubility and dissolution behavior. Int. J. Pharm. 2020, 581, 119284. [Google Scholar] [CrossRef] [PubMed]
- Saboo, S.; Moseson, D.E.; Kestur, U.S.; Taylor, L.S. Patterns of drug release as a function of drug loading from amorphous solid dispersions: A comparison of five different polymers. Eur. J. Pharm. Sci. 2020, 155, 105514. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Brewster, M.E.; Augustijns, P. Evaluation of gastrointestinal drug supersaturation and precipitation: Strategies and issues. Int. J. Pharm. 2013, 453, 25–35. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Augustijns, P.; Brewster, M.E. Supersaturating drug delivery systems: Fast is not necessarily good enough. J. Pharm. Sci. 2012, 101, 7–9. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Zhang, G.G.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef]
- Peng, R.; Huang, J.; He, L.; Zhao, L.; Wang, C.; Wei, W.; Xia, T.; Mao, Y.; Wen, Y.; Wang, L.; et al. Polymer/lipid interplay in altering in vitro supersaturation and plasma concentration of a model poorly soluble drug. Eur. J. Pharm. Sci. 2020, 146, 105262. [Google Scholar] [CrossRef]
- Laitinen, R.; Lobmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef]
- Ahmad, N.; Ahmad, R.; Alam, M.A.; Ahmad, F.J.; Amir, M.; Pottoo, F.H.; Sarafroz, M.; Jafar, M.; Umar, K. Daunorubicin oral bioavailability enhancement by surface coated natural biodegradable macromolecule chitosan based polymeric nanoparticles. Int. J. Biol. Macromol. 2019, 128, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.C.; Pikal, M.J.; Taylor, L.S. A spectroscopic investigation of hydrogen bond patterns in crystalline and amorphous phases in dihydropyridine calcium channel blockers. Pharm. Res. 2002, 19, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Kestur, U.S.; Taylor, L.S. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. CrystEngComm 2010, 12, 2390. [Google Scholar] [CrossRef]
- Cun, D.; Zhang, C.; Bera, H.; Yang, M. Particle engineering principles and technologies for pharmaceutical biologics. Adv. Drug Deliv. Rev. 2021, 174, 140–167. [Google Scholar] [CrossRef]
- Seedher, N.; Bhatia, S. Solubility enhancement of Cox-2 inhibitors using various solvent systems. AAPS PharmSciTech 2003, 4, E33. [Google Scholar] [CrossRef] [Green Version]
- Cirri, M.; Mura, P.; Rabasco, A.M.; Gines, J.M.; Moyano, J.R.; Gonzalez-Rodriguez, M.L. Characterization of ibuproxam binary and ternary dispersions with hydrophilic carriers. Drug Dev. Ind. Pharm. 2004, 30, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.S.; Kumar, V.; Singh, U.P.; Bhat, H.R.; Mazumder, B. Physicochemical characterization and in vitro dissolution studies of solid dispersions of ketoprofen with PVP K30 and d-mannitol. Saudi Pharm. J. 2013, 21, 77–84. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | AUCspring-parachute (μg·h/mL) | Tmax (h) | Cmax (μg/mL) | C36h (μg/mL) |
|---|---|---|---|---|
| FEL | 24.80 | 15 | 1.14 | 0.76 |
| FELPM | 27.01 | 14.5 | 1.39 | 0.77 |
| FELPA | 36.34 | 16.5 | 1.62 | 0.96 |
| faSDDSSE | 58.41 | 6.5 | 2.23 | 1.42 |
| faSDDSQC | 48.63 | 12 | 2.15 | 1.17 |
| faSDDSHME | 70.52 | 7.5 | 2.65 | 1.70 |
| Concentration (mg/mL) of Pre-dissolved Soluplus® | Initial FEL Concentration (μg/mL) in Supersaturated Solution | Half-Time (min) of FEL Crystallization from a Supersaturated Sate |
|---|---|---|
| 0 | 55.56 | 8.38 ± 0.11 |
| 0.15 | 55.56 | 35.08 ± 0.41 |
| 0.20 | 55.56 | 44.41 ± 1.84 |
| 0.25 | 55.56 | 50.40 ± 7.73 |
| 0.30 | 55.56 | 89.68 ± 6.08 |
| Concentration (mg/mL) of Soluplus® | Drug | |
|---|---|---|
| 0.1 | FEL | −4.06 ± 0.31 |
| 0.3 | FEL | −6.98 ± 0.39 |
| 0.5 | FEL | −8.82 ± 0.55 |
| 0.7 | FEL | −9.76 ± 0.50 |
| 0.9 | FEL | −10.47 ± 0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Ma, Y.; Zhang, Y.; Zhang, H.; Zuo, L.; Hao, C.; Yu, W.; Lin, X.; Zhang, Y.; Qi, X.; et al. Tailored Supersaturable Immediate Release Behaviors of Hypotensive Supersaturating Drug-Delivery Systems Combined with Hot-Melt Extrusion Technique and Self-Micellizing Polymer. Polymers 2022, 14, 4800. https://doi.org/10.3390/polym14224800
Yu H, Ma Y, Zhang Y, Zhang H, Zuo L, Hao C, Yu W, Lin X, Zhang Y, Qi X, et al. Tailored Supersaturable Immediate Release Behaviors of Hypotensive Supersaturating Drug-Delivery Systems Combined with Hot-Melt Extrusion Technique and Self-Micellizing Polymer. Polymers. 2022; 14(22):4800. https://doi.org/10.3390/polym14224800
Chicago/Turabian StyleYu, Huan, Yinghui Ma, Yanfei Zhang, Huifeng Zhang, Lili Zuo, Chengyi Hao, Weilun Yu, Xiaoying Lin, Yong Zhang, Xianrong Qi, and et al. 2022. "Tailored Supersaturable Immediate Release Behaviors of Hypotensive Supersaturating Drug-Delivery Systems Combined with Hot-Melt Extrusion Technique and Self-Micellizing Polymer" Polymers 14, no. 22: 4800. https://doi.org/10.3390/polym14224800