
Integrated Approach to Eco-Friendly Thermoplastic Composites Based on Chemically Recycled PET Co-Polymers Reinforced with Treated Banana Fibres
 , , and
, , and
Abstract
:1. Introduction
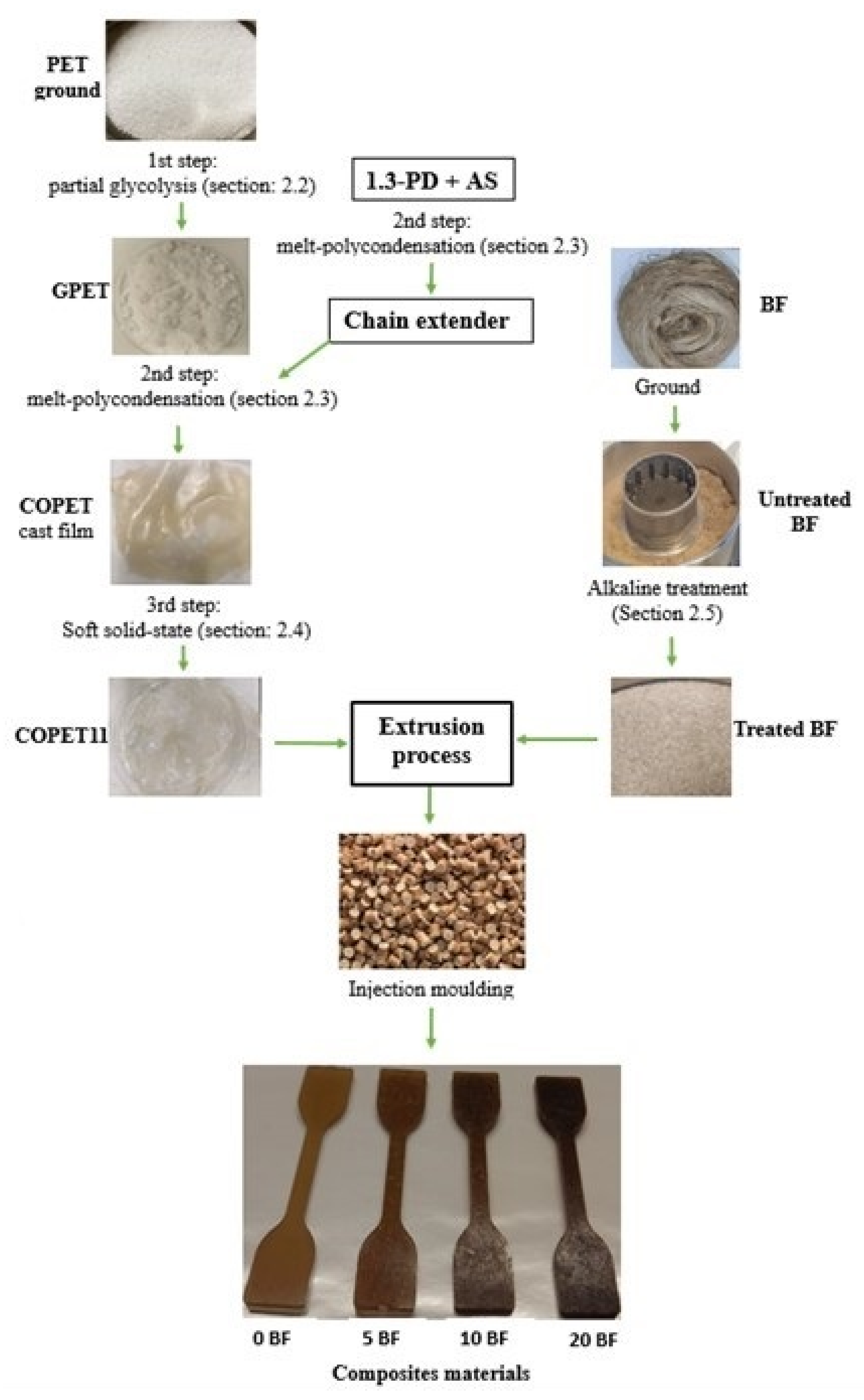
2. Materials and Methods
2.1. Materials
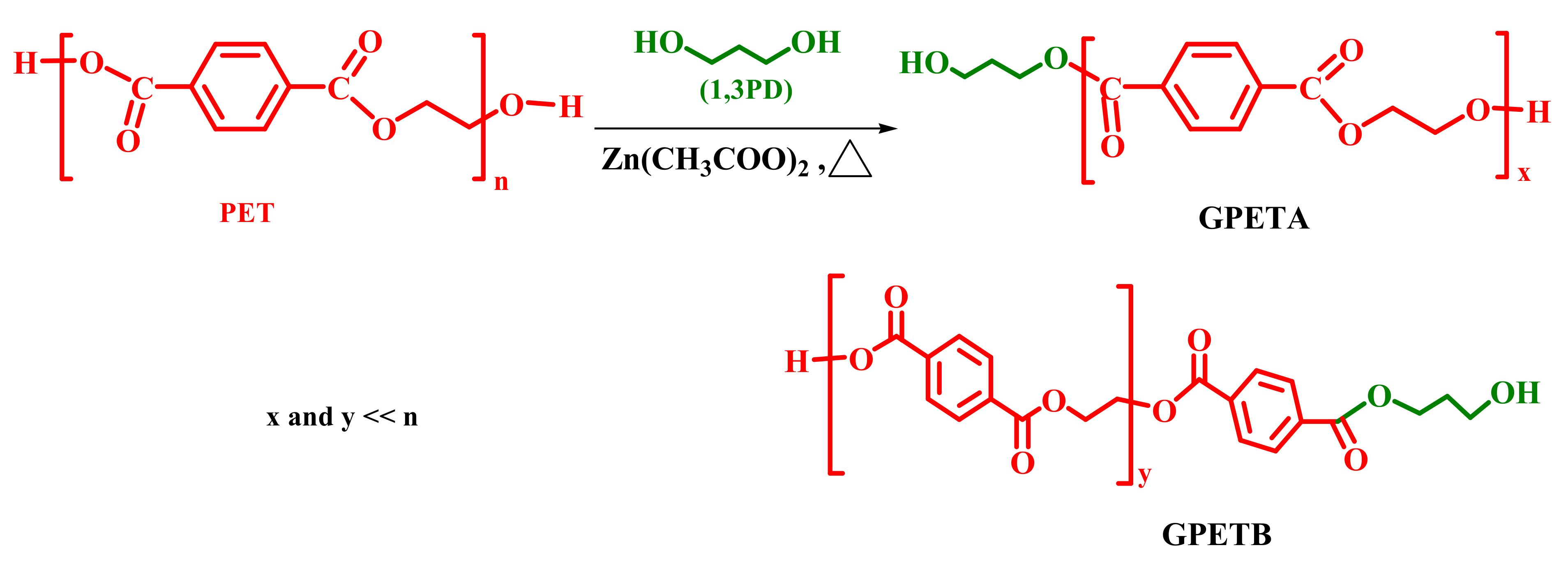
2.2. Glycolysis Reaction
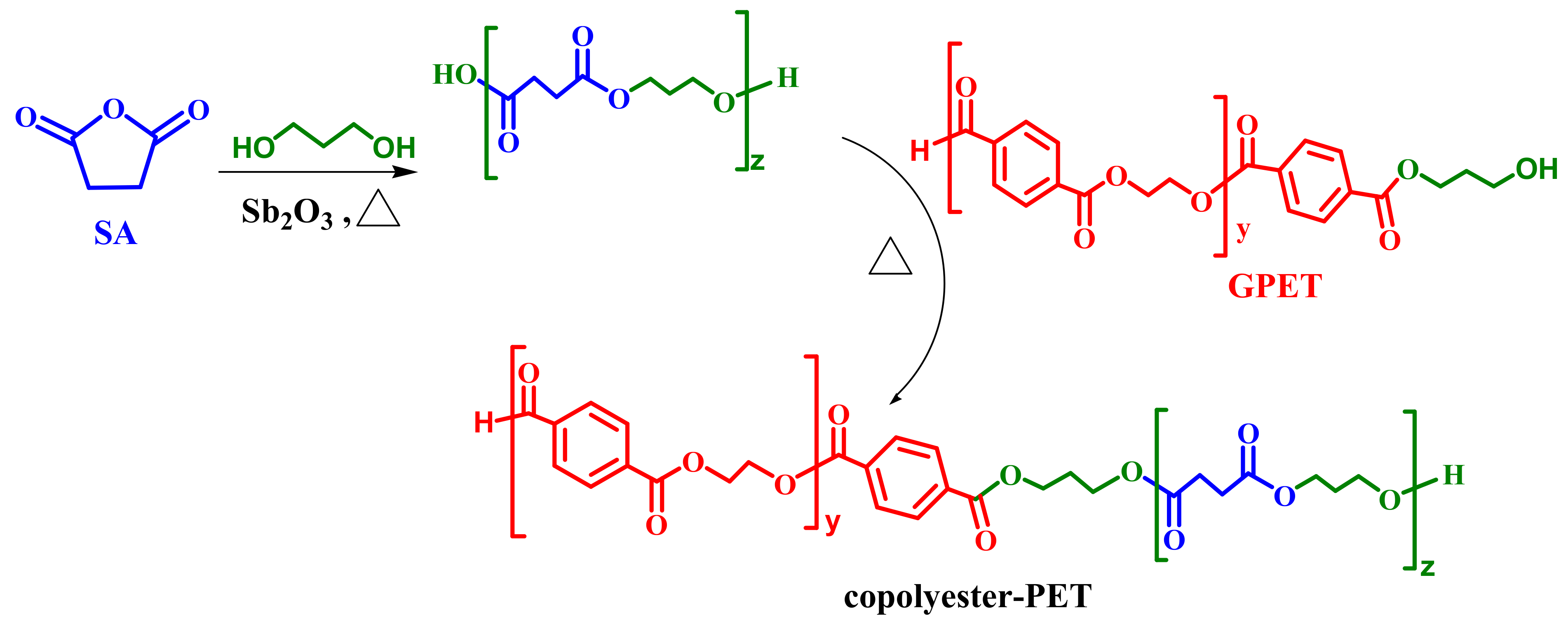
2.3. Co-Polyester Melt Condensation
2.4. Molar Mass Build-Up by Soft Solid-State Polycondensation (S3P)
2.5. Banana Fibres Treatment
2.6. Composites Processing
2.7. Characterisation Techniques
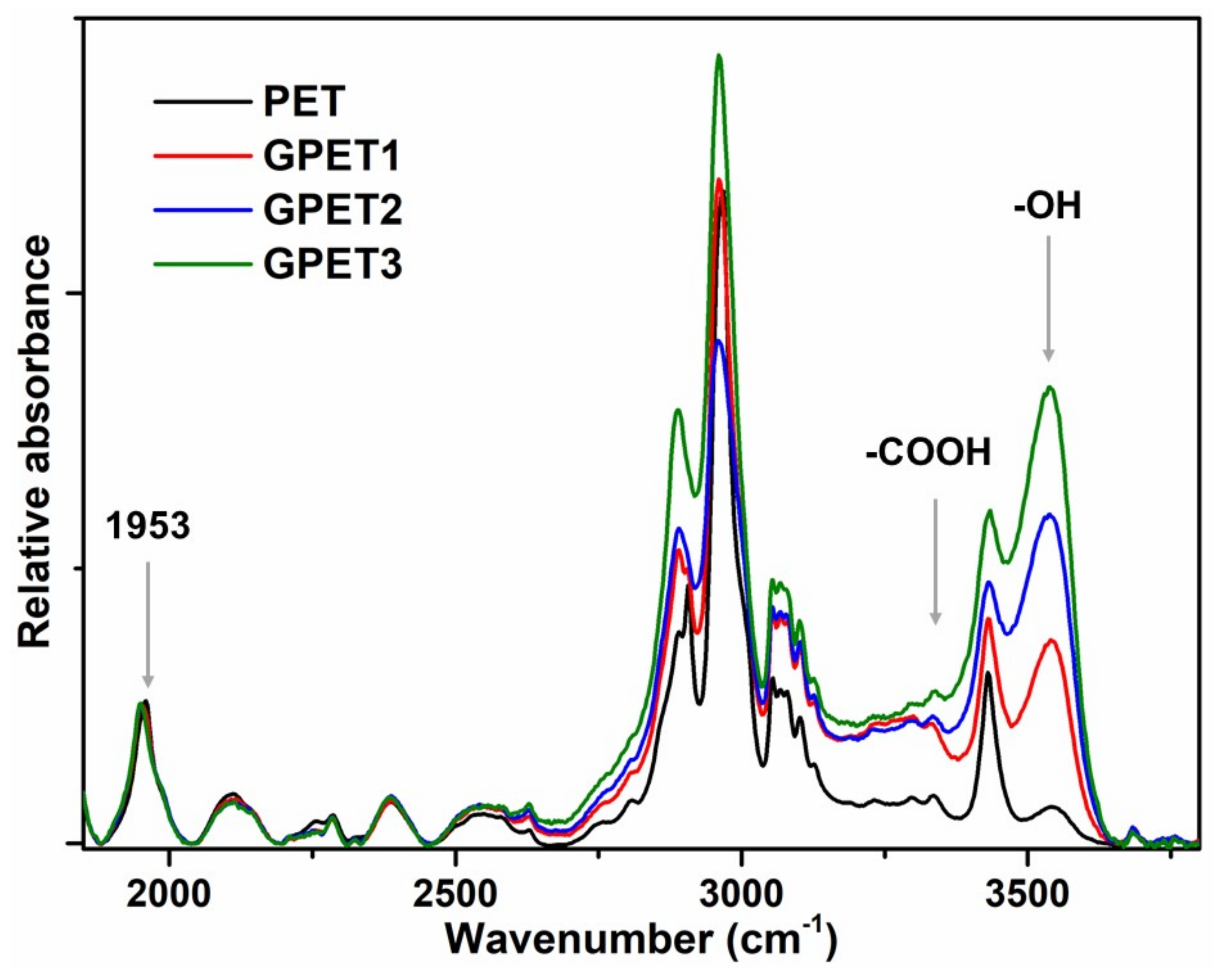
2.7.1. Fourier Transform Infra-Red Spectroscopy (FTIR) Analyses
2.7.2. Intrinsic Viscosity (IV)
2.7.3. Differential Scanning Calorimetry (DSC) Analyses
2.7.4. Thermogravimetric Analysis (TGA)
2.7.5. Dynamic Mechanical Analysis (DMA) and Tensile Tests
2.7.6. Scanning Electron Microscopy (SEM)
3. Results and Discussion
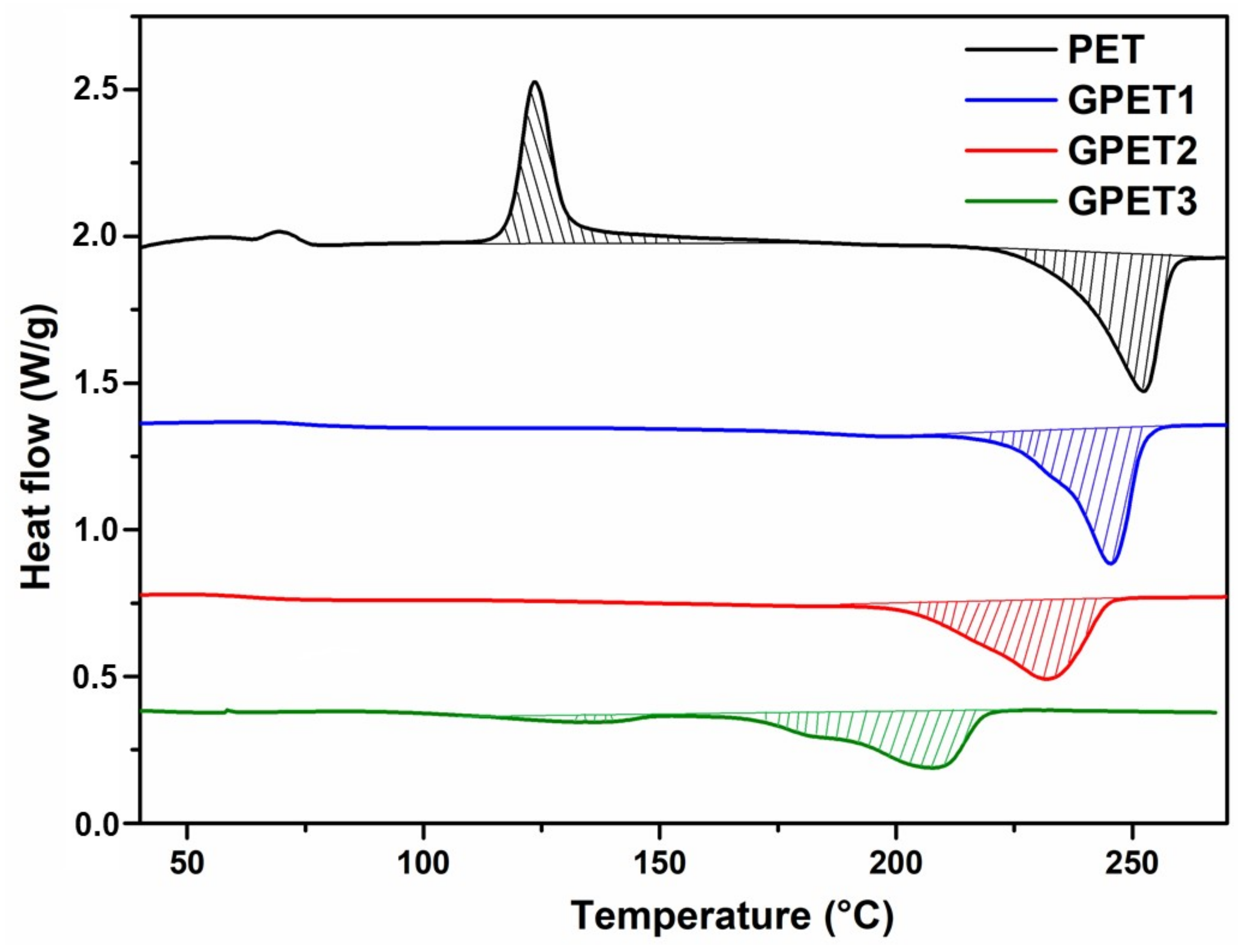
3.1. PET Glycolysis
3.2. Melt Reaction of Glycolysed PET (GPET3)
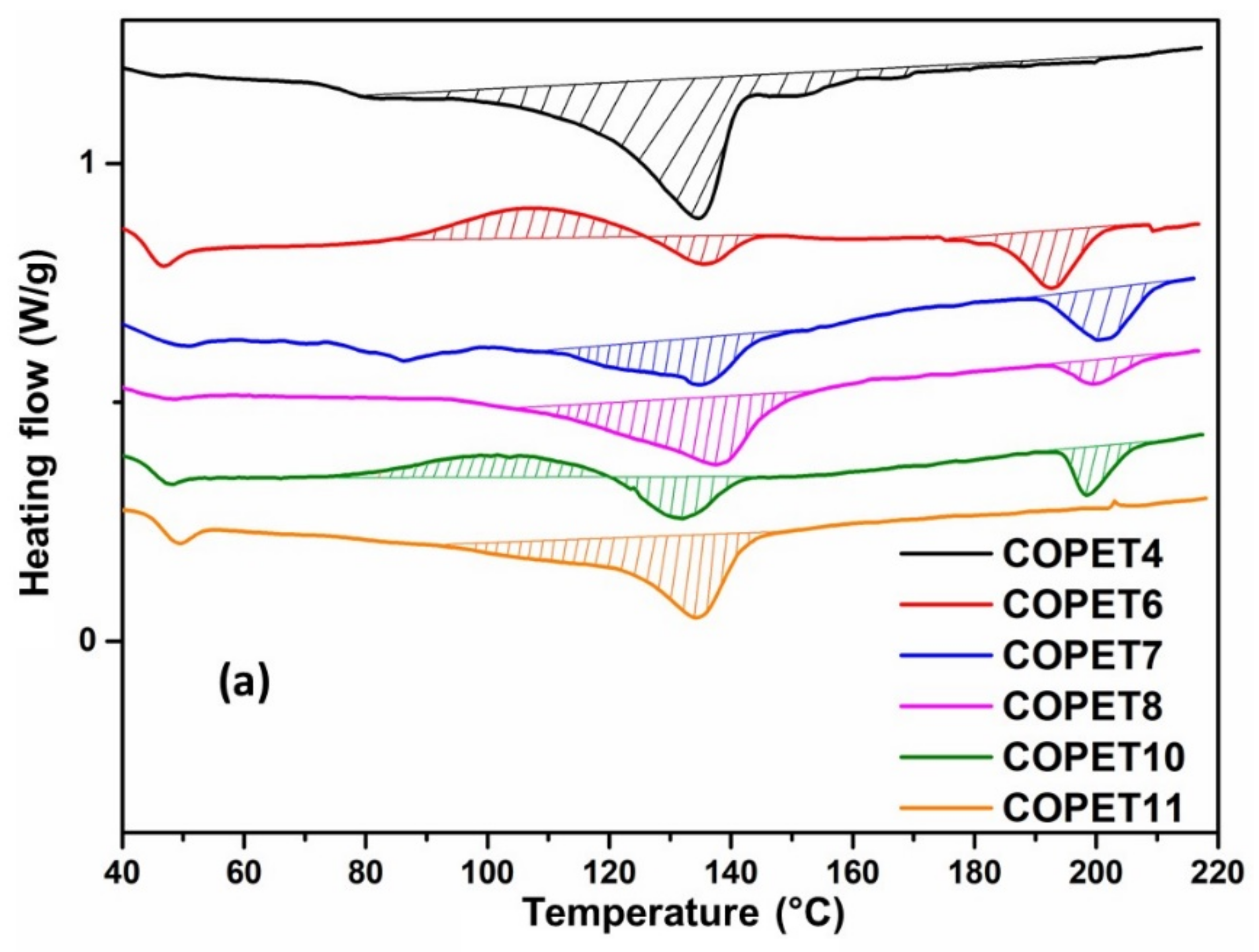
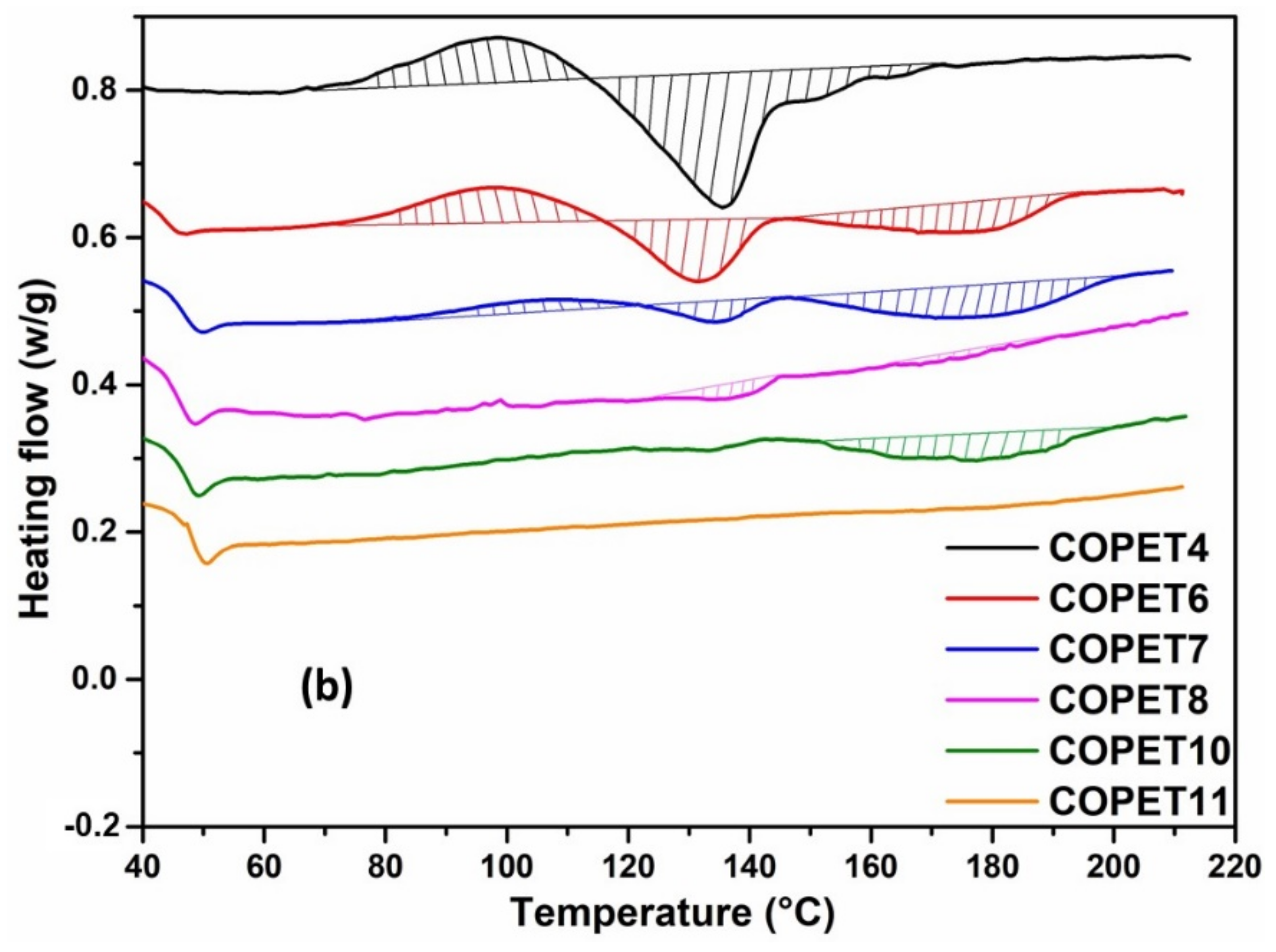
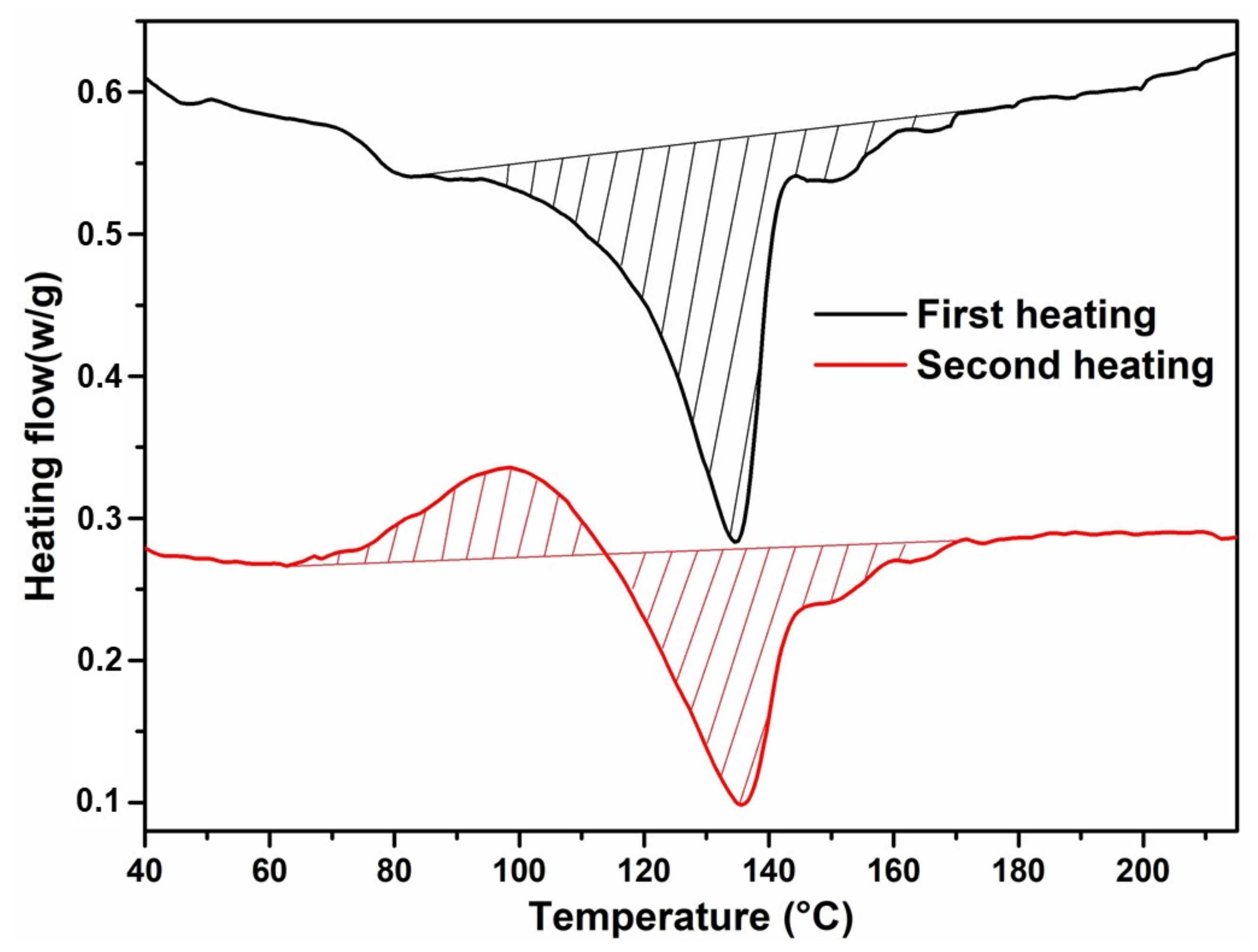
3.3. S3P and SSP of Melt Polymerised Co-Polyester COPET4
3.4. Banana Fibre Treatment and Morphology
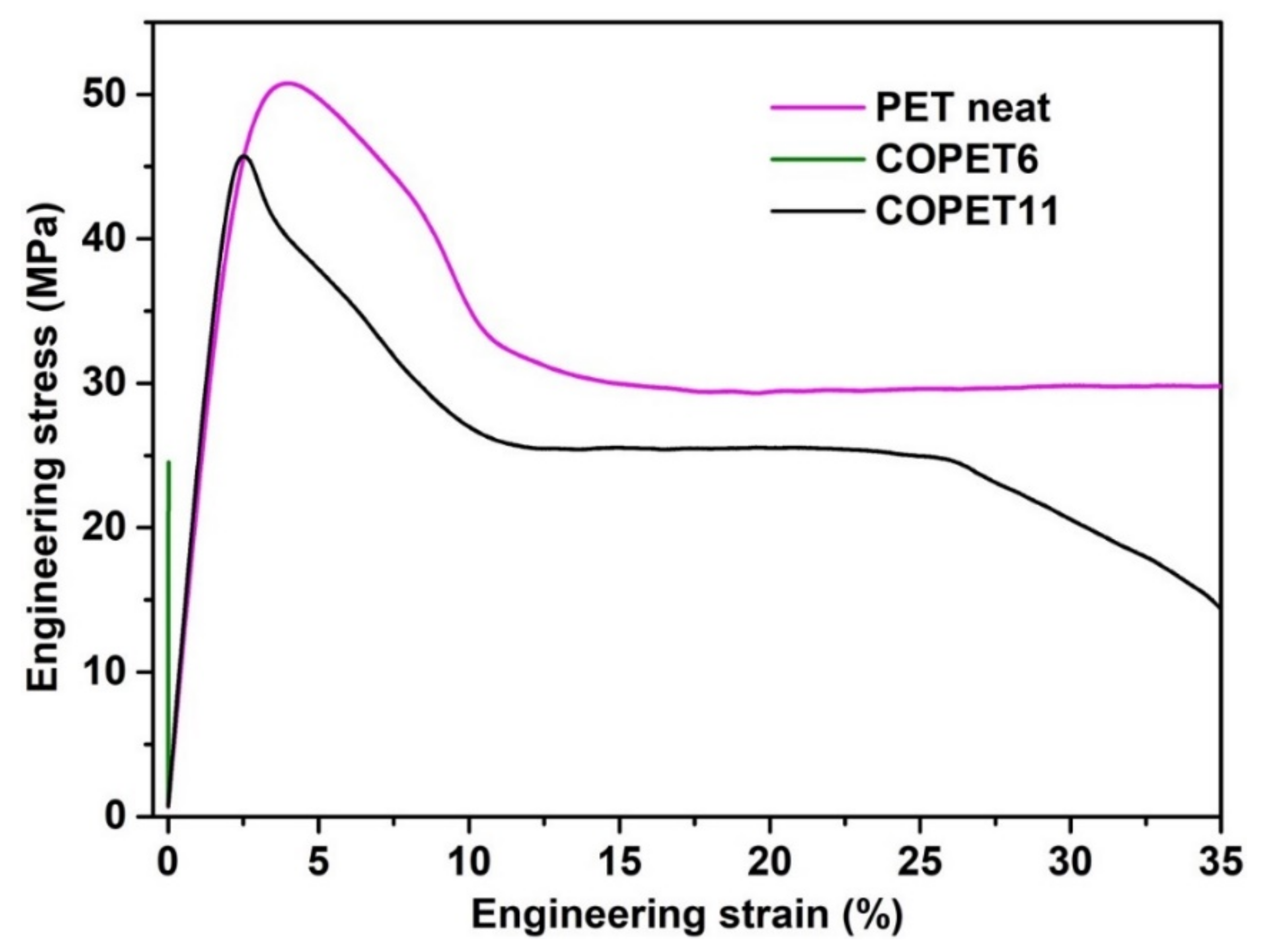
3.5. Tensile Properties of Co-Polyesters Compared with PET
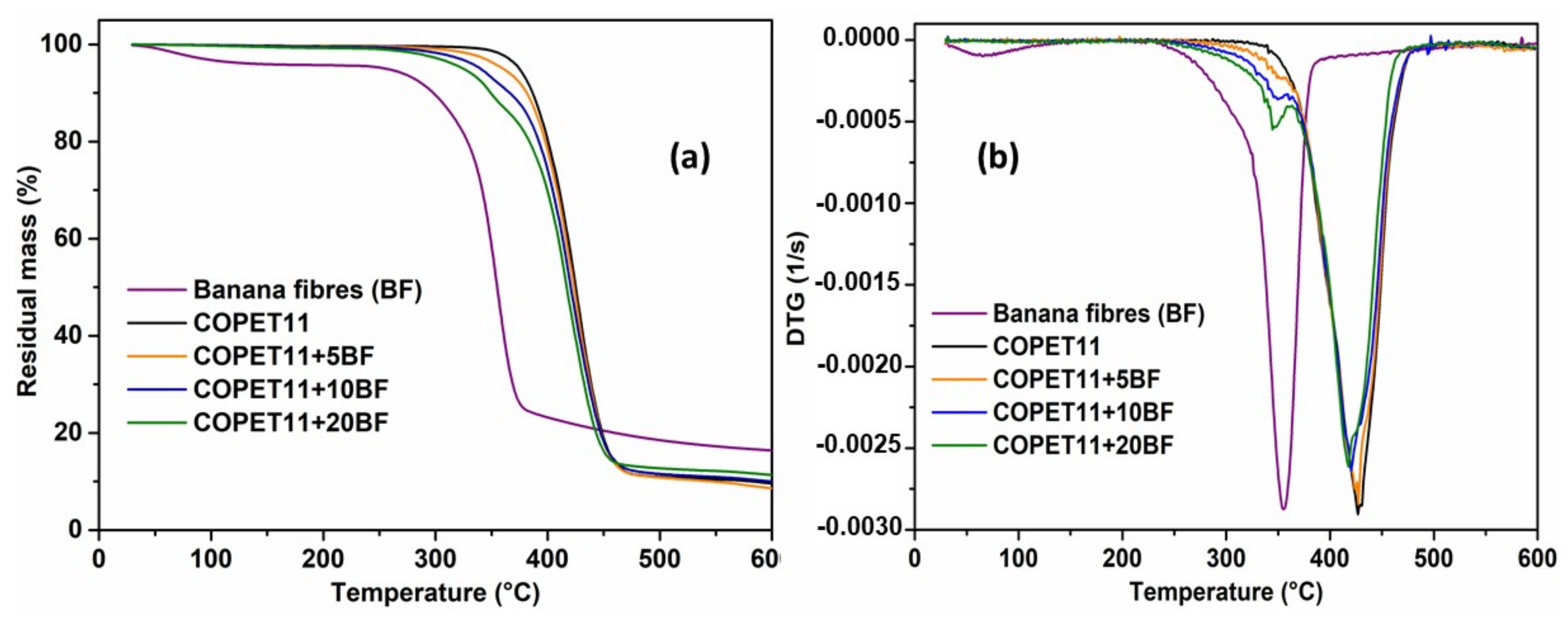
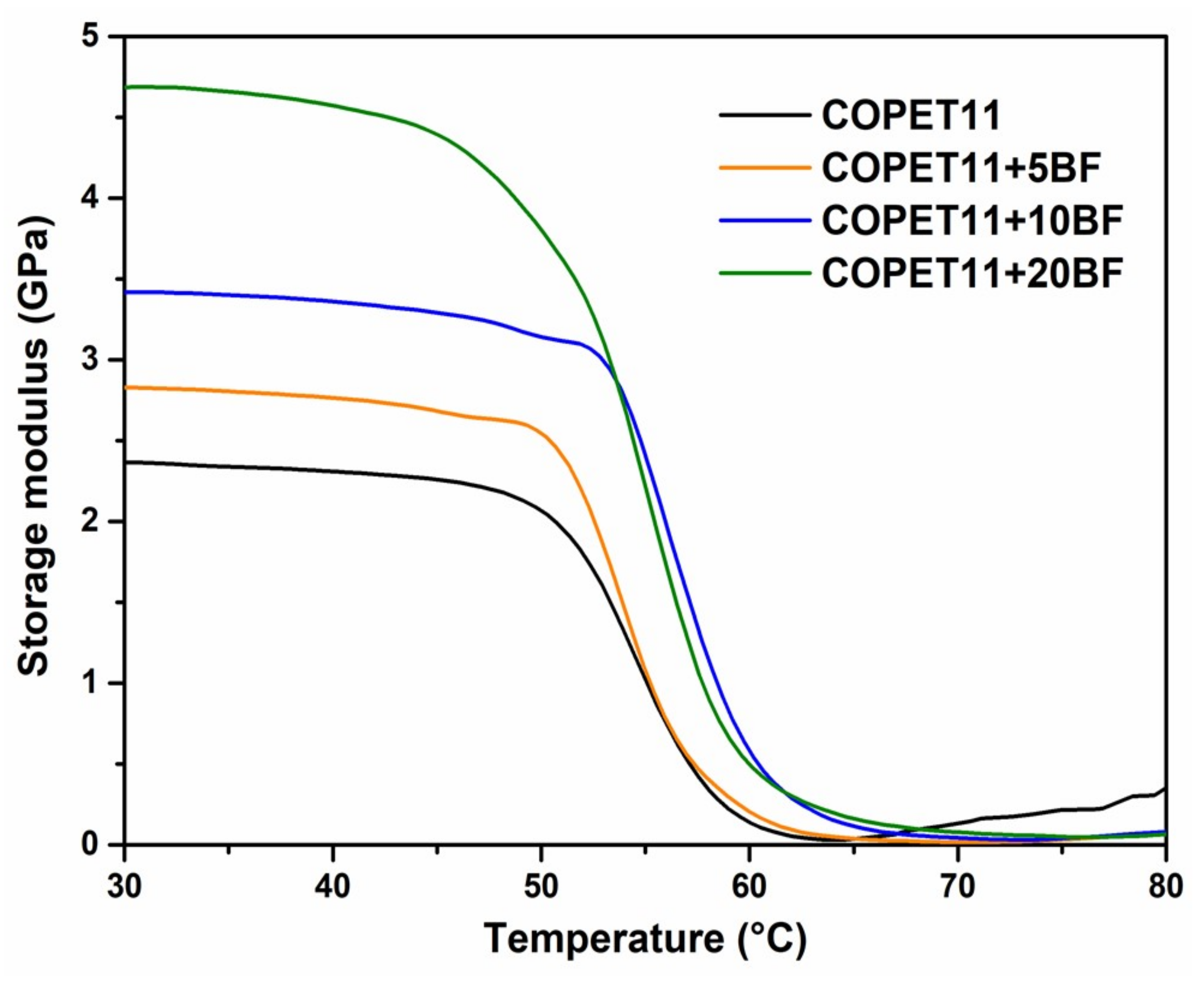
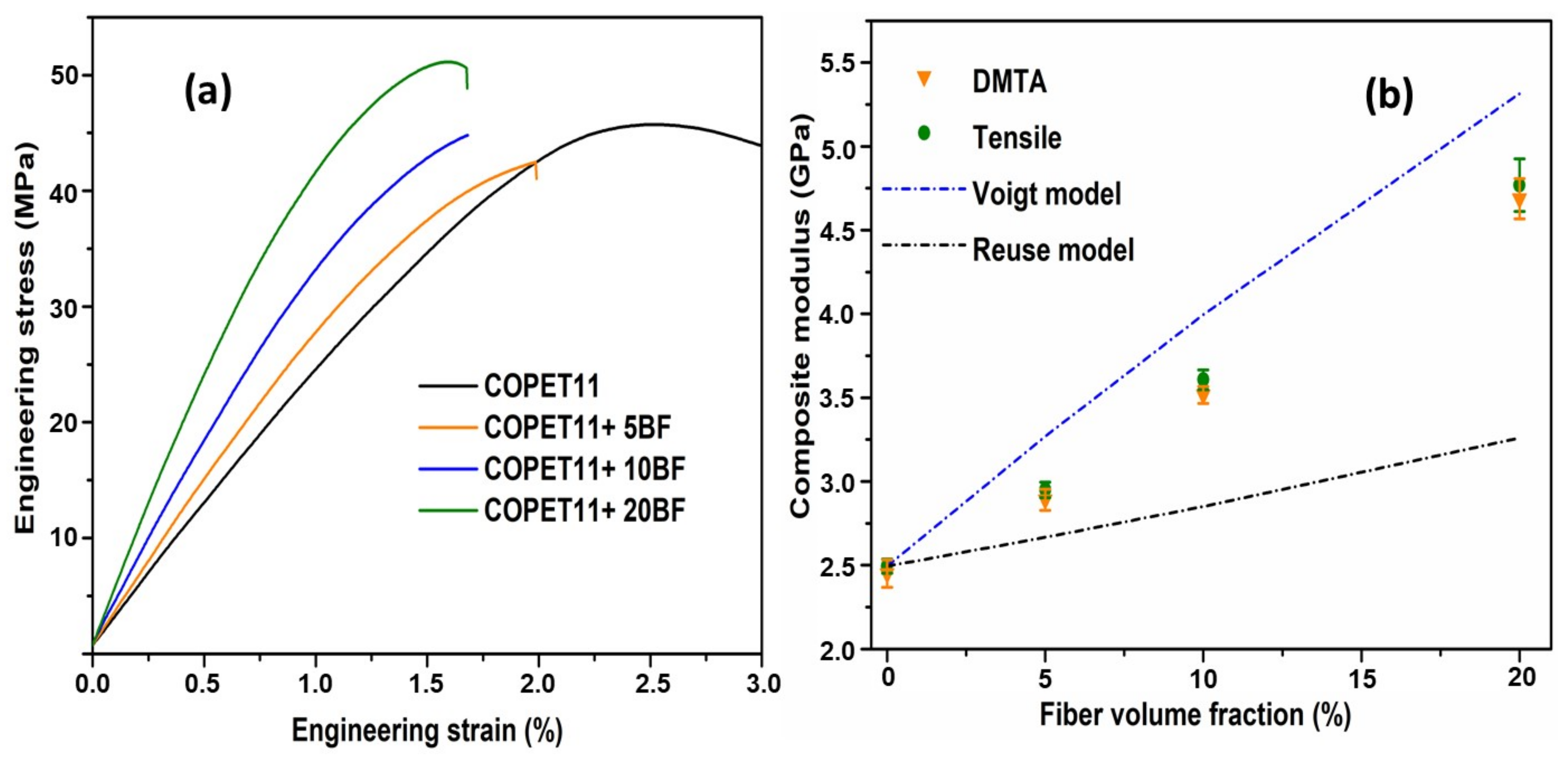
3.6. Properties and Morphology of the Composites
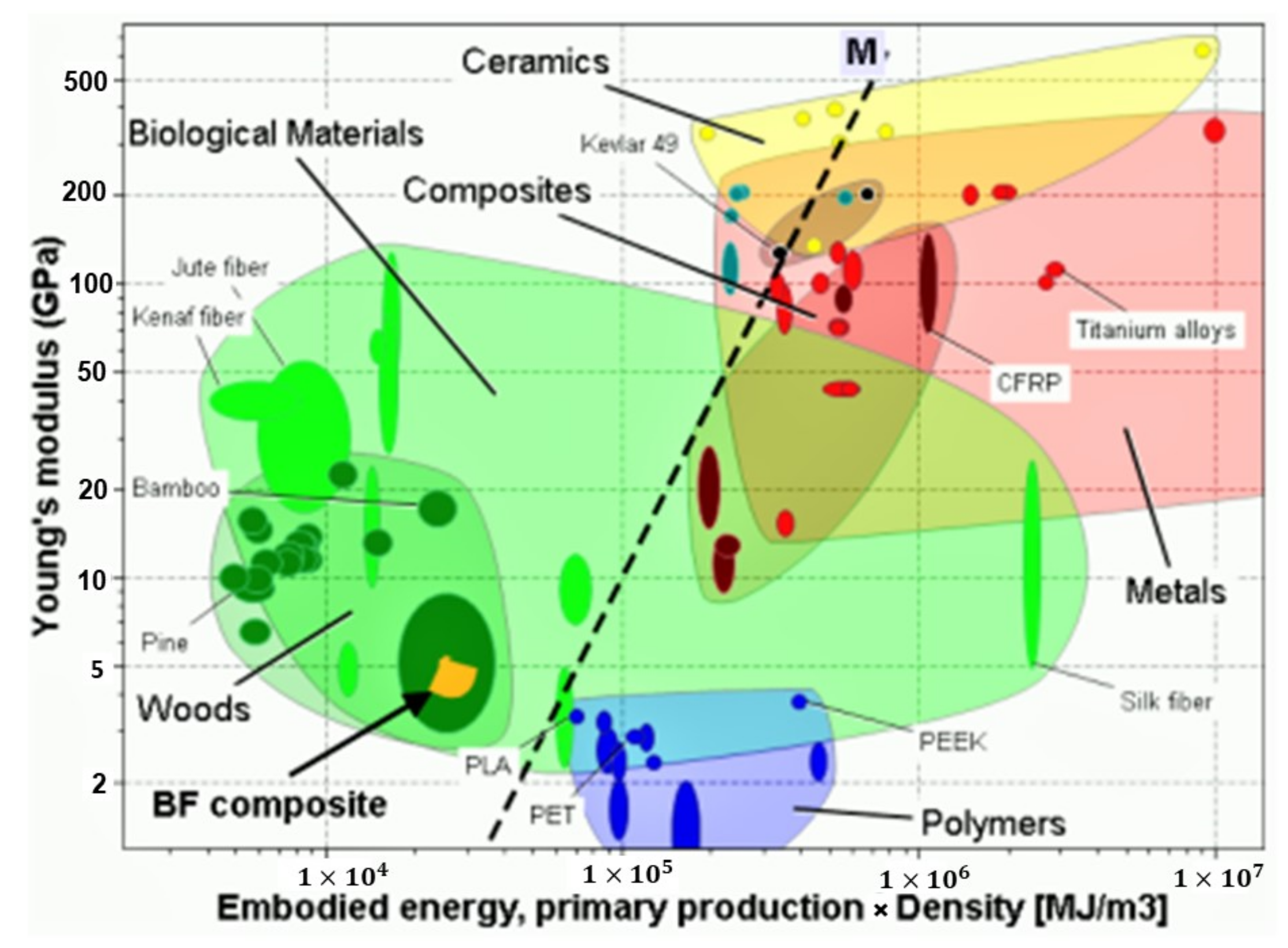
3.7. Elementary Sustainability Analysis of the COPET+BF Composites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santos, P.; Sérgio, H.P. Mechanical properties of polypropylene reinforced with recycled-pet fibres. J. Mater. Process. Technol. 2003, 144, 517–520. [Google Scholar] [CrossRef]
- Kim, J.; Jeong, D.; Son, C.; Lee, Y.; Kim, E.; Moon, I. Synthesis and applications of unsaturated polyester resins based on PET waste. Korean J. Chem. Eng. 2007, 24, 1076–1083. [Google Scholar] [CrossRef]
- Farahat, M.S.; Abdel-Azim, A.A.; Abdel-raowf, A.E. Modified unsaturated polyester resins synthesized from poly (ethylene terephthalate) waste, 1 Synthesis and curing characteristics. Macromol. Mater. Eng. 2000, 283, 1–6. [Google Scholar] [CrossRef]
- Webb, H.K.; Arnott, J.; Crawford, R.J.; Ivanova, E.P. Plastic degradation and its environmental implications with special reference to poly(ethylene terephthalate). Polymers 2013, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Plastics Europe. Plastics the Fact 2021. Plast. Eur. Mark. Res. Gr. Conversio Mark. Strateg. GmbH. 2021, 1–34. Available online: https://plasticseurope.org (accessed on 10 August 2021).
- Linli, G.; Xiao, Z.; Pan, H.; Xu, W.; Wang, Y.; Wang, X. Efficiently production of micron-sized polyethylene terephthalate (PET) powder from waste polyester fibre by physicochemical method. Adv. Powder Technol. 2021, 32, 630–636. [Google Scholar] [CrossRef]
- Ghosal, K.; Nayak, C. Recent advances in chemical recycling of polyethylene terephthalate waste into value added products for sustainable coating solutions-hope vs. hype. Mater. Adv. 2022, 3, 1974–1992. [Google Scholar] [CrossRef]
- Sarda, P.; Hanan, J.C.; Lawrence, J.G.; Allahkarami, M. Sustainability performance of polyethylene terephthalate, clarifying challenges and opportunities. J. Polym. Sci. 2022, 60, 7–31. [Google Scholar] [CrossRef]
- Cheang, C.C.; Ma, Y.; Fok, L. Occurrence and composition of microplastics in the seabed sediments of the coral communities in proximity of a metropolitan area. Int. J. Environ. Res. Public Health 2018, 15, 2270. [Google Scholar] [CrossRef] [Green Version]
- Benyathiar, P.; Kumar, P.; Carpenter, G.; Brace, J.; Mishra, D.K. Polyethylene Terephthalate (PET) Bottle-to-Bottle Recycling for the Beverage Industry: A Review. Polymers 2022, 14, 2366. [Google Scholar] [CrossRef]
- Rorrer, N.A.; Nicholson, S.; Carpenter, A.; Biddy, M.J.; Grundl, N.J.; Beckham, G.T. Combining Reclaimed PET with Bio-based Monomers Enables Plastics Upcycling. Joule 2019, 3, 1006–1027. [Google Scholar] [CrossRef]
- Tapia, J.J.B.; Valdez, M.H.; Cortez, J.C.; García, V.M.D.; Barrios, L.H. Improving the Rheological and Mechanical Properties of Recycled PET Modified by Macromolecular Chain Extenders Synthesized by Controlled Radical Polymerization. J. Polym. Environ. 2018, 26, 4221–4232. [Google Scholar] [CrossRef]
- Candal, M.V.; Safari, M.; Fernández, M.; Otaegi, I.; Múgica, A.; Zubitur, M.; Gonzalo Gerrica-echevarria, G.; Sebastián, V.; Irusta, S.; Loaeza, D.; et al. Structure and properties of reactively extruded opaque post-consumer recycled PET. Polymers 2021, 13, 3531. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Vassiliou, A.A.; Karavelidis, V.D.; Koumbis, A.; Bikiaris, D.N. Novel poly(propylene terephthalate-co-succinate) random copolymers: Synthesis, solid structure, and enzymatic degradation study. Macromolecules 2008, 41, 1675–1684. [Google Scholar] [CrossRef]
- Um, H.J.; Hwang, Y.T.; Choi, K.H.; Kim, H.S. Effect of crystallinity on the mechanical behavior of carbon fiber reinforced polyethylene-terephthalate (CF/PET) composites considering temperature conditions. Compos. Sci. Technol. 2021, 207, 108745. [Google Scholar] [CrossRef]
- Monti, M.; Scrivani, M.T.; Kociolek, I.; Larsen, A.G.; Olafsen, K.; Lambertini, V. Enhanced impact strength of recycled PET/glass fiber composites. Polymers 2021, 13, 1471. [Google Scholar] [CrossRef]
- Karimah, A.; Ridho, R.; Munawar, S.; Adi, S.; Ismadi; Damayanti, R.; Subiyanto, B.; Fatriasari, W.; Fudholi, A. A review on natural fibers for development of eco-friendly bio-composite: Characteristics, and utilizations. J. Mater. Res. Technol. 2021, 13, 2442–2458. [Google Scholar] [CrossRef]
- Dépigny, S.; Wils, E.D.; Tixier, P.; Keng, M.N.; Cillas, C.; Lescot, T.; Jagoret, P. Plantain productivity: Insights from Cameroonian cropping systems. Agric. Syst. 2018, 168, 1–10. [Google Scholar] [CrossRef]
- Komal, U.K.; Lila, M.K.; Singh, I. PLA/banana fiber based sustainable biocomposites: A manufacturing perspective. Compos. Part B Eng. 2020, 180, 107535. [Google Scholar] [CrossRef]
- Neelamana, I.K.; Thomas, S.; Parameswaranpillai, J. Characteristics of Banana Fibers and Banana Fiber Reinforced Phenol Formaldehyde Composites-Macroscale to Nanoscale. J. Appl. Polym. Sci. 2013, 130, 1239–1246. [Google Scholar] [CrossRef]
- Aging, T.; Panowicz, R.; Konarzewski, M.; Durejko, T.; Szala, M.; Łazi, M. Properties of Polyethylene Terephthalate (PET) after Thermo-Oxidative Aging. Materials 2021, 14, 3833. [Google Scholar]
- Wu, D.; Chen, F.; Li, R.; Shi, Y. Reaction kinetics and simulations for solid-state polymerization of poly(ethylene terephthalate). Macromolecules 1997, 30, 6737–6742. [Google Scholar] [CrossRef]
- Sango, T.; Cheumani, Y.M.; Duchatel, L.; Marin, A.; Ndikontar; Lefebvre, J.M. Industrial Crops & Products Step–wise multi–scale deconstruction of banana pseudo–stem (Musa acuminata) biomass and morpho–mechanical characterization of extracted long fi bres for sustainable applications. Ind. Crop. Prod. 2018, 122, 657–668. [Google Scholar] [CrossRef]
- Chamas, A.; Moon, H.; Zheng, J.; Qiu, Y.; Tabassum, T.; Jang, J.H.; Abu-Omar, M.; Scott, S.L.; Suh, S. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng. 2020, 8, 3494–3511. [Google Scholar] [CrossRef] [Green Version]
- Aravindh, M.; Sathish, S.; Ranga, R.R.; Karthick, A.; Mohanavel, V.; Patil, P.P.; Muhibbullah, M.; Osman, S.M. A Review on the Effect of Various Chemical Treatments on the Mechanical Properties of Renewable Fiber-Reinforced Composites. Adv. Mater. Sci. Eng. 2022, 2022, 2009691. [Google Scholar] [CrossRef]
- Gonçalves, B.M.M.; Camillo, M.O.; Oliveira, M.P.; Carreira, L.G.; Moulin, J.C.; Neto, H.F.; Oliveira, B.F.; Pereira, A.C.; Monteiro, S.N. Surface treatments of coffee husk fiber waste for effective incorporation into polymer biocomposites. Polymers 2021, 13, 3428. [Google Scholar] [CrossRef]
- Al-AbdulRazzak, S.; Lofgren, E.A.; Jabarin, S.A. End-group determination in poly(ethylene terephthalate) by infrared spectroscopy. Polym. Int. 2002, 51, 174–182. [Google Scholar] [CrossRef]
- Van Hoof, F. Polyethylene Terephthalate Catalyzed by Titanium (IV) Butoxide. Ph.D. Thesis, Catholic University of Louvain, Ottignies-Louvain-la-Neuve, Belgium, 2012; pp. 33–35. [Google Scholar]
- Fox, B.; Moad, G.; Van Diepen, G.; Mdc, S.; Cook, W.D. Characterization of poly (ethylene terephthalate) and poly (ethylene terephthalate) blends. Polymer 1997, 38, 3035–3043. [Google Scholar] [CrossRef]
- Kasmi, N.; Majdoub, M.; Papageorgiou, G.Z.; Achilias, D.S.; Bikiaris, D.N. Solid-state polymerization of poly(ethylene furanoate) biobased polyester, I: Effect of catalyst type on molecularweight increase. Polymers 2017, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Weisskopf, K. Characterization of polyethylene terephthalate by gel permeation chromatography (GPC). J. Polym. Sci. Part A Polym. Chem. 1988, 26, 1919–1935. [Google Scholar] [CrossRef]
- Majumdar, A.; Shukla, S.; Singh, A.A.; Arora, S. Resources, Conservation & Recycling Circular fashion: Properties of fabrics made from mechanically recycled poly-ethylene terephthalate (PET) bottles. Resour. Conserv. Recycl. 2020, 161, 104915. [Google Scholar] [CrossRef]
- Thomsen, T.B.; Hunt, C.J.; Meyer, A.S. Influence of substrate crystallinity and glass transition temperature on enzymatic degradation of polyethylene terephthalate (PET). New Biotechnol. 2022, 69, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Nutenki, R.; Van Velthem, P.; Kuete, M.; Lezaak, M.; Destoop, V.; Ballout, W.; Pardoen, T.; Bailly, C. Bio-Sourced Poly l-Lactide-Flax Composites with Close to Maximum Stiffness at Low Fiber Content through Two-Stage Annealing. Macromol. Mater. Eng. 2021, 306, 2100327. [Google Scholar] [CrossRef]
- Al-AbdulRazzak, S.; Jabarin, S.A. Processing characteristics of poly(ethylene terephthalate): Hydrolytic and thermal degradation. Polym. Int. 2002, 51, 164–173. [Google Scholar] [CrossRef]
- Lalhmangaihzuala, S.; Laldinpuii, Z.; Lalmuanpuia, C.; Vanlaldinpuia, K. Glycolysis of poly(Ethylene terephthalate) using biomass-waste derived recyclable heterogeneous catalyst. Polymers 2021, 13, 37. [Google Scholar] [CrossRef]
- Devaux, J.; Godard, P.; Mercier, J.P. Etude cinetique de la degradation du poly(oxytetramethyleneoxyterephtaloyle). Makromol. Chem. 1978, 179, 2201–2209. [Google Scholar] [CrossRef]
- Karanastasis, A.A.; Safin, V.; Pitet, L.M. Bio-Based Upcycling of Poly(ethylene terephthalate) Waste for the Preparation of High-Performance Thermoplastic Copolyesters. Macromolecules 2022, 55, 1042–1049. [Google Scholar] [CrossRef]
- Hablot, E.; Matadi, R.; Ahzi, S.; Avérous, L. Renewable biocomposites of dimer fatty acid-based polyamides with cellulose fibres: Thermal, physical and mechanical properties. Compos. Sci. Technol. 2010, 70, 504–509. [Google Scholar] [CrossRef]
- Dhaka, V.; Singh, S.; Anil, A.G.; Naik, T.S.S.K.; Garg, S.; Samuel, J.; Kumar, M.; Ramamurthy, P.C.; Singh, J. Occurrence, toxicity and remediation of polyethylene terephthalate plastics. A review. Environ. Chem. Lett. 2022, 20, 1777–1800. [Google Scholar] [CrossRef]
- Silva, F.S.; Ribeiro, C.E.G.; Demartini, T.J.C.; Rodríguez, R.J.S. Physical, Chemical, Mechanical, and Microstructural Characterization of Banana Pseudostem Fibers from Musa Sapientum. Macromol. Symp. 2020, 394, 2000052. [Google Scholar] [CrossRef]
- Gunge, A.; Koppad, P.G.; Nagamadhu, M.; Kivade, S.B.; Murthy, K.V.S. Study on mechanical properties of alkali treated plain woven banana fabric reinforced biodegradable composites. Compos. Commun. 2019, 13, 47–51. [Google Scholar] [CrossRef]
- Kupwade-Patil, K.; Wolf, C.; Chin, S.; Ochsendorf, J.; Hajiah, A.E.; Al-Mumin, A.; Büyüköztûrk, O. Impact of Embodied Energy on materials/buildings with partial replacement of ordinary Portland Cement (OPC) by natural Pozzolanic Volcanic Ash. J. Clean. Prod. 2018, 177, 547–554. [Google Scholar] [CrossRef]
- Ashby, M.F. Materials Selection in Mechanical Design, 4th ed.; Elsevier Ltd.: Oxford, UK, 2015; Volume 7, pp. 437–458. [Google Scholar]
- Papadaki, D.; Nikolaou, D.A.; Assimakopoulos, M.N. Circular Environmental Impact of Recycled Building Materials and Residential Renewable Energy. Sustainability 2022, 14, 4039. [Google Scholar] [CrossRef]
- Mehta, A.; Gaur, U.; Wunderlich, B. Equilibrium Melting Parameters of Poly ( ethy1ene Terephthalate). Polymer 1978, 16, 289–296. [Google Scholar] [CrossRef]
- Cecci, R.R.R.; Passos, A.A.; Aguiar, N.T.C.; Silva, L.A. Banana pseudostem fibers characterization and comparison with reported data on jute and sisal fibers. SN Appl. Sci. 2020, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Manna, S.; Chowdhury, R.; Sen, R.; Roy, D.; Adhikari, B. Bioresource Technology Enhancement of tensile strength of lignocellulosic jute fibers by alkali-steam treatment. Bioresour. Technol. 2010, 101, 3182–3187. [Google Scholar] [CrossRef]
- Haque, M.; Hasan, M.; Islam, S.; Ali, E. Bioresource Technology Physico-mechanical properties of chemically treated palm and coir fiber reinforced polypropylene composites. Bioresour. Technol. 2009, 100, 4903–4906. [Google Scholar] [CrossRef]
- Bozaci, E.; Sever, K.; Sarikanat, M.; Seki, Y.; Demir, A.; Ozdogan, E.; Tavman, I. Effects of the atmospheric plasma treatments on surface and mechanical properties of flax fiber and adhesion between fiber-matrix for composite materials. Compos. Part B Eng. 2013, 45, 565–572. [Google Scholar] [CrossRef]
- Parre, A.; Karthikeyan, B.; Balaji, A.; Udhayasankar, R. Proceedings Investigation of chemical, thermal and morphological properties of untreated and NaOH treated banana fiber. Mater. Today Proc. 2020, 22, 347–352. [Google Scholar] [CrossRef]
- Gassan, J.; Bledzki, A.K. Thermal Degradation of Flax and Jute Fibers. Appl. Polym. Sci. 2001, 82, 1417–1422. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-PET Oligomer | Molar Ratio Co-Diol/PET | Approx. Reacted Glycol (%) | Analysis of Glycolysed Co-PET | DSC Analysis | ||||
|---|---|---|---|---|---|---|---|---|
| Acid Value (µeq/g) | Hydroxyl Value (µeq/g) | Mn (g/mol) | Tg (°C) | Peak Tm (°C) | χ (%) | |||
| PET | - | - | 42 | 43 | 23,500 | 76 | 250 | 78.0 |
| GPET1 | 10 | 40.8 | 120 | 225 | 5800 | 76 | 245 | 33.0 |
| GPET2 | 20 | 32.2 | 114 | 430 | 3700 | 76 | 231 | 30.0 |
| GPET3 | 30 | 15 | 125 | 512 | 3125 | 76 | 207 | 25.0 |
| Co-Polyester Acronym | Glycolysed PET: Diol: Succinic Anhydride | FTIR | DSC Analysis Second Heating | ||||
|---|---|---|---|---|---|---|---|
| Hydroxyl Value (µeq/g) | Acid Value (µeq/g) | Mn PET (g/mol) | Tg (°C) | Peak Tm (°C) | χ (%) | ||
| COPET1 | 1:6:7 | 77 | 84 | 12,400 | 28 | 161 | 13.0 |
| COPET2 | 1:6:8 | 205 | 52 | 7800 | 25 | 186 | 12.0 |
| COPET3 | 1:6:10 | 362 | 64 | 4700 | 22 | 157 | 16.0 |
| COPET4 precipitated | 1:2:3 | 212 | 165 | 5300 | 48 | 135 | 10.0 |
| COPET4 cast film | “ | “ | “ | “ | 48 | 135 | 24.0 |
| Sample | T SSP (°C) | Intrinsic Viscosity (dL/g) * | MW from IV (g/mol) | Chain Ends from FTIR | DSC Analysis First Heating | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| [COOH] (μeq/g) | [OH] (μeq/g) | Mn (g/mol) | Tg (°C) | Tm1 (°C) | Tm2 (°C) | χm2 (%) | ||||
| COPET4-FILM | - | 0.243 | 17,600 | 165 | 212 | 5300 | 48 | 134 | 152 (shoulder) | 24 |
| COPET5 | 2 h @ 160 | 0.252 | 18,500 | 158 | 124 | 7100 | 48 | - | 182.5 | 17.0 |
| COPET6 | +2 h @ 170 | 0.265 | 19,900 | 29 | 155 | 10,300 | 48 | 134 | 193 | 6.0 |
| COPET7 | +2 h @ 180 | 0.314 | 25,000 | 25 | 168 | 10,400 | 48 | 134 | 202 | 7.0 |
| COPET8 | +2 h @ 190 | 0.327 | 26,500 | 18 | 112 | 15,400 | 48 | 134 | 200 | 2.0 |
| Sample | T SSP (°C) | SA Additional Amount (% wt) | Reduced Viscosity (dL/g) | MW from IV (g/mol) | Chain Ends (FTIR) | DSC Analysis First Heating | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| [COOH] (μeq/g) | [OH] (μeq/g) | Tg (°C) | Tm1 (°C) | Tm2 (°C) | χm2(%) | |||||
| PET | 0.53 | 49,700 | 42 | 43 | 78 | 250 | 8.0 | |||
| COPET6 | 170 | - | 0.265 | 19,900 | 29 | 155 | 48 | 134 | 193 | 6.0 |
| COPET6_AS1 | 0.5 | 0.265 | 19,900 | - | - | - | 134 | 197 | 7.0 | |
| COPET6_AS2 | 1 | 0.265 | 19,900 | - | - | - | 134 | 193 | 5.0 | |
| COPET10 | 170 | 0.5 | 0,293 | 22,800 | 22 | 212 | 48 | 134 | 198 | 3.0 |
| COPET11 | 170 | 1 | 0,350 | 29,100 | 40 | 119 | 48 | 134 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuete, M.A.; Van Velthem, P.; Ballout, W.; Nysten, B.; Devaux, J.; Ndikontar, M.K.; Pardoen, T.; Bailly, C. Integrated Approach to Eco-Friendly Thermoplastic Composites Based on Chemically Recycled PET Co-Polymers Reinforced with Treated Banana Fibres. Polymers 2022, 14, 4791. https://doi.org/10.3390/polym14224791
Kuete MA, Van Velthem P, Ballout W, Nysten B, Devaux J, Ndikontar MK, Pardoen T, Bailly C. Integrated Approach to Eco-Friendly Thermoplastic Composites Based on Chemically Recycled PET Co-Polymers Reinforced with Treated Banana Fibres. Polymers. 2022; 14(22):4791. https://doi.org/10.3390/polym14224791
Chicago/Turabian StyleKuete, Martial Aime, Pascal Van Velthem, Wael Ballout, Bernard Nysten, Jacques Devaux, Maurice Kor Ndikontar, Thomas Pardoen, and Christian Bailly. 2022. "Integrated Approach to Eco-Friendly Thermoplastic Composites Based on Chemically Recycled PET Co-Polymers Reinforced with Treated Banana Fibres" Polymers 14, no. 22: 4791. https://doi.org/10.3390/polym14224791