

Crystallization of D-A Conjugated Polymers: A Review of Recent Research


Abstract
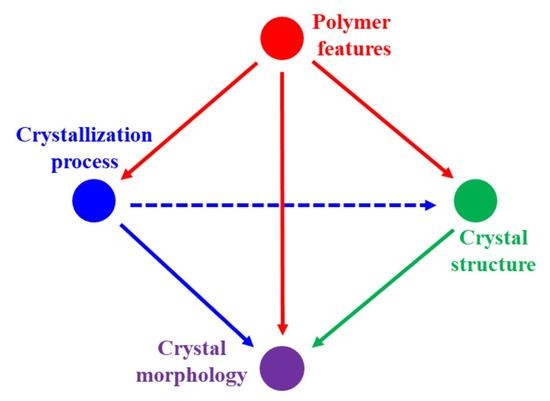
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
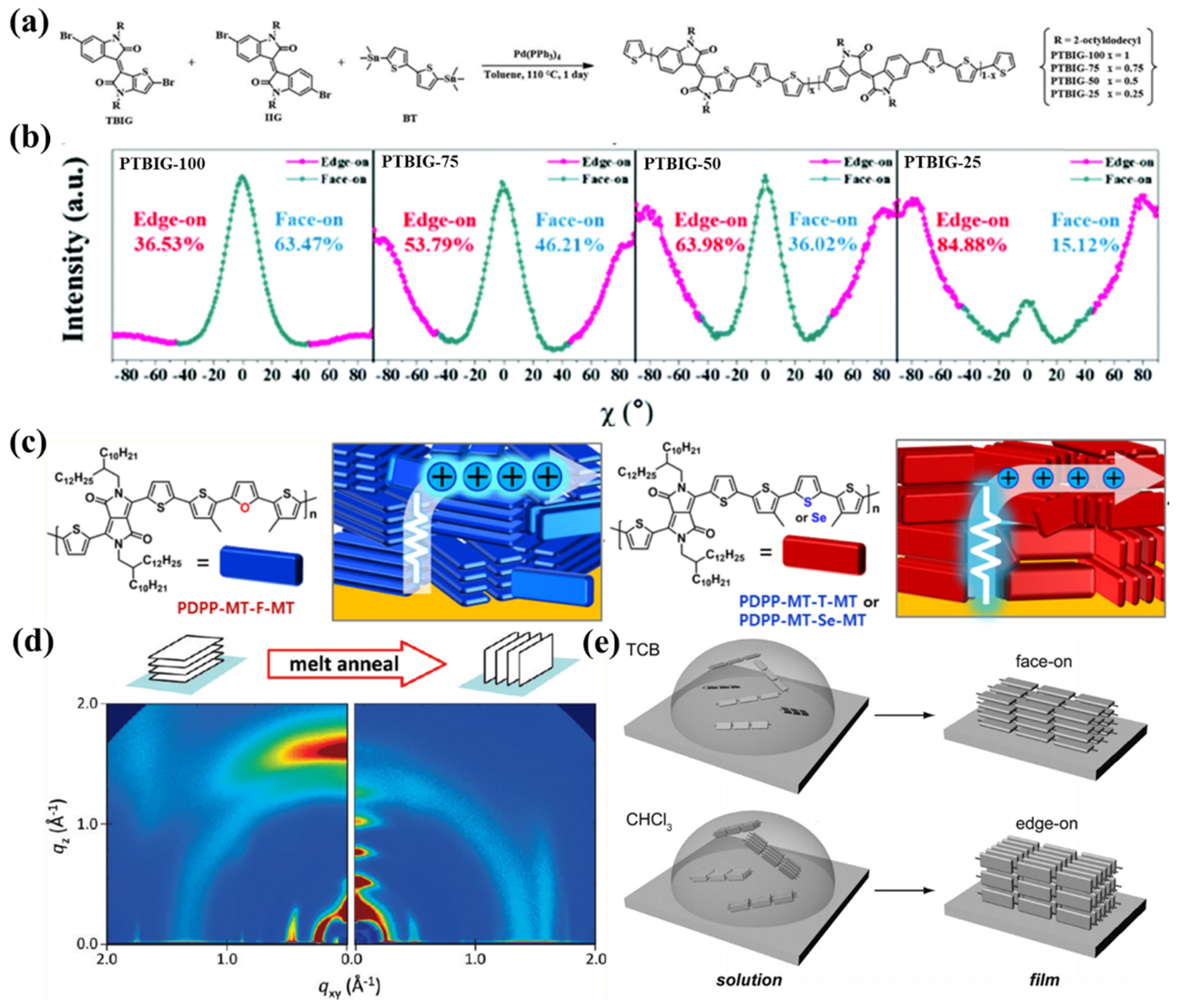{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Features of D-A Conjugated Polymers
2.1. The Regioregularity (RR) of D-A Conjugated Polymers
2.2. The Stiffness of the Backbone
2.3. The Anisotropic Interchain Interactions
2.4. The Role of Alkyl Side Chains
3. The Crystallization Process of D-A Conjugated Polymers
3.1. The Chain Conformation in the Solution
3.2. The Nucleation Process
3.3. The Growth Process
4. The Crystal Structure
4.1. The Molecular Orientation
4.2. Polymorphism
5. The Crystal Morphology
5.1. Locally Ordered Structures
5.2. Fibrils
5.3. Nanowires
5.4. Rhizoid Crystals
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, L.; Gao, M.; Hou, J. Advances and prospective in thermally stable nonfullerene polymer solar cells. Sci. China Chem. 2021, 64, 1875–1887. [Google Scholar] [CrossRef]
- Li, X.; Li, P.; Wu, Z.; Luo, D.; Yu, H.-Y.; Lu, Z.-H. Review and perspective of materials for flexible solar cells. Mater. Rep. Energy 2021, 1, 100001. [Google Scholar] [CrossRef]
- Ashizawa, M.; Zheng, Y.; Tran, H.; Bao, Z. Intrinsically stretchable conjugated polymer semiconductors in field effect transistors. Prog. Polym. Sci. 2020, 100, 101181. [Google Scholar] [CrossRef]
- Zhu, H.; Shin, E.S.; Liu, A.; Ji, D.; Xu, Y.; Noh, Y.Y. Printable Semiconductors for Backplane TFTs of Flexible OLED Displays. Adv. Funct. Mater. 2019, 30, 1904588. [Google Scholar] [CrossRef]
- Mdluli, S.B.; Ramoroka, M.E.; Yussuf, S.T.; Modibane, K.D.; John-Denk, V.S.; Iwuoha, E.I. pi-Conjugated Polymers and Their Application in Organic and Hybrid Organic-Silicon Solar Cells. Polymers 2022, 14, 716. [Google Scholar] [CrossRef]
- Murad, A.R.; Iraqi, A.; Aziz, S.B.; Abdullah, S.N.; Brza, M.A. Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review. Polymers 2020, 12, 2627. [Google Scholar] [CrossRef]
- Heeger, A.J. Semiconducting polymers: The Third Generation. Chem. Soc. Rev. 2010, 39, 2354. [Google Scholar] [CrossRef]
- Liu, F.; Gu, Y.; Jung, J.W.; Jo, W.H.; Russell, T.P. On the morphology of polymer-based photovoltaics. J. Polym. Sci. Part B Polym. Phys. 2012, 50, 1018–1044. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Duan, C.; Li, M.; Hu, Z.; Wang, J.; Liu, F.; Li, N.; Brabec, C.J.; Janssen, R.A.J.; et al. Morphology Optimization via Side Chain Engineering Enables All-Polymer Solar Cells with Excellent Fill Factor and Stability. J. Am. Chem. Soc. 2018, 140, 8934–8943. [Google Scholar] [CrossRef] [Green Version]
- Noriega, R.; Rivnay, J.; Vandewal, K.; Koch, F.P.V.; Stingelin, N.; Smith, P.; Toney, M.F.; Salleo, A. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 2013, 12, 1038–1044. [Google Scholar] [CrossRef]
- Xia, T.; Cai, Y.; Fu, H.; Sun, Y. Optimal bulk-heterojunction morphology enabled by fibril network strategy for high-performance organic solar cells. Sci. China Chem. 2019, 62, 662–668. [Google Scholar] [CrossRef]
- Zhou, K.; Xin, J.; Ma, W. Hierarchical Morphology Stability under Multiple Stresses in Organic Solar Cells. ACS Energy Lett. 2019, 4, 447–455. [Google Scholar] [CrossRef]
- Zhou, K.; Xian, K.; Ye, L. Morphology control in high-efficiency all-polymer solar cells. InfoMat 2022, 4, e12270. [Google Scholar] [CrossRef]
- Chang, M.; Lim, G.T.; Park, B.; Reichmanis, E. Control of Molecular Ordering, Alignment, and Charge Transport in Solution-Processed Conjugated Polymer Thin Films. Polymers 2017, 9, 212. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Hu, W. Multilevel Investigation of Charge Transport in Conjugated Polymers. Acc. Chem. Res. 2016, 49, 2435–2443. [Google Scholar] [CrossRef]
- Gu, K.; Loo, Y.L. The Polymer Physics of Multiscale Charge Transport in Conjugated Systems. J. Polym. Sci. Part B Polym. Phys. 2019, 57, 1559–1571. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Park, C.; Sin, D.H.; Park, J.H.; Cho, K. Recent Advances in Morphology Optimization for Organic Photovoltaics. Adv. Mater. 2018, 30, 1800453. [Google Scholar] [CrossRef]
- Yao, Z.-F.; Wang, J.-Y.; Pei, J. High-performance polymer field-effect transistors: From the perspective of multi-level microstructures. Chem. Sci. 2021, 12, 1193–1205. [Google Scholar] [CrossRef]
- Groves, C. Suppression of geminate charge recombination in organic photovoltaic devices with a cascaded energy heterojunction. Energy Environ. Sci. 2013, 6, 1546. [Google Scholar] [CrossRef] [Green Version]
- Sweetnam, S.; Graham, K.R.; Ngongang Ndjawa, G.O.; Heumuller, T.; Bartelt, J.A.; Burke, T.M.; Li, W.; You, W.; Amassian, A.; McGehee, M.D. Characterization of the polymer energy landscape in polymer:fullerene bulk heterojunctions with pure and mixed phases. J. Am. Chem. Soc. 2014, 136, 14078–14088. [Google Scholar] [CrossRef]
- Cao, X.; Xie, L.; Zhu, X.; Lv, J.; Fan, H. The principles of selecting green solvent additives for optimizing the phase separation structure of polymer solar cells based on PTB7:PC71BM. Eur. Polym. J. 2022, 180, 111603. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, Q.; Zhou, K.; Yu, X.; Liu, J.; Han, Y.; Xie, Z. Improve exciton generation and dissociation by increasing fullerene content in the mixed phase of P3HT/fullerene. Colloids Surf. A 2016, 506, 723–731. [Google Scholar] [CrossRef]
- McDowell, C.; Abdelsamie, M.; Toney, M.F.; Bazan, G.C. Solvent Additives: Key Morphology-Directing Agents for Solution-Processed Organic Solar Cells. Adv. Mater. 2018, 30, 1707114. [Google Scholar] [CrossRef]
- Liu, F.; Gu, Y.; Wang, C.; Zhao, W.; Chen, D.; Briseno, A.L.; Russell, T.P. Efficient polymer solar cells based on a low bandgap semi-crystalline DPP polymer-PCBM blends. Adv. Mater. 2012, 24, 3947–3951. [Google Scholar] [CrossRef]
- Franeker, J.J.; Turbiez, M.; Li, W.; Wienk, M.M.; Janssen, R.A. A real-time study of the benefits of co-solvents in polymer solar cell processing. Nat. Commun. 2015, 6, 6229. [Google Scholar] [CrossRef] [Green Version]
- Xia, D.; Li, C.; Li, W. Crystalline Conjugated Polymers for Organic Solar Cells: From Donor, Acceptor to Single-Component. Chem. Rec. 2019, 19, 962–972. [Google Scholar] [CrossRef]
- Kim, M.; Ryu, S.U.; Park, S.A.; Choi, K.; Kim, T.; Chung, D.; Park, T. Donor–Acceptor-Conjugated Polymer for High-Performance Organic Field-Effect Transistors: A Progress Report. Adv. Funct. Mater. 2019, 30, 1904545. [Google Scholar] [CrossRef]
- PanFeng, G.; LiYong, W.; HaiYan, F.; Yuan, D. Synthesis, characterizations and photovoltaic applications of a thickness-insensitive benzodifuran based copolymer. Eur. Polym. J. 2022, 172, 111189. [Google Scholar] [CrossRef]
- Ma, Y.-F.; Zhang, Y.; Zhang, H.-L. Solid additives in organic solar cells: Progress and perspectives. J. Mater. Chem. C 2022, 10, 2364–2374. [Google Scholar] [CrossRef]
- Yu, R.; Wei, X.; Wu, G.; Tan, Z.A. Layer-by-layered organic solar cells: Morphology optimizing strategies and processing techniques. Aggregate 2021, 3, e107. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, H.; Li, W.; Lin, Y.-F.; Balestra, F.; Ghibaudo, G.; Noh, Y.-Y. Exploring the Charge Transport in Conjugated Polymers. Adv. Mater. 2017, 29, 1702729. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, Y.; Yu, X.; Xing, R.; Liu, J.; Han, Y. Structure and Morphology Control in Thin Films of Conjugated Polymers for an Improved Charge Transport. Polymers 2013, 5, 1272–1324. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Boutilier, M.S.H.; Kidambi, P.R.; Jang, D.; Hadjiconstantinou, N.G.; Karnik, R. Fundamental transport mechanisms, fabrication and potential applications of nanoporous atomically thin membranes. Nat. Nanotechnol. 2017, 12, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, D.; Zhao, K.; Han, Y. Optimizing Morphology to Trade Off Charge Transport and Mechanical Properties of Stretchable Conjugated Polymer Films. Macromolecules 2021, 54, 3907–3926. [Google Scholar] [CrossRef]
- Li, Z.; Chueh, C.-C.; Jen, A.K.-Y. Recent advances in molecular design of functional conjugated polymers for high-performance polymer solar cells. Prog. Polym. Sci. 2019, 99, 101175. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chem. Rev. 2009, 109, 5868–5923. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Jiang, Y.; Sui, Y.; Zhang, J.; Tian, H.; Han, Y.; Deng, Y.; Hu, W.; Geng, Y. Donor–Acceptor Conjugated Polymers Based on Bisisoindigo: Energy Level Modulation toward Unipolar n-Type Semiconductors. Macromolecules 2018, 51, 8652–8661. [Google Scholar] [CrossRef]
- Tanaka, H.; Wakamatsu, A.; Kondo, M.; Kawamura, S.; Kuroda, S.-i.; Shimoi, Y.; Park, W.-T.; Noh, Y.-Y.; Takenobu, T. Microscopic observation of efficient charge transport processes across domain boundaries in donor-acceptor-type conjugated polymers. Commun. Phys. 2019, 2, 96. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Cui, S.; Li, J.; Wei, X.; Zheng, M. Diketopyrrolopyrrole Based Organic Semiconductor Materials for Field-Effect Transistors. Front. Chem. 2021, 9, 671294. [Google Scholar] [CrossRef]
- Lei, T.; Wang, J.-Y.; Pei, J. Roles of Flexible Chains in Organic Semiconducting Materials. Chem. Mater. 2013, 26, 594–603. [Google Scholar] [CrossRef]
- Mei, J.; Bao, Z. Side Chain Engineering in Solution-Processable Conjugated Polymers. Chem. Mater. 2013, 26, 604–615. [Google Scholar] [CrossRef]
- He, Y.; Kukhta, N.A.; Marks, A.; Luscombe, C.K. The effect of side chain engineering on conjugated polymers in organic electrochemical transistors for bioelectronic applications. J. Mater. Chem. C 2022, 10, 2314–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-G.; Li, Y. Side-chain engineering of high-efficiency conjugated polymer photovoltaic materials. Sci. China Chem. 2014, 58, 192–209. [Google Scholar] [CrossRef]
- Cui, C.; Wong, W.Y. Effects of Alkylthio and Alkoxy Side Chains in Polymer Donor Materials for Organic Solar Cells. Macromol. Rapid Commun. 2016, 37, 287–302. [Google Scholar] [CrossRef]
- Meng, B.; Liu, J.; Wang, L. Oligo(ethylene glycol) as side chains of conjugated polymers for optoelectronic applications. Polym. Chem. 2020, 11, 1261–1270. [Google Scholar] [CrossRef]
- Bunn, C.W. Molecular Structure and the Crystallinity of Long-Chain Polymers. J. Appl. Phys. 1954, 25, 820–825. [Google Scholar] [CrossRef]
- Mandelkern, L. The crystallization of flexible polymer molecules. Chem. Rev. 1955, 56, 903–958. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Park, J.S.; Lee, J.-W.; Kim, F.S.; Kim, H.J.; Kim, B.J. Regioregularity-control of conjugated polymers: From synthesis and properties, to photovoltaic device applications. J. Mater. Chem. A 2022, 10, 2672–2696. [Google Scholar]
- Liu, Y.; Lu, H.; Li, M.; Zhang, Z.; Feng, S.; Xu, X.; Wu, Y.; Bo, Z. Enhancing the Performance of Non-Fullerene Organic Solar Cells Using Regioregular Wide-Bandgap Polymers. Macromolecules 2018, 51, 8646–8651. [Google Scholar] [CrossRef]
- Zhong, H.; Ye, L.; Chen, J.-Y.; Jo, S.B.; Chueh, C.-C.; Carpenter, J.H.; Ade, H.; Jen, A.K.Y. A regioregular conjugated polymer for high performance thick-film organic solar cells without processing additive. J. Mater. Chem. A 2017, 5, 10517–10525. [Google Scholar] [CrossRef]
- Fu, H.; Li, Y.; Yu, J.; Wu, Z.; Fan, Q.; Lin, F.; Woo, H.Y.; Gao, F.; Zhu, Z.; Jen, A.K. High Efficiency (15.8%) All-Polymer Solar Cells Enabled by a Regioregular Narrow Bandgap Polymer Acceptor. J. Am. Chem. Soc. 2021, 143, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.; Kim, H.; Lee, D.; Jang, S.; Ban, L.; Lim, B.; Lee, J.; Lee, Y. Regioregular D1-A-D2-A Terpolymer with Controlled Thieno [3,4-b]thiophene Orientation for High-Efficiency Polymer Solar Cells Processed with Nonhalogenated Solvents. Macromolecules 2016, 49, 3328–3335. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, Z.; Zhang, G.; McDowell, C.; Luo, P.; Jia, X.; Ford, M.J.; Wang, M.; Bazan, G.C.; Huang, F.; et al. Toward High Efficiency Polymer Solar Cells: Rearranging the Backbone Units into a Readily Accessible Random Tetrapolymer. Adv. Energy Mater. 2017, 8, 1701668. [Google Scholar] [CrossRef]
- Steyrleuthner, R.; Di Pietro, R.; Collins, B.A.; Polzer, F.; Himmelberger, S.; Schubert, M.; Chen, Z.; Zhang, S.; Salleo, A.; Ade, H.; et al. The role of regioregularity, crystallinity, and chain orientation on electron transport in a high-mobility n-type copolymer. J. Am. Chem. Soc. 2014, 136, 4245–4256. [Google Scholar] [CrossRef] [PubMed]
- Heflin, J.R.; Wong, K.Y.; Zamani-Khamiri, O.; Garito, A.F. Nonlinear optical properties of linear chains and electron-correlation effects. Phys. Rev. B 1988, 38, 1573–1576. [Google Scholar] [CrossRef]
- Zhu, C.; Guo, Z.H.; Mu, A.U.; Liu, Y.; Wheeler, S.E.; Fang, L. Low Band Gap Coplanar Conjugated Molecules Featuring Dynamic Intramolecular Lewis Acid-Base Coordination. J. Org. Chem. 2016, 81, 4347–4352. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Fang, L. Locking the Coplanar Conformation of pi-Conjugated Molecules and Macromolecules Using Dynamic Noncovalent Bonds. Macromol. Rapid Commun. 2018, 39, 1700241. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Yao, Z.-F.; Lu, Y.; Ding, L.; Yu, Z.-D.; You, H.-Y.; Wang, X.-Y.; Zhou, Y.-Y.; Zou, L.; Wang, J.-Y.; et al. Precise tracking and modulating aggregation structures of conjugated copolymers in solutions. Polym. Chem. 2020, 11, 3716–3722. [Google Scholar] [CrossRef]
- Lei, T.; Dou, J.H.; Cao, X.Y.; Wang, J.Y.; Pei, J. Electron-deficient poly(p-phenylene vinylene) provides electron mobility over 1 cm(2) V(-1) s(-1) under ambient conditions. J. Am. Chem. Soc. 2013, 135, 12168–12171. [Google Scholar] [CrossRef]
- Lei, T.; Xia, X.; Wang, J.Y.; Liu, C.J.; Pei, J. “Conformation locked” strong electron-deficient poly(p-phenylene vinylene) derivatives for ambient-stable n-type field-effect transistors: Synthesis, properties, and effects of fluorine substitution position. J. Am. Chem. Soc. 2014, 136, 2135–2141. [Google Scholar] [CrossRef]
- Fei, Z.; Boufflet, P.; Wood, S.; Wade, J.; Moriarty, J.; Gann, E.; Ratcliff, E.L.; McNeill, C.R.; Sirringhaus, H.; Kim, J.S.; et al. Influence of Backbone Fluorination in Regioregular Poly(3-alkyl-4-fluoro)thiophenes. J. Am. Chem. Soc. 2015, 137, 6866–6879. [Google Scholar] [CrossRef] [PubMed]
- Onwubiko, A.; Yue, W.; Jellett, C.; Xiao, M.; Chen, H.Y.; Ravva, M.K.; Hanifi, D.A.; Knall, A.C.; Purushothaman, B.; Nikolka, M.; et al. Fused electron deficient semiconducting polymers for air stable electron transport. Nat. Commun. 2018, 9, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Yu, Z.D.; Zhang, R.Z.; Yao, Z.F.; You, H.Y.; Jiang, L.; Un, H.I.; Dong, B.W.; Xiong, M.; Wang, J.Y.; et al. Rigid Coplanar Polymers for Stable n-Type Polymer Thermoelectrics. Angew. Chem. Int. Ed. 2019, 58, 11390–11394. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kalin, A.J.; Yuan, T.; Al-Hashimi, M.; Fang, L. Fully conjugated ladder polymers. Chem. Sci. 2017, 8, 2503–2521. [Google Scholar] [CrossRef]
- Teo, Y.C.; Lai, H.W.H.; Xia, Y. Synthesis of Ladder Polymers: Developments, Challenges, and Opportunities. Chemistry 2017, 23, 14101–14112. [Google Scholar] [CrossRef] [PubMed]
- Kuei, B.; Gomez, E.D. Chain conformations and phase behavior of conjugated polymers. Soft Matter 2016, 13, 49–67. [Google Scholar] [CrossRef]
- Zhang, S.; Ocheje, M.U.; Huang, L.; Galuska, L.; Cao, Z.; Luo, S.; Cheng, Y.H.; Ehlenberg, D.; Goodman, R.B.; Zhou, D.; et al. The Critical Role of Electron-Donating Thiophene Groups on the Mechanical and Thermal Properties of Donor–Acceptor Semiconducting Polymers. Adv. Electron. Mater. 2019, 5, 1800899. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Li, Z.; Zhang, S.; Galuska, L.; Li, T.; Do, C.; Xia, W.; Hong, K.; Gu, X. Decoupling Poly(3-alkylthiophenes)’ Backbone and Side-Chain Conformation by Selective Deuteration and Neutron Scattering. Macromolecules 2020, 53, 11142–11152. [Google Scholar] [CrossRef]
- Zhao, K.; Zhang, Q.; Chen, L.; Zhang, T.; Han, Y. Nucleation and Growth of P(NDI2OD-T2) Nanowires via Side Chain Ordering and Backbone Planarization. Macromolecules 2021, 54, 2143–2154. [Google Scholar] [CrossRef]
- Zhang, W.; Gomez, E.D.; Milner, S.T. Predicting Chain Dimensions of Semiflexible Polymers from Dihedral Potentials. Macromolecules 2014, 47, 6453–6461. [Google Scholar] [CrossRef]
- Cao, X.; Zhao, K.; Chen, L.; Liu, J.; Han, Y. Conjugated polymer single crystals and nanowires. Polym. Crystal. 2019, 2, e10064. [Google Scholar] [CrossRef]
- Hu, W. The physics of polymer chain-folding. Phys. Rep. 2018, 747, 1–50. [Google Scholar] [CrossRef]
- Lauritzen, J.I.; Hoffman, J.D. Theory of Formation of Polymer Crystals with Folded Chains in Dilute Solution. J. Res. Natl. Bur. Stand. A Phys. Chem. 1960, 64, 73–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Arif, M.; Zou, J.; Khondaker, S.I.; Zhai, L. Controlling Poly(3-hexylthiophene) Crystal Dimension: Nanowhiskers and Nanoribbons. Macromolecules 2009, 42, 9390–9393. [Google Scholar] [CrossRef]
- Tang, X.; Chen, W.; Li, L. The Tough Journey of Polymer Crystallization: Battling with Chain Flexibility and Connectivity. Macromolecules 2019, 52, 3575–3591. [Google Scholar] [CrossRef]
- Natta, G.; Corradini, P. Conformation of linear chains and their mode of packing in the crystal state. J. Polym. Sci. 1959, 39, 29–46. [Google Scholar] [CrossRef]
- Lim, A.L.; Liu, F.; Ferdous, S.; Muthukumar, M.; Briseno, A.L. Polymer semiconductor crystals. Mater. Today 2010, 13, 14–24. [Google Scholar] [CrossRef]
- Briseno, A.L.; Mannsfeld, S.C.B.; Jenekhe, S.A.; Bao, Z.; Xia, Y. Introducing organic nanowire transistors. Mater. Today 2008, 11, 38–47. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L.; Xing, R.; Liu, J.; Han, Y. Simultaneous control over both molecular order and long-range alignment in films of the donor-acceptor copolymer. Langmuir 2015, 31, 469–479. [Google Scholar] [CrossRef]
- Um, H.A.; Lee, D.H.; Heo, D.U.; Yang, D.S.; Shin, J.; Baik, H.; Cho, M.J.; Choi, D.H. High aspect ratio conjugated polymer nanowires for high performance field-effect transistors and phototransistors. ACS Nano 2015, 9, 5264–5274. [Google Scholar] [CrossRef]
- Xiao, C.; Zhao, G.; Zhang, A.; Jiang, W.; Janssen, R.A.; Li, W.; Hu, W.; Wang, Z. High Performance Polymer Nanowire Field-Effect Transistors with Distinct Molecular Orientations. Adv. Mater. 2015, 27, 4963–4968. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, D.H.; Yang, D.S.; Heo, D.U.; Kim, K.H.; Shin, J.; Kim, H.J.; Baek, K.Y.; Lee, K.; Baik, H.; et al. Novel polymer nanowire crystals of diketopyrrolopyrrole-based copolymer with excellent charge transport properties. Adv. Mater. 2013, 25, 4102–4106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, H.; Jiang, S.; Zhao, G.; Shi, Q.; Tan, J.; Jiang, L.; Hu, W.; Zhan, X. High Performance Nanocrystals of a Donor–Acceptor Conjugated Polymer. Chem. Mater. 2013, 25, 2649–2655. [Google Scholar] [CrossRef]
- Ahn, K.S.; Jo, H.; Kim, J.B.; Seo, I.; Lee, H.H.; Lee, D.R. Structural Transition and Interdigitation of Alkyl Side Chains in the Conjugated Polymer Poly(3-hexylthiophene) and Their Effects on the Device Performance of the Associated Organic Field-Effect Transistor. ACS Appl. Mater. Inter. 2020, 12, 1142–1150. [Google Scholar] [CrossRef]
- Wu, Z.; Petzold, A.; Henze, T.; Thurn-Albrecht, T.; Lohwasser, R.H.; Sommer, M.; Thelakkat, M. Temperature and Molecular Weight Dependent Hierarchical Equilibrium Structures in Semiconducting Poly(3-hexylthiophene). Macromolecules 2010, 43, 4646–4653. [Google Scholar] [CrossRef]
- Li, Q.Y.; Yao, Z.F.; Wu, H.T.; Luo, L.; Ding, Y.F.; Yang, C.Y.; Wang, X.Y.; Shen, Z.; Wang, J.Y.; Pei, J. Regulation of High Miscibility for Efficient Charge-Transport in n-Doped Conjugated Polymers. Angew. Chem. Int. Ed. 2022, 134, e202200221. [Google Scholar]
- Yiu, A.T.; Beaujuge, P.M.; Lee, O.P.; Woo, C.H.; Toney, M.F.; Frechet, J.M. Side-chain tunability of furan-containing low-band-gap polymers provides control of structural order in efficient solar cells. J. Am. Chem. Soc. 2012, 134, 2180–2185. [Google Scholar] [CrossRef]
- Shin, J.; Park, G.E.; Lee, D.H.; Um, H.A.; Lee, T.W.; Cho, M.J.; Choi, D.H. Bis(thienothiophenyl) diketopyrrolopyrrole-based conjugated polymers with various branched alkyl side chains and their applications in thin-film transistors and polymer solar cells. ACS. Appl. Mater. Inter. 2015, 7, 3280–3288. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Ding, Z.; Liu, J.; Wang, L. Diketopyrrolopyrrole-based Conjugated Polymers Bearing Branched Oligo(Ethylene Glycol) Side Chains for Photovoltaic Devices. Angew. Chem. Int. Ed. 2016, 55, 10376–10380. [Google Scholar] [CrossRef]
- Meng, B.; Song, H.; Chen, X.; Xie, Z.; Liu, J.; Wang, L. Replacing Alkyl with Oligo(ethylene glycol) as Side Chains of Conjugated Polymers for Close π–π Stacking. Macromolecules 2015, 48, 4357–4363. [Google Scholar] [CrossRef]
- Dou, J.-H.; Zheng, Y.-Q.; Lei, T.; Zhang, S.-D.; Wang, Z.; Zhang, W.-B.; Wang, J.-Y.; Pei, J. Systematic Investigation of Side-Chain Branching Position Effect on Electron Carrier Mobility in Conjugated Polymers. Adv. Funct. Mater. 2014, 24, 6270–6278. [Google Scholar] [CrossRef]
- Lei, T.; Dou, J.H.; Pei, J. Influence of alkyl chain branching positions on the hole mobilities of polymer thin-film transistors. Adv. Mater. 2012, 24, 6457–6461. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, H.; Han, C.C. Temperature Induced Structure Evolution of Regioregular Poly(3-hexylthiophene) in Dilute Solution and its Influence on Thin Film Morphology. Macromolecules 2010, 43, 10031–10037. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, H.; Han, C.C. Unimer–Aggregate Equilibrium to Large Scale Association of Regioregular Poly(3-hexylthiophene) in THF Solution. Macromolecules 2011, 44, 5020–5026. [Google Scholar] [CrossRef]
- Cao, X.; Chen, L.; Zhao, K.; Liu, J.; Han, Y. Diketopyrrolopyrrole-based polymer nanowires: Control of chain conformation and nucleation. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 833–841. [Google Scholar] [CrossRef]
- Cao, X.; Li, M.; Liu, J.; Wang, H.; Zhou, K.; Han, Y. Control over fibril width via different solubility additives for diketopyrrolopyrrole-based photovoltaic devices. Org. Electron. 2015, 24, 280–287. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 2014, 5, 5293. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Y.; Di Virgilio, L.; Yao, Z.F.; Yu, Z.D.; Wang, X.Y.; Zhou, Y.Y.; Li, Q.Y.; Lu, Y.; Zou, L.; Wang, H.I.; et al. Correlating Charge Transport Properties of Conjugated Polymers in Solution Aggregates and Thin-Film Aggregates. Angew. Chem. Int. Ed. 2021, 60, 20483–20488. [Google Scholar] [CrossRef]
- Yao, Z.F.; Wang, Z.Y.; Wu, H.T.; Lu, Y.; Li, Q.Y.; Zou, L.; Wang, J.Y.; Pei, J. Ordered Solid-State Microstructures of Conjugated Polymers Arising from Solution-State Aggregation. Angew. Chem. Int. Ed. 2020, 59, 17467–17471. [Google Scholar] [CrossRef]
- Zhou, Y.-Y.; Wang, Z.-Y.; Yao, Z.-F.; Yu, Z.-D.; Lu, Y.; Wang, X.-Y.; Liu, Y.; Li, Q.-Y.; Zou, L.; Wang, J.-Y.; et al. Systematic Investigation of Solution-State Aggregation Effect on Electrical Conductivity in Doped Conjugated Polymers. CCS Chem. 2021, 3, 2994–3004. [Google Scholar] [CrossRef]
- Steyrleuthner, R.; Schubert, M.; Howard, I.; Klaumunzer, B.; Schilling, K.; Chen, Z.; Saalfrank, P.; Laquai, F.; Facchetti, A.; Neher, D. Aggregation in a high-mobility n-type low-bandgap copolymer with implications on semicrystalline morphology. J. Am. Chem. Soc. 2012, 134, 18303–18317. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Tassone, C.J.; Niskala, J.R.; Yiu, A.T.; Lee, O.P.; Weiss, T.M.; Wang, C.; Frechet, J.M.; Beaujuge, P.M.; Toney, M.F. A mechanistic understanding of processing additive-induced efficiency enhancement in bulk heterojunction organic solar cells. Adv. Mater. 2014, 26, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, W.; Dou, L.; Chen, C.C.; Chang, W.H.; Liu, Y.; Li, G.; Yang, Y. Elucidating double aggregation mechanisms in the morphology optimization of diketopyrrolopyrrole-based narrow bandgap polymer solar cells. Adv. Mater. 2014, 26, 3142–3147. [Google Scholar] [CrossRef] [PubMed]
- Panzer, F.; Bassler, H.; Kohler, A. Temperature Induced Order-Disorder Transition in Solutions of Conjugated Polymers Probed by Optical Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Shimomura, T.; Miura, T. Simulation Study of the Effect of the Side-Chain Structure on the Initial Nucleation Process of Polythiophene Derivatives. J. Phys. Chem. B 2017, 121, 1108–1117. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Z.-Y.; Yao, Z.-F.; Liu, N.-F.; Wang, X.-Y.; Zhou, Y.-Y.; Luo, L.; Shen, Z.; Wang, J.-Y.; Pei, J. Controllable Transformation between the Kinetically and Thermodynamically Stable Aggregates in a Solution of Conjugated Polymers. Macromolecules 2021, 54, 5815–5824. [Google Scholar] [CrossRef]
- Muthukumar, M. Nucleation in polymer crystallization. Adv. Chem. Phys. 2004, 128, 1–63. [Google Scholar]
- Erdemir, D.; Lee, A.Y.; Myerson, A.S. Nucleation of Crystals from Solution: Classical and Two-Step Models. Acc. Chem. Res. 2009, 42, 621–629. [Google Scholar] [CrossRef]
- Wu, T.; Chandran, S.; Zhang, Y.; Zheng, T.; Pfohl, T.; Xu, J.; Reiter, G. Primary Nucleation in Metastable Solutions of Poly(3-hexylthiophene). Macromolecules. 2022, 55, 3325–3334. [Google Scholar] [CrossRef]
- Zhang, F.; Mohammadi, E.; Luo, X.; Strzalka, J.; Mei, J.; Diao, Y. Critical Role of Surface Energy in Guiding Crystallization of Solution-Coated Conjugated Polymer Thin Films. Langmuir 2018, 34, 1109–1122. [Google Scholar] [CrossRef]
- Franeker, J.J.; Heintges, G.H.; Schaefer, C.; Portale, G.; Li, W.; Wienk, M.M.; Schoot, P.; Janssen, R.A. Polymer Solar Cells: Solubility Controls Fiber Network Formation. J. Am. Chem. Soc. 2015, 137, 11783–11794. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Du, Z.; Chen, L.; Zhao, K.; Li, H.; Liu, J.; Han, Y. Long diketopyrrolopyrrole-based polymer nanowires prepared by decreasing the aggregate speed of the polymer in solution. Polymer 2017, 118, 135–142. [Google Scholar] [CrossRef]
- Cao, X.; Hu, Y.; Wang, R.; Lu, Y.; Ou, B.; Liao, B.; Fan, H.; Guo, Y.; Liu, Q. Understanding the crystallization process of a diketopyrrolopyrrole-based conjugated polymer in blend films. J. Polym. Sci. 2021, 59, 925–934. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Lauritzen, J.I. Crystallization of bulk polymers with chain folding: Theory of growth of lamellar spherulites. J. Res. Natl. Bur. Stand. A Phys. Chem. 1961, 65A, 297–336. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.D.; Miller, R.L. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: Theory and experiment. Polymer 1997, 38, 3151–3212. [Google Scholar] [CrossRef]
- Markov, I.V. Crystal Growth for Beginners, 2nd ed.; World Scientific: Singapore, 2003. [Google Scholar]
- Cao, X.; Fan, H. Formation of D-A conjugated polymer crystals: Diffusion and conformational transition theory. Polymer 2022, 243, 124606. [Google Scholar] [CrossRef]
- Osaka, I.; Takimiya, K. Backbone orientation in semiconducting polymers. Polymer 2015, 59, A1–A15. [Google Scholar] [CrossRef]
- Brinkmann, M.; Hartmann, L.; Biniek, L.; Tremel, K.; Kayunkid, N. Orienting semi-conducting pi-conjugated polymers. Macromol. Rapid Commun. 2014, 35, 9–26. [Google Scholar] [CrossRef]
- Liu, X.; Yan, Y.; Zhang, Q.; Zhao, K.; Han, Y. n-Type D-A Conjugated Polymers: Relationship Between Microstructure and Electrical/Mechanical Performance. Chem. Res. Chin. Univ. 2021, 37, 1019–1030. [Google Scholar] [CrossRef]
- Agbolaghi, S.; Abbaspoor, S.; Massoumi, B.; Sarvari, R.; Sattari, S.; Aghapour, S.; Charoughchi, S. Conversion of Face-On Orientation to Edge-On/Flat-On in Induced-Crystallization of Poly(3-hexylthiophene) via Functionalization/Grafting of Reduced Graphene Oxide with Thiophene Adducts. Macromol. Chem. Phys. 2018, 219, 1700484. [Google Scholar] [CrossRef]
- Brinkmann, M. Structure and morphology control in thin films of regioregular poly(3-hexylthiophene). J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1218–1233. [Google Scholar] [CrossRef]
- Rahimi, K.; Botiz, I.; Stingelin, N.; Kayunkid, N.; Sommer, M.; Koch, F.P.; Nguyen, H.; Coulembier, O.; Dubois, P.; Brinkmann, M.; et al. Controllable processes for generating large single crystals of poly(3-hexylthiophene). Angew. Chem. Int. Ed. 2012, 51, 11131–11135. [Google Scholar] [CrossRef]
- Ma, Z.; Geng, Y.; Yan, D. Extended-chain lamellar packing of poly(3-butylthiophene) in single crystals. Polymer 2007, 48, 31–34. [Google Scholar] [CrossRef]
- Molina-Lopez, F.; Wu, H.-C.; Wang, G.-J.N.; Yan, H.; Shaw, L.; Xu, J.; Toney, M.F.; Bao, Z. Enhancing Molecular Alignment and Charge Transport of Solution-Sheared Semiconducting Polymer Films by the Electrical-Blade Effect. Adv. Electron. Mater. 2018, 4, 1800110. [Google Scholar] [CrossRef]
- Chang, M.; Choi, D.; Egap, E. Macroscopic Alignment of One-Dimensional Conjugated Polymer Nanocrystallites for High-Mobility Organic Field-Effect Transistors. ACS Appl. Mater. Inter. 2016, 8, 13484–13491. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, M.; He, C.; Shi, W.; Deng, Y.; Han, Y.; Ye, L.; Geng, Y. Unraveling the Molar Mass Dependence of Shearing-Induced Aggregation Structure of a High-Mobility Polymer Semiconductor. Adv. Mater. 2022, 34, e2108255. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Yoon, M.; Lee, J. Enhanced Performance of Cyclopentadithiophene-Based Donor-Acceptor-Type Semiconducting Copolymer Transistors Obtained by a Wire Bar-Coating Method. Polymers 2021, 14, 2. [Google Scholar] [CrossRef]
- Lee, J.; Shin, E.-S.; Kim, Y.-J.; Noh, Y.-Y.; Yang, C. Controlling the ambipolarity of thieno-benzoisoindigo polymer-based transistors: The balance of face-on and edge-on populations. J. Mater. Chem. C 2020, 8, 296–302. [Google Scholar] [CrossRef]
- Lee, H.W.; Kim, H.S.; Kim, D.; Yoon, M.; Lee, J.; Hwang, D.-H. Comparative Study of Charge-Transport Behavior of Edge-on- and Face-on-Oriented Diketopyrrolopyrrole-Based Conjugated Copolymers Bearing Chalcogenophene Units. Chem. Mater. 2022, 34, 314–324. [Google Scholar] [CrossRef]
- Rivnay, J.; Steyrleuthner, R.; Jimison, L.H.; Casadei, A.; Chen, Z.; Toney, M.F.; Facchetti, A.; Neher, D.; Salleo, A. Drastic Control of Texture in a High Performance n-Type Polymeric Semiconductor and Implications for Charge Transport. Macromolecules 2011, 44, 5246–5255. [Google Scholar] [CrossRef]
- Schuettfort, T.; Thomsen, L.; McNeill, C.R. Observation of a Distinct Surface Molecular Orientation in Films of a High Mobility Conjugated Polymer. J. Am. Chem. Soc. 2013, 135, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; An, C.; Marszalek, T.; Baumgarten, M.; Yan, H.; Müllen, K.; Pisula, W. Controlling the Surface Organization of Conjugated Donor–Acceptor Polymers by their Aggregation in Solution. Adv. Mater. 2016, 28, 9430–9438. [Google Scholar] [CrossRef] [PubMed]
- Ihn, K.J.; Moulton, J.; Smith, P. Whiskers of Poly(3-alkylthiophene)s. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 735–742. [Google Scholar] [CrossRef]
- Yao, Z.F.; Zheng, Y.Q.; Dou, J.H.; Lu, Y.; Ding, Y.F.; Ding, L.; Wang, J.Y.; Pei, J. Approaching Crystal Structure and High Electron Mobility in Conjugated Polymer Crystals. Adv. Mater. 2021, 33, e2006794. [Google Scholar] [CrossRef]
- Li, M.; Balawi, A.H.; Leenaers, P.J.; Ning, L.; Heintges, G.H.L.; Marszalek, T.; Pisula, W.; Wienk, M.M.; Meskers, S.C.J.; Yi, Y.; et al. Impact of polymorphism on the optoelectronic properties of a low-bandgap semiconducting polymer. Nat. Commun. 2019, 10, 2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremel, K.; Fischer, F.S.U.; Kayunkid, N.; Pietro, R.D.; Tkachov, R.; Kiriy, A.; Neher, D.; Ludwigs, S.; Brinkmann, M. Charge Transport Anisotropy in Highly Oriented Thin Films of the Acceptor Polymer P(NDI2OD-T2). Adv. Energy Mater. 2014, 4, 1301659. [Google Scholar] [CrossRef]
- Trefz, D.; Gross, Y.M.; Dingler, C.; Tkachov, R.; Hamidi-Sakr, A.; Kiriy, A.; McNeill, C.R.; Brinkmann, M.; Ludwigs, S. Tuning Orientational Order of Highly Aggregating P(NDI2OD-T2) by Solvent Vapor Annealing and Blade Coating. Macromolecules 2018, 52, 43–54. [Google Scholar] [CrossRef]
- Fischer, F.S.U.; Kayunkid, N.; Trefz, D.; Ludwigs, S.; Brinkmann, M. Structural Models of Poly(cyclopentadithiophene-alt-benzothiadiazole) with Branched Side Chains: Impact of a Single Fluorine Atom on the Crystal Structure and Polymorphism of a Conjugated Polymer. Macromolecules 2015, 48, 3974–3982. [Google Scholar] [CrossRef]
- Schulz, G.L.; Fisher, F.S.U.; Trefz, D.; Melnyk, A.; Hamidi-Sakr, A.; Brinkmann, M.; Andrienko, D.; Ludwigs, S. The PCPDTBT Family: Correlations between Chemical Structure, Polymorphism, and Device Performance. Macromolecules 2017, 50, 1402. [Google Scholar] [CrossRef]
- Zhao, K.; Zhang, T.; Zhang, L.; Li, J.; Li, H.; Wu, F.; Chen, Y.; Zhang, Q.; Han, Y. Role of Molecular Weight in Microstructural Transition and Its Correlation to the Mechanical and Electrical Properties of P(NDI2OD-T2) Thin Films. Macromolecules 2021, 54, 10203–10215. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Zhao, K.; Zhang, L.; Zhang, Q.; Yu, X.; Tian, H.; Han, Y. Increasing the Charge Transport of P(NDI2OD-T2) by Improving the Polarization of the NDI2OD Unit along the Backbone Direction and Preaggregation via H-Bonding. Macromolecules 2022, 55, 2497–2508. [Google Scholar] [CrossRef]
- Li, H.; Liu, X.; Jin, T.; Zhao, K.; Zhang, Q.; He, C.; Yang, H.; Chen, Y.; Huang, J.; Yu, X.; et al. Optimizing the Intercrystallite Connection of a Donor-Acceptor Conjugated Semiconductor Polymer by Controlling the Crystallization Rate via Temperature. Macromol. Rapid Commun. 2022, 43, e2200084. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, H.; Zhang, L.; Wang, S.; Chen, Y.; Zhang, Q.; Zhang, J.; Tian, H.; Han, Y. Optimizing the Crystallization Behavior and Film Morphology of Donor–Acceptor Conjugated Semiconducting Polymers by Side-Chain–Solvent Interaction in Nonpolar Solvents. Macromolecules 2021, 54, 10557–10573. [Google Scholar] [CrossRef]
- Wang, S.; Kappl, M.; Liebewirth, I.; Muller, M.; Kirchhoff, K.; Pisula, W.; Mullen, K. Organic field-effect transistors based on highly ordered single polymer fibers. Adv. Mater. 2012, 24, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-Q.; Yao, Z.-F.; Dou, J.-H.; Wang, Y.; Ma, W.; Zou, L.; Nikzad, S.; Li, Q.-Y.; Sun, Z.-H.; Yu, Z.-A.; et al. Influence of solution-state aggregation on conjugated polymer crystallization in thin films and microwire crystals. Giant 2021, 7, 100064. [Google Scholar] [CrossRef]
- Vicsek, T. Pattern Formation in Diffusion-Limited Aggregation. Phys. Rev. Lett. 1984, 53, 2281–2284. [Google Scholar] [CrossRef]
- Nie, W.-C.; Xiao, Q.; Wu, J.-M.; Song, F.; Wang, X.-L.; Wang, Y.-Z. Dendritic crystallization and morphology control of random poly(p-dioxanone-co-butylene-co-succinate) copolyesters. Eur. Polym. J. 2018, 108, 76–84. [Google Scholar] [CrossRef]
- Reiter, G. Model Experiments for a Molecular Understanding of Polymer Crystallization. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 1869–1877. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Cao, X.; Fan, H. Crystallization of D-A Conjugated Polymers: A Review of Recent Research. Polymers 2022, 14, 4612. https://doi.org/10.3390/polym14214612
Hu Y, Cao X, Fan H. Crystallization of D-A Conjugated Polymers: A Review of Recent Research. Polymers. 2022; 14(21):4612. https://doi.org/10.3390/polym14214612
Chicago/Turabian StyleHu, Yibo, Xinxiu Cao, and Hui Fan. 2022. "Crystallization of D-A Conjugated Polymers: A Review of Recent Research" Polymers 14, no. 21: 4612. https://doi.org/10.3390/polym14214612