
Effect of Iodine Filler on Photoisomerization Kinetics of Photo-Switchable Thin Films Based on PEO-BDK-MR

 , , ,
, , ,
Abstract
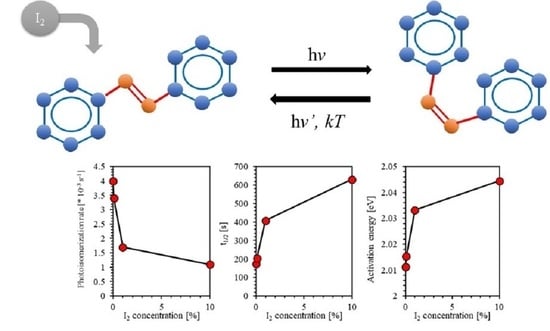
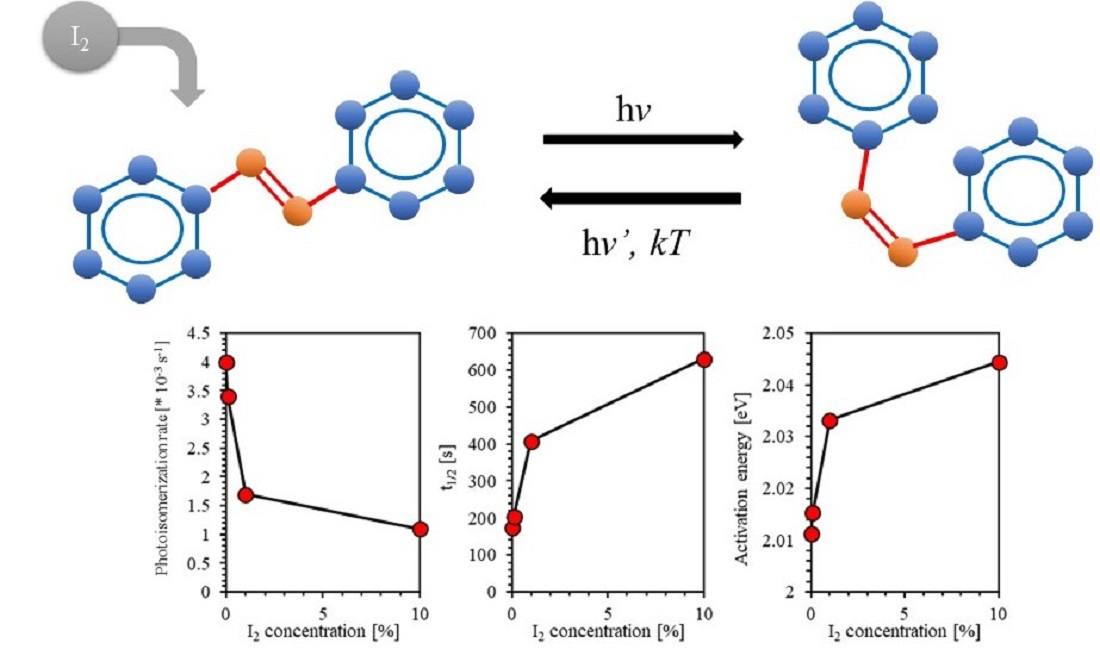
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
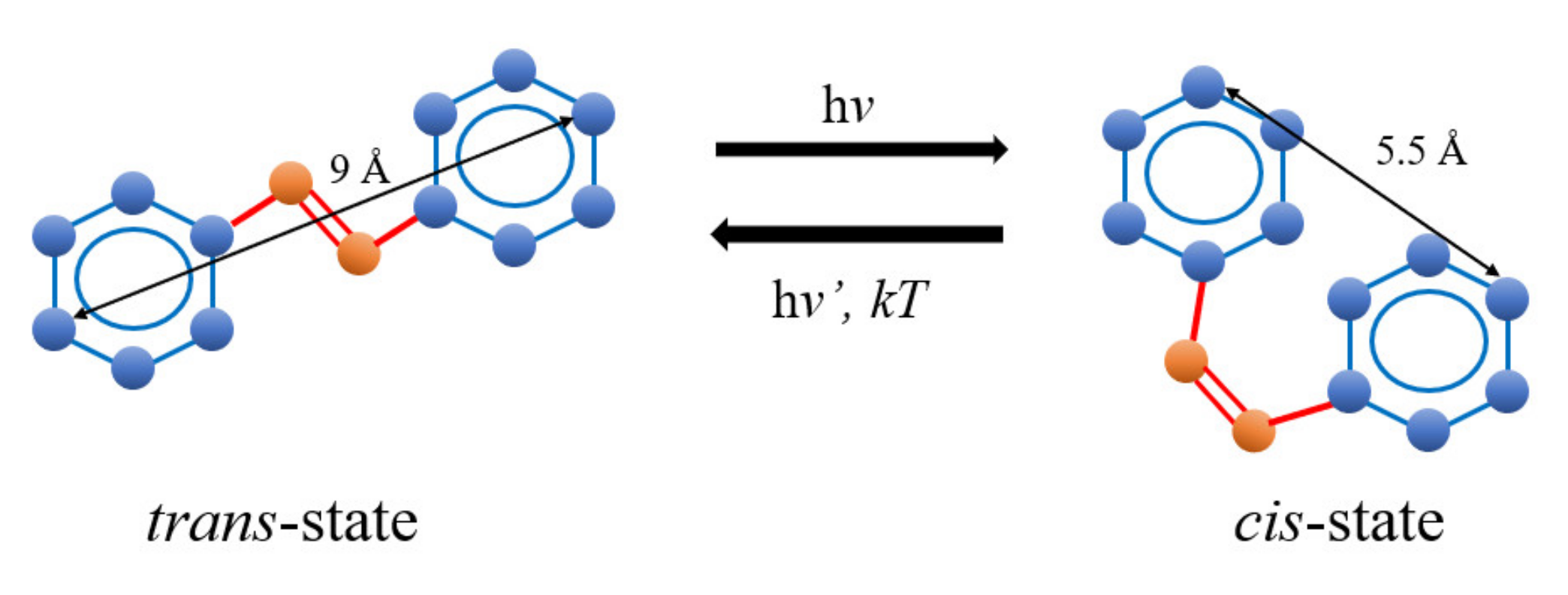
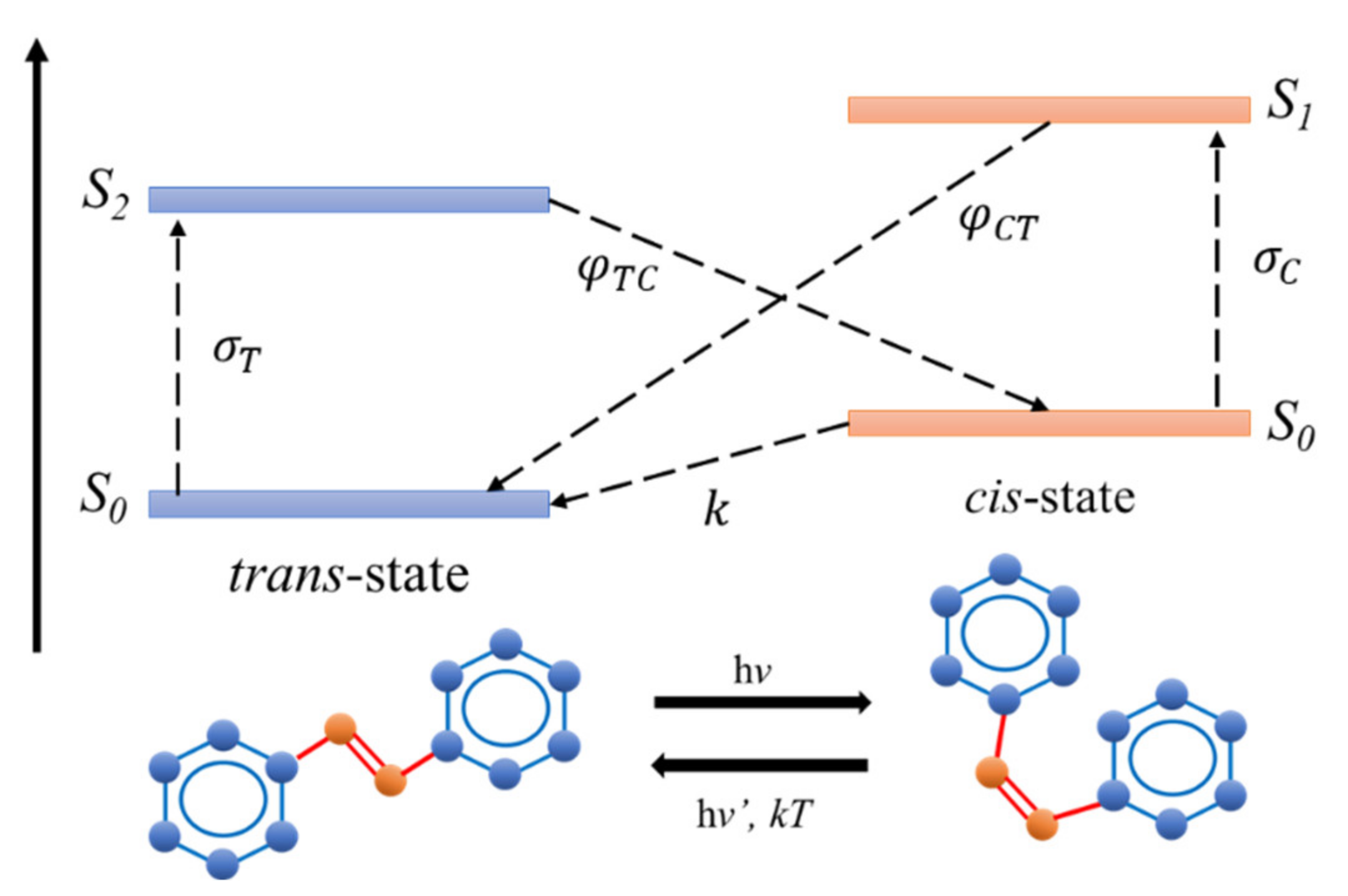
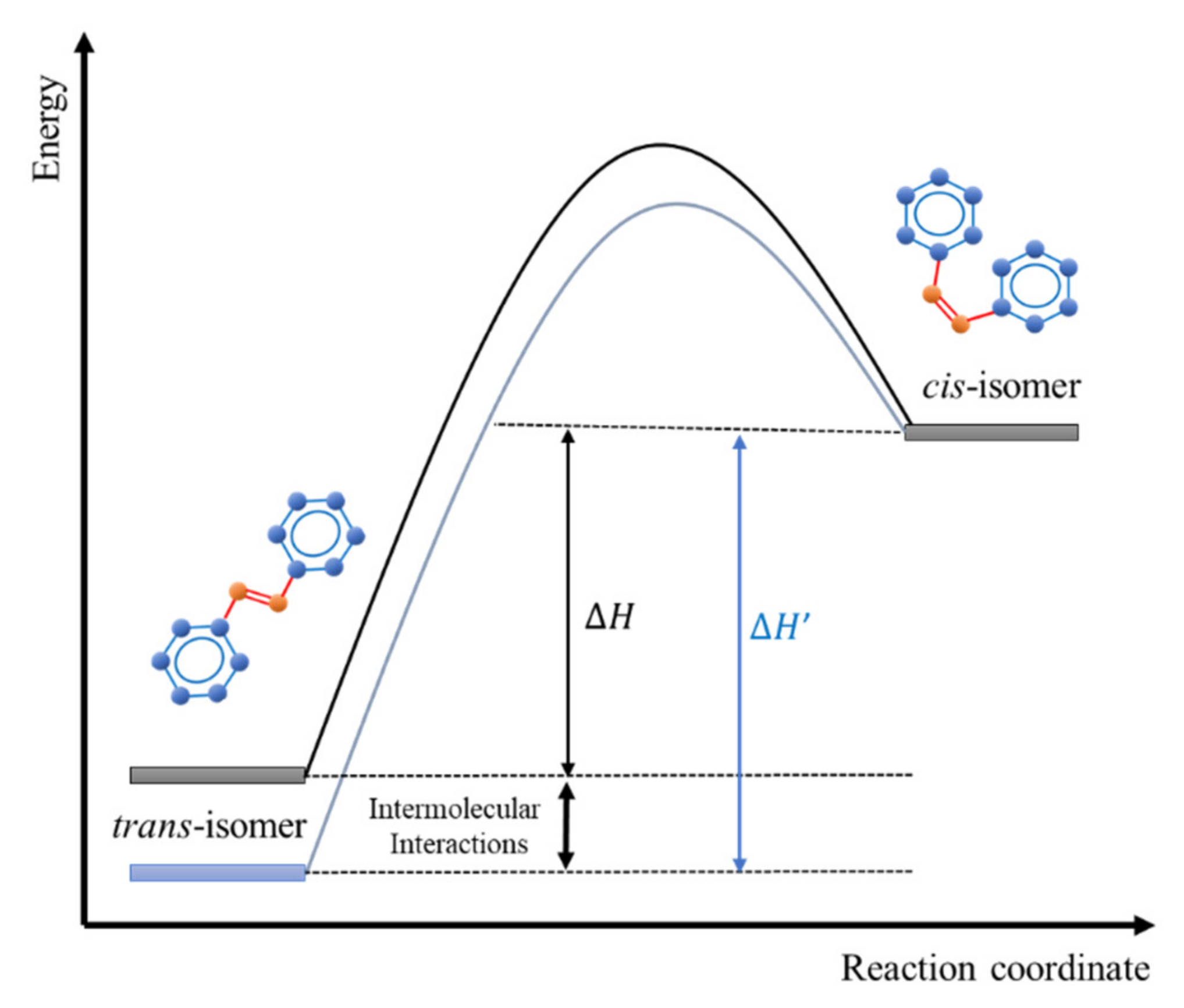
2. Theoretical Background
3. Experimental Details
3.1. Materials
- 300.000 g/mol Polyethylene Oxide (PEO) (–CH2CH2O–)n
- 256.30 g/mol Benzyl Dimethyl Ketal (BDK)
- 126.90 g/mol Iodine (I2)
- 32.04 g/mol Absolute Methanol (CH3OH, with a purity of 99.8%)The above materials were purchased from Sigma-Aldrich Co., Inc., Munich, Germany.
- SCP SCIENCE (Montreal, QC, Canada) supplied Methyl-Red (MR) (C15H15N3O3) of pH level between 4.2 and 4.6 powder.
3.2. Synthesis of (PEO-BDK-MR)/I2 Complex Composite Thin Films
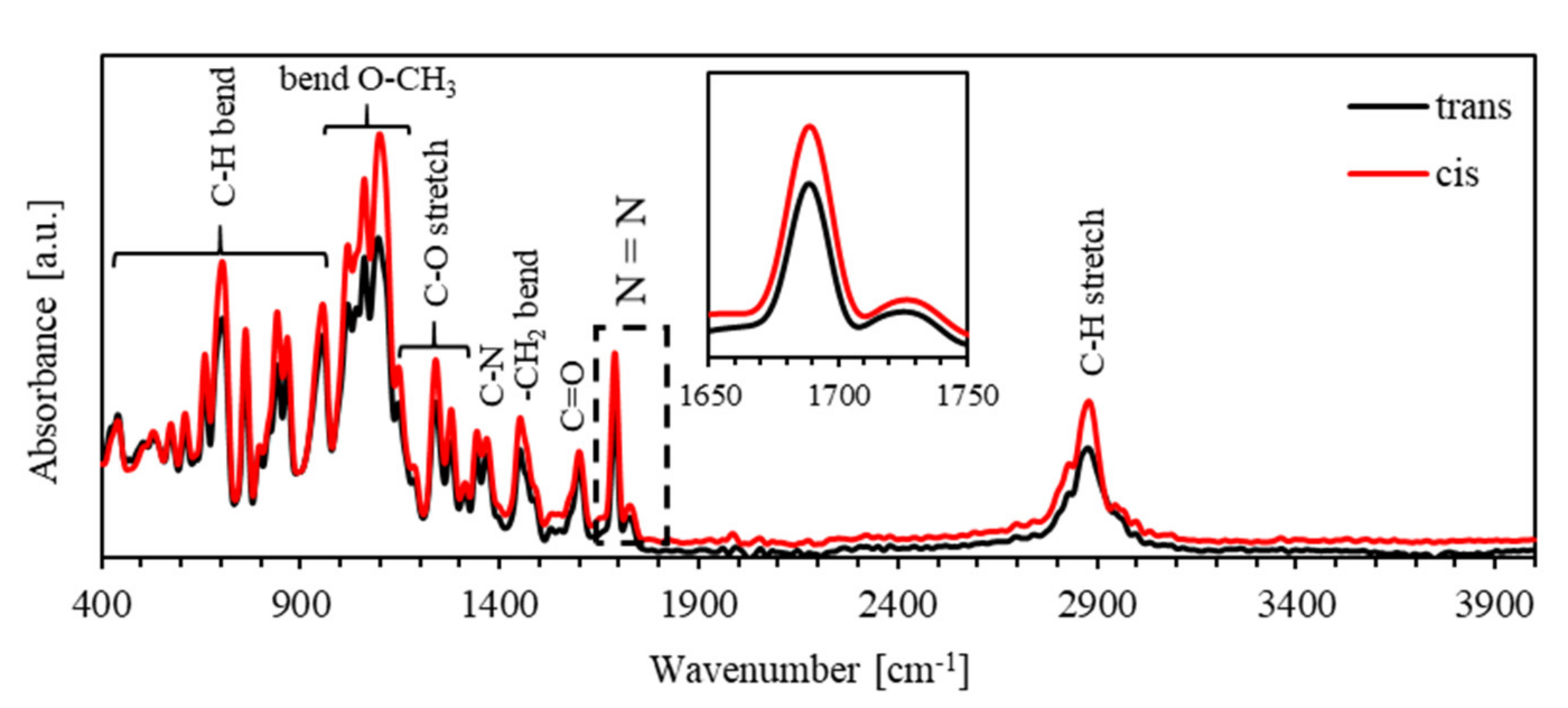
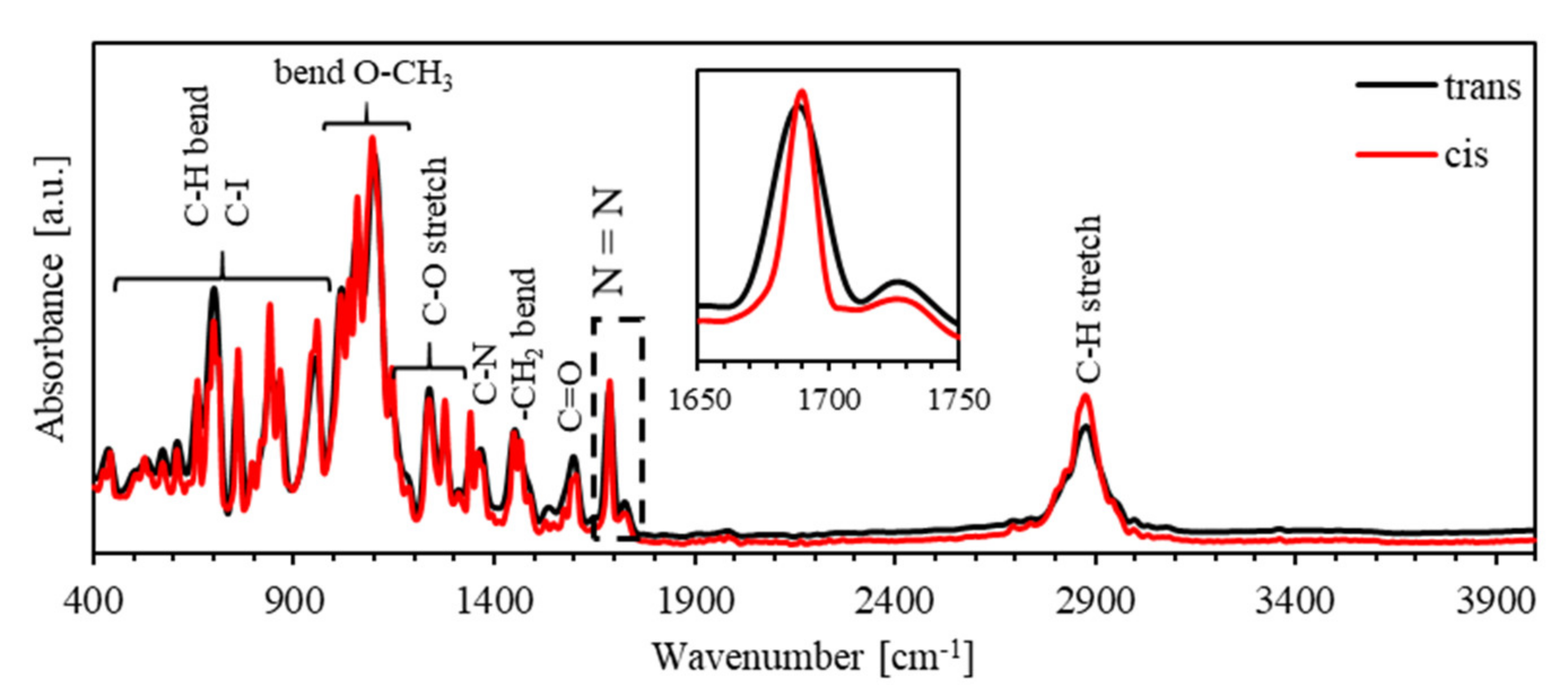
3.3. Characterization
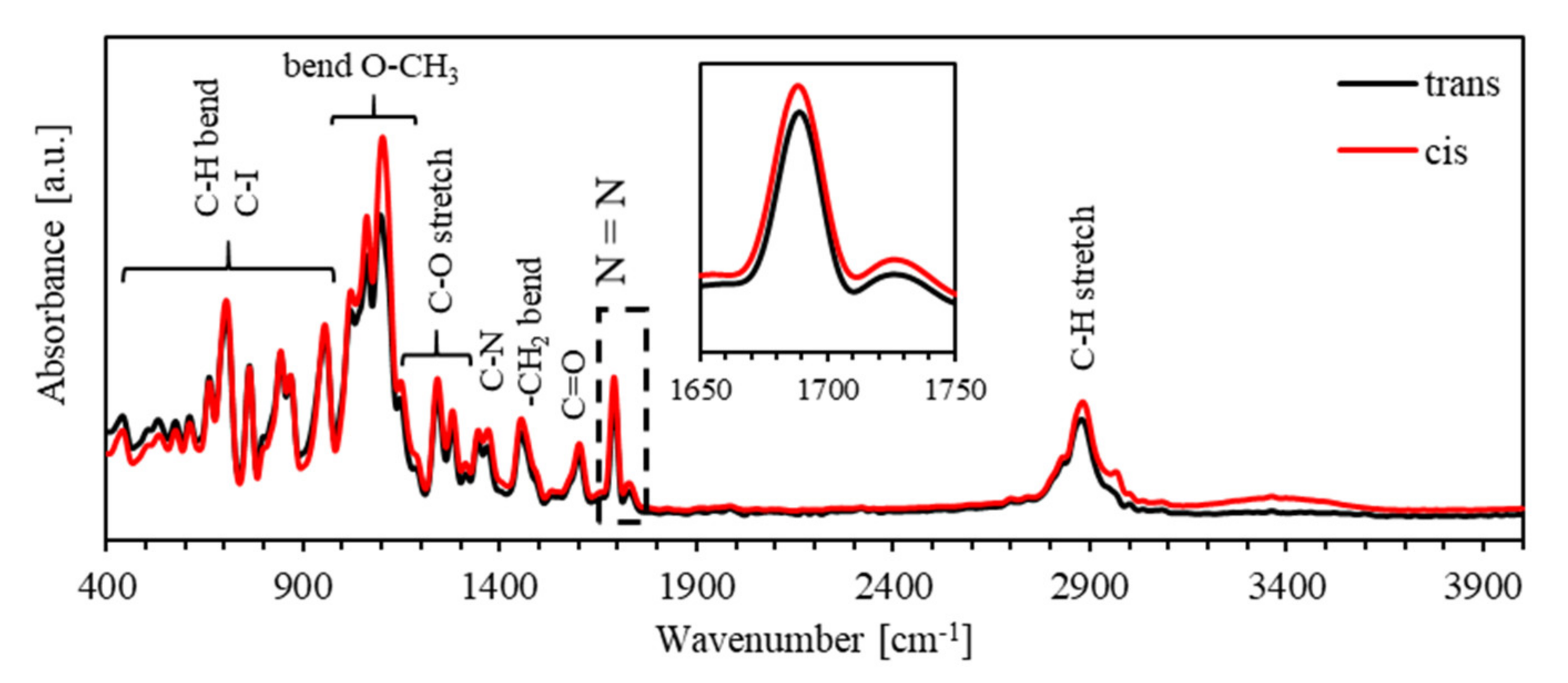
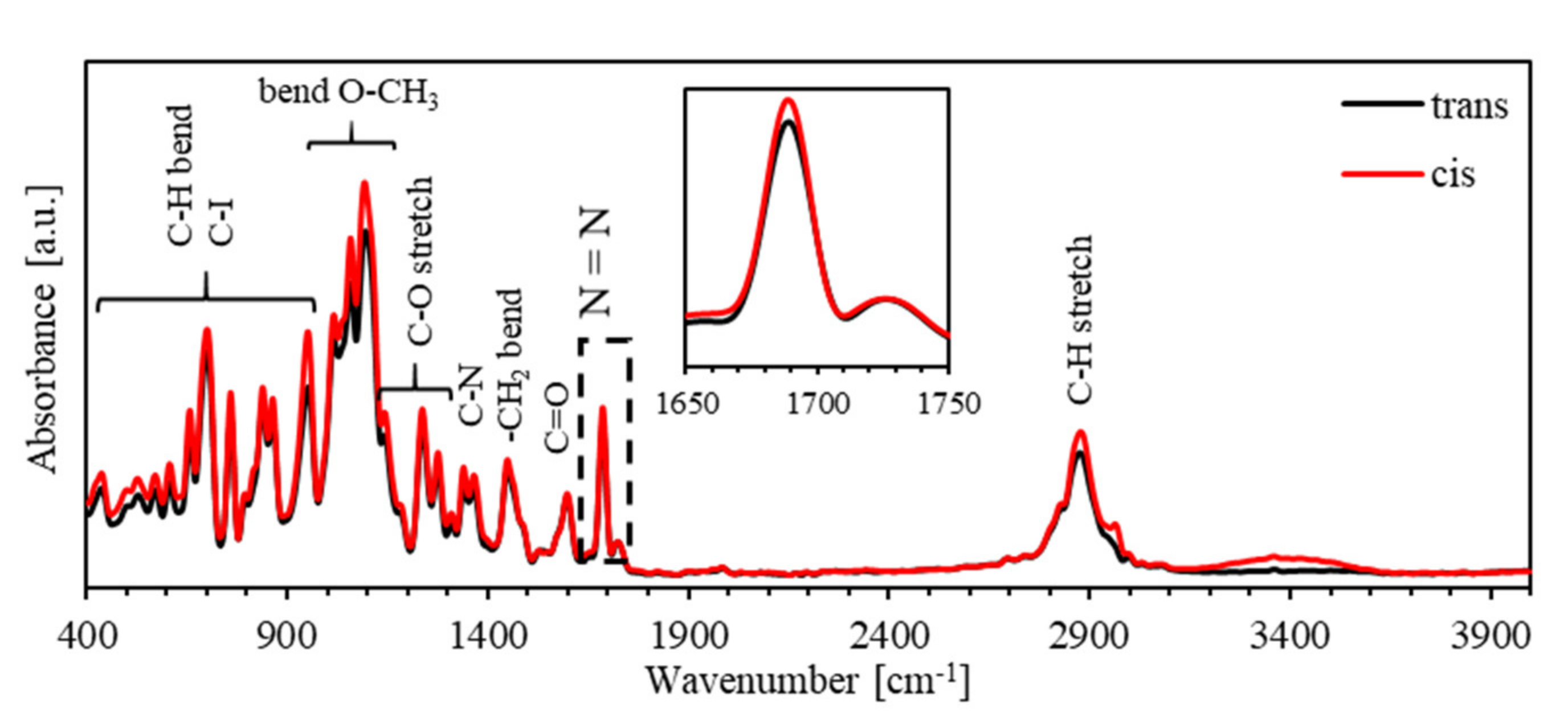
4. Results and Discussion
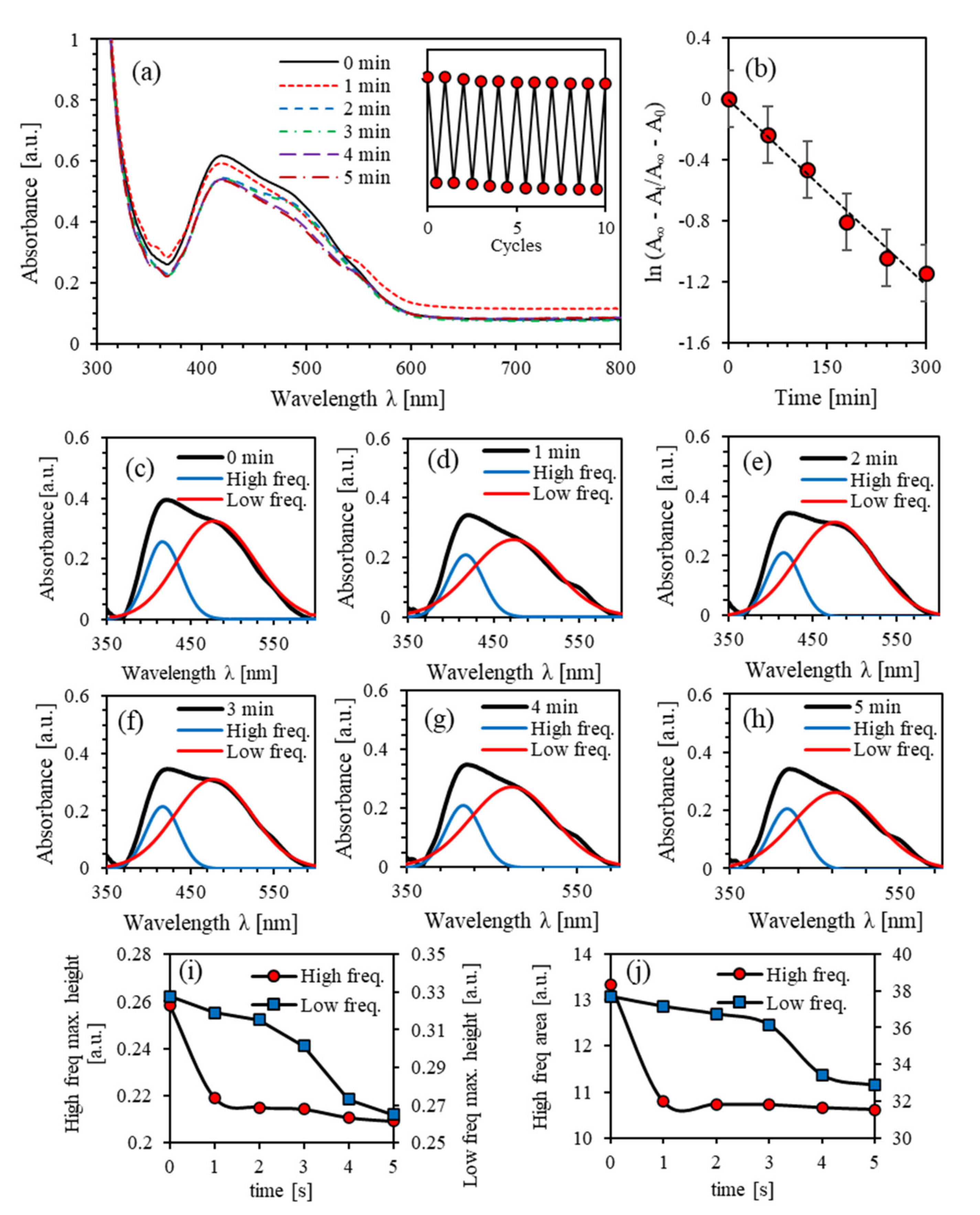
4.1. Photoisomerization Kinetics of PEO-BDK-MR
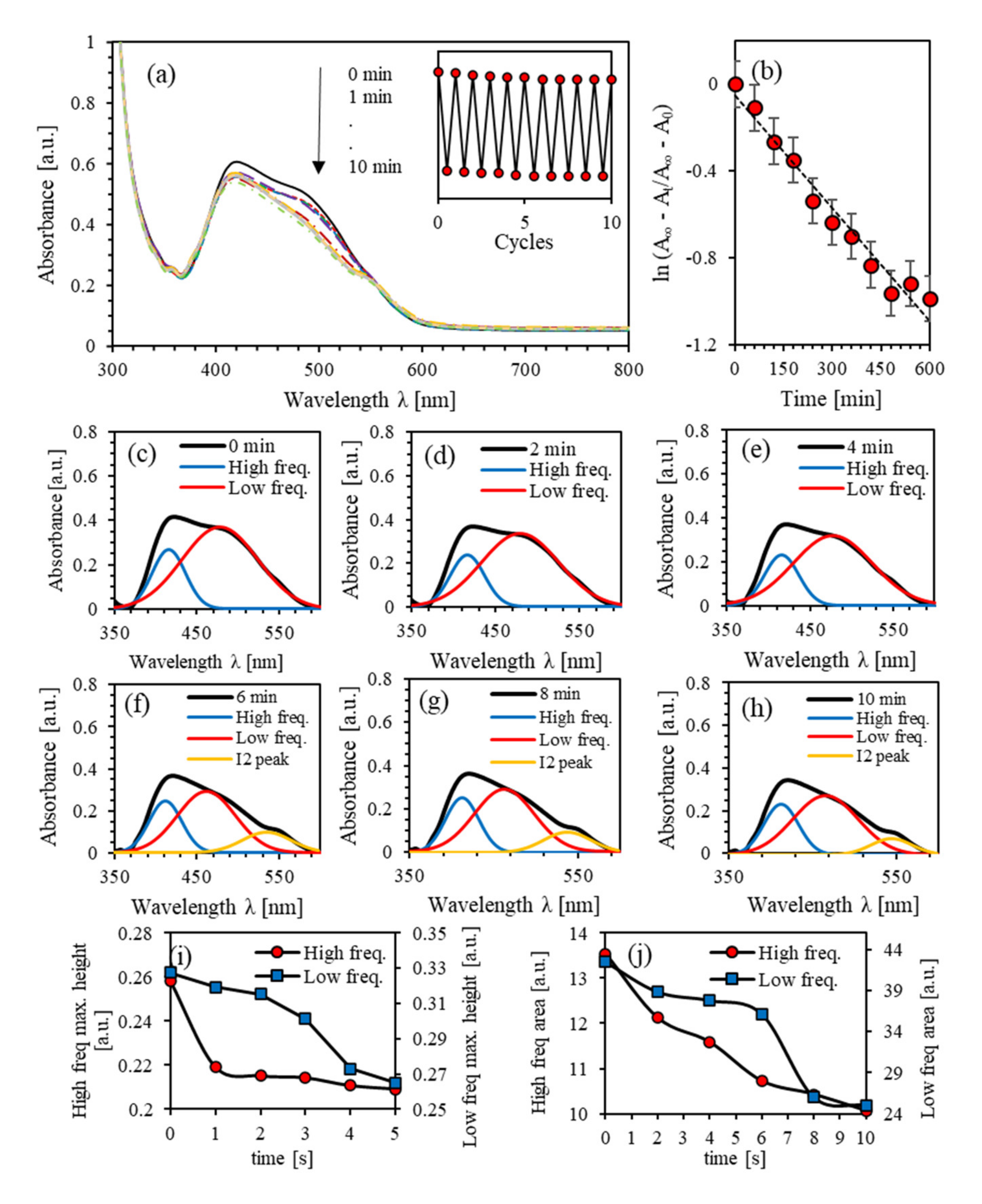
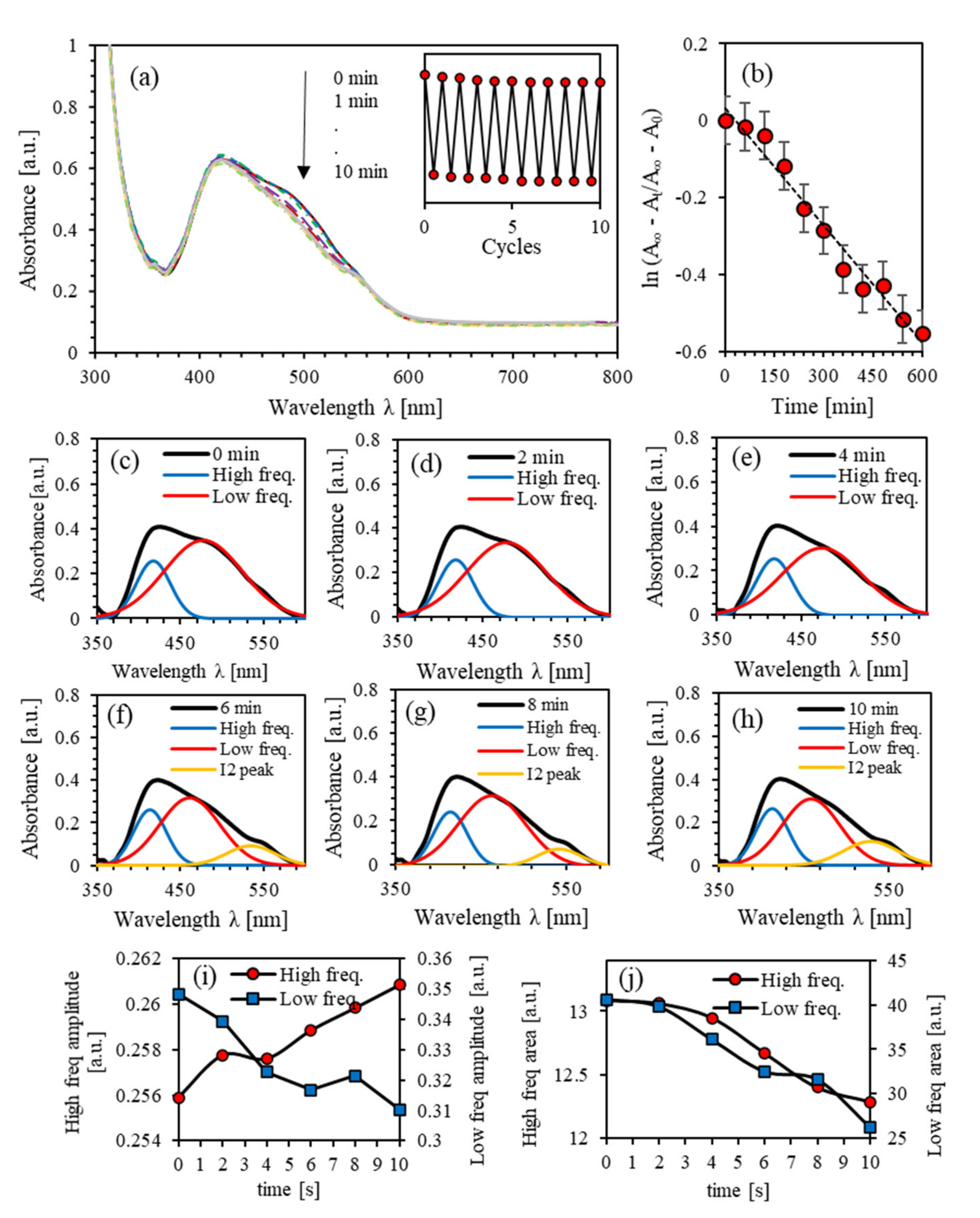
4.2. Photoisomerization Kinetics of (PEO-BDK-MR)/I2 (0.1%)
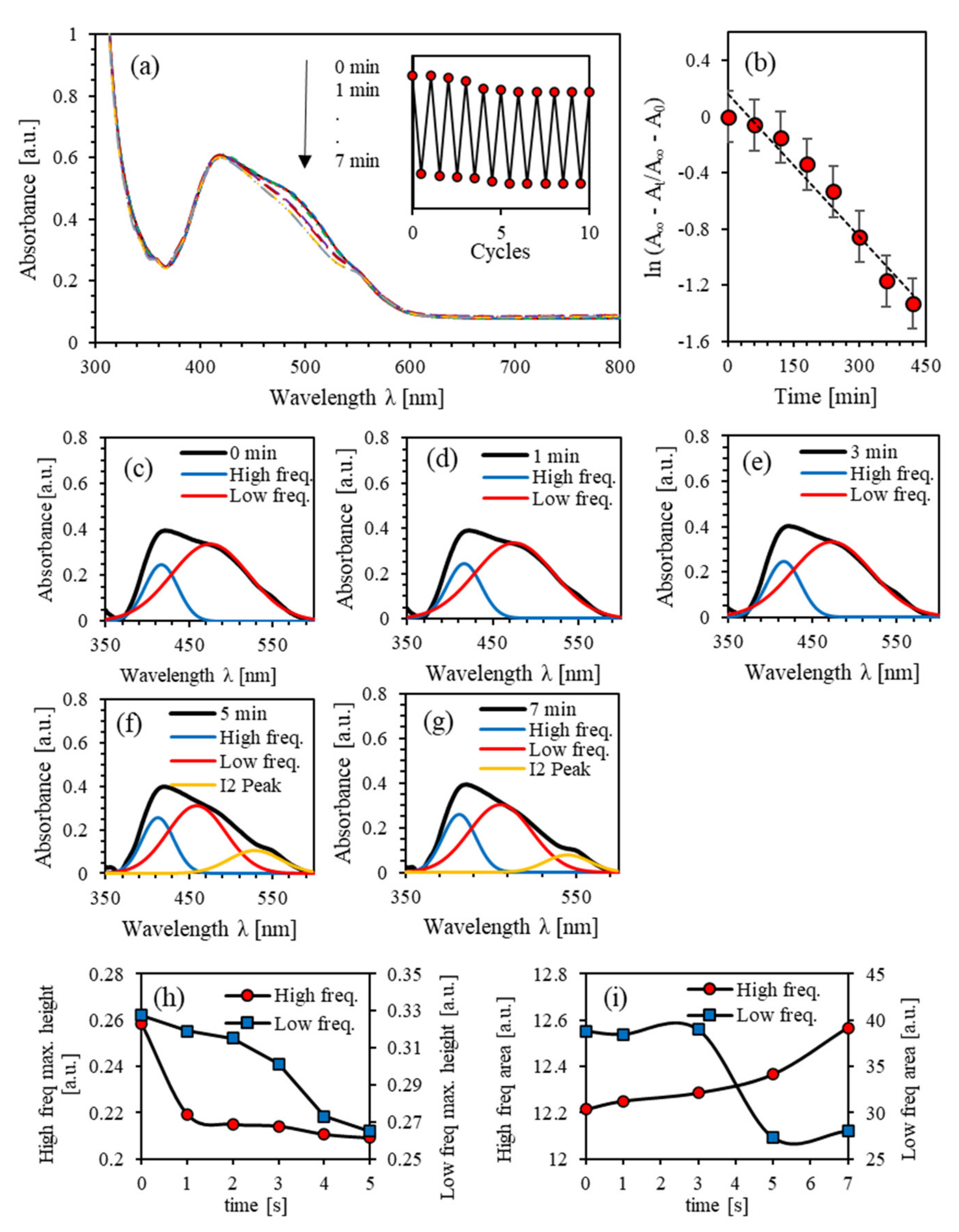
4.3. Photoisomerization Kinetics of (PEO-BDK-MR)/I2 (1.0%)
4.4. Photoisomerization Kinetics of (PEO-BDK-MR)/I2 (10.0%)
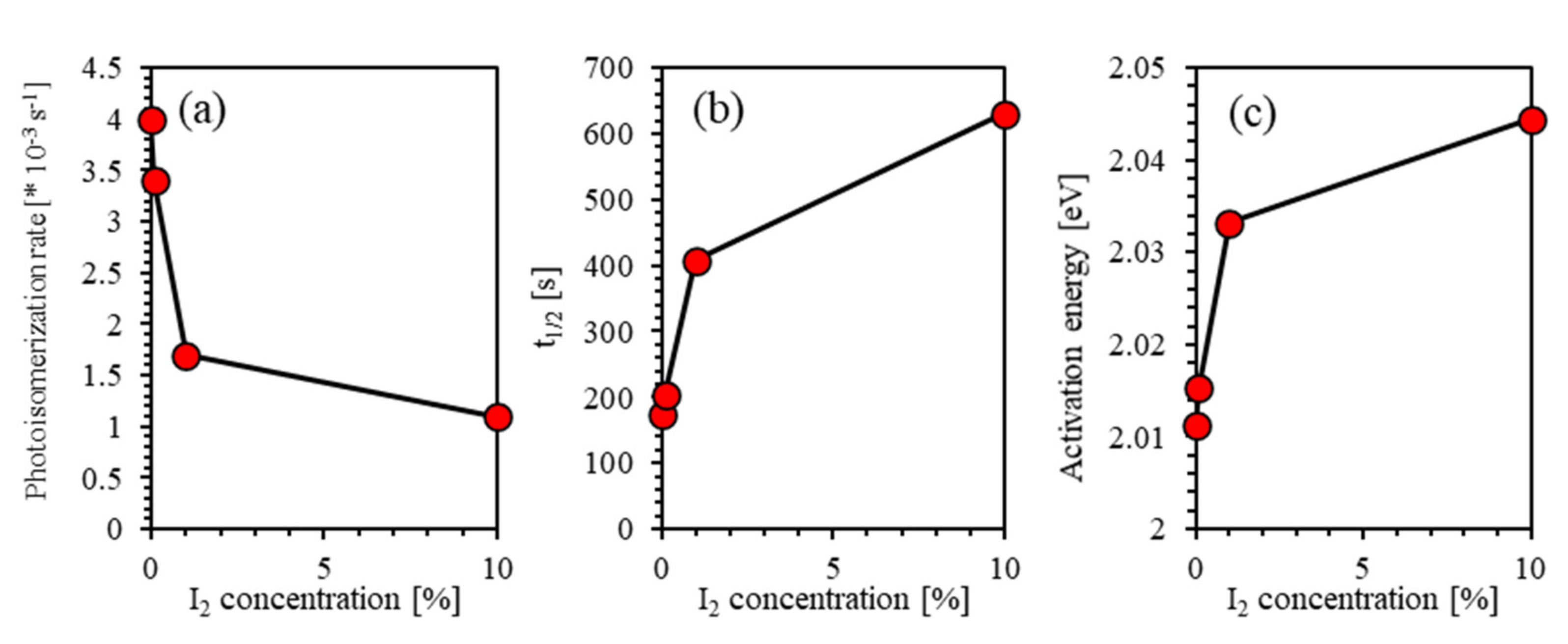
4.5. Effect of Iodine Filler on Photoisomerization Kinetics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.-Y.; He, Y.; Wang, Z.; Xu, J.; Xie, M.; Tao, P.; Ji, D.; Moth-Poulsen, K.; Li, T. Photochemical Phase Transitions Enable Coharvesting of Photon Energy and Ambient Heat for Energetic Molecular Solar Thermal Batteries That Upgrade Thermal Energy. J. Am. Chem. Soc. 2020, 142, 12256–12264. [Google Scholar] [CrossRef]
- Petersen, A.U.; Hofmann, A.I.; Fillols, M.; Mansø, M.; Jevric, M.; Wang, Z.; Sumby, C.J.; Müller, C.; Moth-Poulsen, K. Solar Energy Storage by Molecular Norbornadiene–Quadricyclane Photoswitches: Polymer Film Devices. Adv. Sci. 2019, 6, 1900367. [Google Scholar] [CrossRef] [Green Version]
- Mulatihan, D.N.; Guo, T.; Zhao, Y. Azobenzene Photoswitch for Isomerization-Dependent Cancer Therapy via Azo-Combretastatin A4 and Phototrexate. Photochem. Photobiol. 2020, 96, 1163–1168. [Google Scholar] [CrossRef]
- Matera, C.; Gomila-Juaneda, A.; Camarero, N.; Libergoli, M.; Soler, C.; Gorostiza, P. Photoswitchable Antimetabolite for Targeted Photoactivated Chemotherapy. J. Am. Chem. Soc. 2018, 140, 15764–15773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masutani, K.; Morikawa, M.-A.; Kimizuka, N. A liquid azobenzene derivative as a solvent-free solar thermal fuel. Chem. Commun. 2014, 50, 15803–15806. [Google Scholar] [CrossRef]
- Tebikachew, B.E.; Edhborg, F.; Kann, N.; Albinsson, B.; Moth-Poulsen, K. Turn-off mode fluorescent norbornadiene-based photoswitches. Phys. Chem. Chem. Phys. 2018, 20, 23195–23201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Roffey, A.; Losantos, R.; Lennartson, A.; Jevric, M.; Petersen, A.U.; Quant, M.; Dreos, A.; Wen, X.; Sampedro, D.; et al. Macroscopic heat release in a molecular solar thermal energy storage system. Energy Environ. Sci. 2018, 12, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Udmark, J.; Börjesson, K.; Rodrigues, R.; Roffey, A.; Abrahamsson, M.; Nielsen, M.B.; Moth-Poulsen, K. Evaluating dihydroazulene/vinylheptafulvene photoswitches for solar energy storage applications. ChemSusChem 2017, 10, 3049. [Google Scholar] [CrossRef] [PubMed]
- Moth-Poulsen, K.; Ćoso, D.; Börjesson, K.; Vinokurov, N.; Meier, S.K.; Majumdar, A.; Vollhardt, K.P.C.; Segalman, R.A. Molecular solar thermal (MOST) energy storage and release system. Energy Environ. Sci. 2012, 5, 8534–8537. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hughes, R.P.; Aprahamian, I. Near-Infrared Light Activated Azo-BF2 Switches. J. Am. Chem. Soc. 2014, 136, 13190–13193. [Google Scholar] [CrossRef]
- Zhang, Z.; He, Y.; Zhou, Y.; Yu, C.; Han, L.; Li, T. Pyrazolylazophenyl Ether-Based Photoswitches: Facile Synthesis, (Near-)Quantitative Photoconversion, Long Thermal Half-Life, Easy Functionalization, and Versatile Applications in Light-Responsive Systems. Chem. A Eur. J. 2019, 25, 13402–13410. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Marazzi, M.; Frutos, L.M.; Sampedro, D. E/Z Photochemical switches: Syntheses, properties and applications. RSC Adv. 2013, 3, 6241–6266. [Google Scholar] [CrossRef]
- Cembran, A.; Bernardi, F.; Garavelli, M.; Gagliardi, A.L.; Orlandi, G. On the Mechanism of the cis−trans Isomerization in the Lowest Electronic States of Azobenzene: S0, S1, and T1. J. Am. Chem. Soc. 2004, 126, 3234–3243. [Google Scholar] [CrossRef] [Green Version]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef]
- Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Nather, C.; Renth, F.; Temps, F. Highly efficient reversible Z− E photoisomerization of a bridged azobenzene with visible light through resolved S1 (nπ*) absorption bands. J. Am. Chem. Soc. 2009, 131, 15594–15595. [Google Scholar] [CrossRef]
- Bléger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef]
- Al-Bataineh, Q.M.; Ahmad, A.A.; Alsaad, A.M.; Telfah, A. New Insight on Photoisomerization Kinetics of Photo-Switchable Thin Films Based on Azobenzene/Graphene Hybrid Additives in Polyethylene Oxide. Polymer 2020, 12, 2954. [Google Scholar] [CrossRef]
- Al-Bataineh, Q.M.; Ahmad, A.A.; Alsaad, A.M.; Qattan, I.A.; Bani-Salameh, A.A.; Telfah, A.D. Kinematics of Photoisomerization Processes of PMMA-BDK-MR Polymer Composite Thin Films. Polymer 2020, 12, 1275. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Alsaad, A.M.; Al-Bataineh, Q.M.; Al-Akhras, M.-A.H.; Albataineh, Z.; Alizzy, K.A.; Daoud, N.S. Synthesis and characterization of ZnO NPs-doped PMMA-BDK-MR polymer-coated thin films with UV curing for optical data storage applications. Polym. Bull. 2021, 78, 1189–1211. [Google Scholar] [CrossRef]
- Alsaad, A.M.; Al-Bataineh, Q.M.; Telfah, M.; Ahmad, A.A.; AlBataineh, Z.; Telfah, A. Optical properties and photo-isomerization processes of PMMA–BDK–MR nanocomposite thin films doped by silica nanoparticles. Polym. Bull. 2020, 1–17. [Google Scholar] [CrossRef]
- Le, T.-H.; Kim, Y.; Yoon, H. Electrical and Electrochemical Properties of Conducting Polymers. Polymer 2017, 9, 150. [Google Scholar] [CrossRef]
- Palacio, G.; Pulcinelli, S.H.; Mahiou, R.; Boyer, D.; Chadeyron, G.; Santilli, C.V. Coupling Photoluminescence and Ionic Conduction Properties Using the Different Coordination Sites of Ureasil–Polyether Hybrid Materials. ACS Appl. Mater. Interfaces 2018, 10, 37364–37373. [Google Scholar] [CrossRef]
- Morgenstern, K. Isomerization reactions on single adsorbed molecules. Acc. Chem. Res. 2009, 42, 213–223. [Google Scholar] [CrossRef]
- Henzl, J.; Mehlhorn, M.; Gawronski, H.; Rieder, K.H.; Morgenstern, K. Reversible cis–trans isomerization of a single azobenzene molecule. Angew. Chem. Int. Ed. 2006, 45, 603–606. [Google Scholar] [CrossRef]
- Choi, B.-Y.; Kahng, S.-J.; Kim, S.; Kim, H.; Kim, H.W.; Song, Y.J.; Ihm, J.; Kuk, Y. Conformational Molecular Switch of the Azobenzene Molecule: A Scanning Tunneling Microscopy Study. Phys. Rev. Lett. 2006, 96, 156106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshima, H.; Ojima, N.; Uchimoto, H. Mechanical Motion of Azobenzene Crystals upon Photoirradiation. J. Am. Chem. Soc. 2009, 131, 6890–6891. [Google Scholar] [CrossRef]
- Rau, H. Photoisomerization of Azobenzenes. Photoreact. Org. Thin Films 2002, 2, 3–47. [Google Scholar] [CrossRef]
- Shi, Y.; Steier, W.H.; Yu, L.; Chen, M.; Dalton, L.R. Large stable photoinduced refractive index change in a nonlinear optical polyester polymer with disperse red side groups. Appl. Phys. Lett. 1991, 58, 1131–1133. [Google Scholar] [CrossRef]
- Sekkat, Z.; Morichère, D.; Dumont, M.; Loucif-Saìbi, R.; Delaire, J.A. Photoisomerization of azobenzene derivatives in polymeric thin films. J. Appl. Phys. 1992, 71, 1543–1545. [Google Scholar] [CrossRef]
- Roseanne, J.; Repines, S.; Szarka, A.; Hochstrasser, R. Femtosecond laser studies of cw-stilbene photoisomerization reaction. J. Chem. Phys. 1993, 98, 6291–6315. [Google Scholar]
- Nägele, T.; Hoche, R.; Zinth, W.; Wachtveitl, J. Femtosecond photoisomerization of cis-azobenzene. Chem. Phys. Lett. 1997, 272, 489–495. [Google Scholar] [CrossRef]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Coelho, P.J.; Sousa, C.M.; Castro, M.C.R.; Fonseca, A.M.C.; Raposo, M.M.M. Fast thermal cis–trans isomerization of heterocyclic azo dyes in PMMA polymers. Opt. Mater. 2013, 35, 1167–1172. [Google Scholar] [CrossRef] [Green Version]
- Feringa, B.L.; Jager, W.F.; de Lange, B. Organic materials for reversible optical data storage. Tetrahedron 1993, 49, 8267–8310. [Google Scholar] [CrossRef]
- Bortolus, P.; Monti, S. Cis-trans photoisomerization of azobenzene. Solvent and triplet donors effects. J. Phys. Chem. 1979, 83, 648–652. [Google Scholar] [CrossRef]
- Rodier, J.M.; Myers, A.B. cis-Stilbene photochemistry: Solvent dependence of the initial dynamics and quantum yields. J. Am. Chem. Soc. 1993, 115, 10791–10795. [Google Scholar] [CrossRef]
- Grabowski, D.; Chudzicka-Czupała, A.; Chrupała-Pniak, M.; Mello, A.L.; Paruzel-Czachura, M. Work ethic and organizational commitment as conditions of unethical pro-organizational behavior: Do engaged workers break the ethical rules? Int. J. Sel. Assess. 2019, 27, 193–202. [Google Scholar] [CrossRef]
- Athar, M.R.; Shahzad, K.; Ahmad, J.; Ijaz, M.S. Impact of Islamic work ethics on organizational commitment: Mediating role of job satisfaction. J. Islamic Bus. Manag. 2016, 6, 397–416. [Google Scholar]
- Feng, Y.; Liu, H.; Luo, W.; Liu, E.; Zhao, N.; Yoshino, K.; Feng, W. Covalent functionalization of graphene by azobenzene with molecular hydrogen bonds for long-term solar thermal storage. Sci. Rep. 2013, 3, 1–8. [Google Scholar] [CrossRef]
- Kucharski, T.J.; Tian, Y.; Akbulatov, S.; Boulatov, R. Chemical solutions for the closed-cycle storage of solar energy. Energy Environ. Sci. 2011, 4, 4449–4472. [Google Scholar] [CrossRef]
- Kunz, A.; Heindl, A.H.; Dreos, A.; Wang, Z.; Moth-Poulsen, K.; Becker, J.; Wegner, H.A. Intermolecular London Dispersion Interactions of Azobenzene Switches for Tuning Molecular Solar Thermal Energy Storage Systems. ChemPlusChem 2019, 84, 1145–1148. [Google Scholar] [CrossRef] [Green Version]
- Lennartson, A.; Roffey, A.; Moth-Poulsen, K. Designing photoswitches for molecular solar thermal energy storage. Tetrahedron Lett. 2015, 56, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Merino, E. Synthesis of azobenzenes: The coloured pieces of molecular materials. Chem. Soc. Rev. 2011, 40, 3835–3853. [Google Scholar] [CrossRef]
- Schweighauser, L.; Strauss, M.A.; Bellotto, S.; Wegner, H.A. Attraction or Repulsion? London Dispersion Forces Control Azobenzene Switches. Angew. Chem. Int. Ed. 2015, 54, 13436–13439. [Google Scholar] [CrossRef]
- Durgun, E.; Grossman, J.C. Photoswitchable Molecular Rings for Solar-Thermal Energy Storage. J. Phys. Chem. Lett. 2013, 4, 854–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, R.; Hostettler, N.; Neuburger, M.; Wegner, H.A. Oligoazobenzenes-switching in a new dimension. Chimia 2010, 64, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Kucharski, T.J.; Ferralis, N.; Kolpak, A.M.; Zheng, J.O.; Nocera, D.G.; Grossman, J.C. Templated assembly of photoswitches significantly increases the energy-storage capacity of solar thermal fuels. Nat. Chem. 2014, 6, 441–447. [Google Scholar] [CrossRef]
- Pang, W.; Xue, J.; Pang, H. A High Energy Density Azobenzene/Graphene Oxide Hybrid with Weak Nonbonding Interactions for Solar Thermal Storage. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Al-Abdallat, Y.; Jum’h, I.; Al Bsoul, A.; Jumah, R.; Telfah, A. Photocatalytic degradation dynamics of methyl orange using coprecipitation synthesized fe 3 o 4 nanoparticles. Water Air Soil Pollut. 2019, 230, 1–16. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Ozen, A.S.; Doruker, P.; Aviyente, V. Effect of Cooperative Hydrogen Bonding in Azo−Hydrazone Tautomerism of Azo Dyes. J. Phys. Chem. A 2007, 111, 13506–13514. [Google Scholar] [CrossRef]
- Ruzza, P.; Hussain, R.; Biondi, B.; Calderan, A.; Tessari, I.; Bubacco, L.; Siligardi, G. Effects of Trehalose on Thermodynamic Properties of Alpha-synuclein Revealed through Synchrotron Radiation Circular Dichroism. Biomolecules 2015, 5, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.J.; Kim, N.; Lee, M. Photophysical properties and photoisomerization processes of Methyl Red embedded in rigid polymer. Appl. Opt. 1995, 34, 138–143. [Google Scholar] [CrossRef]
- Abu-Jamous, A.; Zihlif, A. Study of the electrical conduction in poly(ethylene oxide) doped with iodine. Phys. B Condens. Matter 2010, 405, 2762–2767. [Google Scholar] [CrossRef]
- Gabes, W.; Stufkens, D. Electronic absorption spectra of symmetrical and asymmetrical trihalide ions. Spectrochim. Acta Part A Mol. Spectrosc. 1974, 30, 1835–1841. [Google Scholar] [CrossRef]
- Zidan, H.M.; Abdelrazek, E.M.; Abdelghany, A.M.; Tarabiah, A.E. Characterization and some physical studies of PVA/PVP filled with MWCNTs. J. Mater. Res. Technol. 2019, 8, 904–913. [Google Scholar] [CrossRef]
- Kim, K.-I.; Kim, D.-A.; Patel, K.D.; Shin, U.S.; Kim, H.-W.; Lee, J.-H.; Lee, H.-H. Carbon nanotube incorporation in PMMA to prevent microbial adhesion. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Seto, M.; Maeda, Y.; Matsuyama, T.; Yamaoka, H.; Sakai, H. Mössbauer spectroscopic study of fullerene C60 doped with iodine. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 1993, 76, 348–349. [Google Scholar] [CrossRef]
- Karlsen, E.M.; Spanget-Larsen, J. FTIR investigation of the reaction between pyridine and iodine in a polyethylene host. Formation of N-iodopyridinium polyiodide. Chem. Phys. Lett. 2009, 473, 227–232. [Google Scholar] [CrossRef]
- Mansø, M.; Petersen, A.U.; Wang, Z.; Erhart, P.; Nielsen, M.B.; Moth-Poulsen, K. Molecular solar thermal energy storage in photoswitch oligomers increases energy densities and storage times. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Bataineh, Q.M.; Ahmad, A.A.; Alsaad, A.M.; Qattan, I.A.; Aljarrah, I.A.; Telfah, A.D. Effect of Iodine Filler on Photoisomerization Kinetics of Photo-Switchable Thin Films Based on PEO-BDK-MR. Polymers 2021, 13, 841. https://doi.org/10.3390/polym13050841
Al-Bataineh QM, Ahmad AA, Alsaad AM, Qattan IA, Aljarrah IA, Telfah AD. Effect of Iodine Filler on Photoisomerization Kinetics of Photo-Switchable Thin Films Based on PEO-BDK-MR. Polymers. 2021; 13(5):841. https://doi.org/10.3390/polym13050841
Chicago/Turabian StyleAl-Bataineh, Qais M., A. A. Ahmad, A. M. Alsaad, I. A. Qattan, Ihsan A. Aljarrah, and Ahmad D. Telfah. 2021. "Effect of Iodine Filler on Photoisomerization Kinetics of Photo-Switchable Thin Films Based on PEO-BDK-MR" Polymers 13, no. 5: 841. https://doi.org/10.3390/polym13050841