
Biobased Thermoplastic Elastomers: Structure-Property Relationship of Poly(hexamethylene 2,5-furanodicarboxylate)-Block-Poly(tetrahydrofuran) Copolymers Prepared by Melt Polycondensation

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
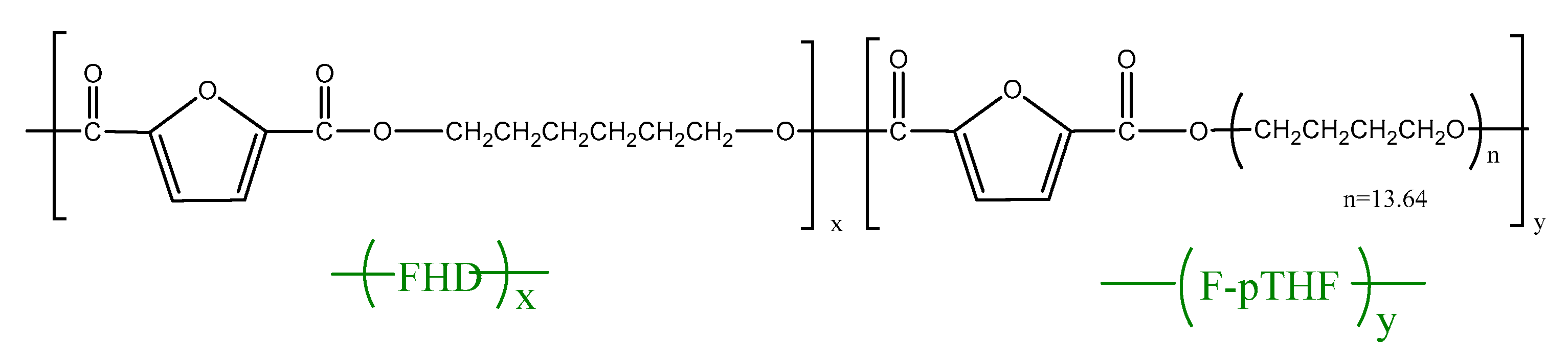
2.1. Synthesis of PHF-b-F-pTHF Copolymers
2.2. Characterization Methods
3. Results and Discussion
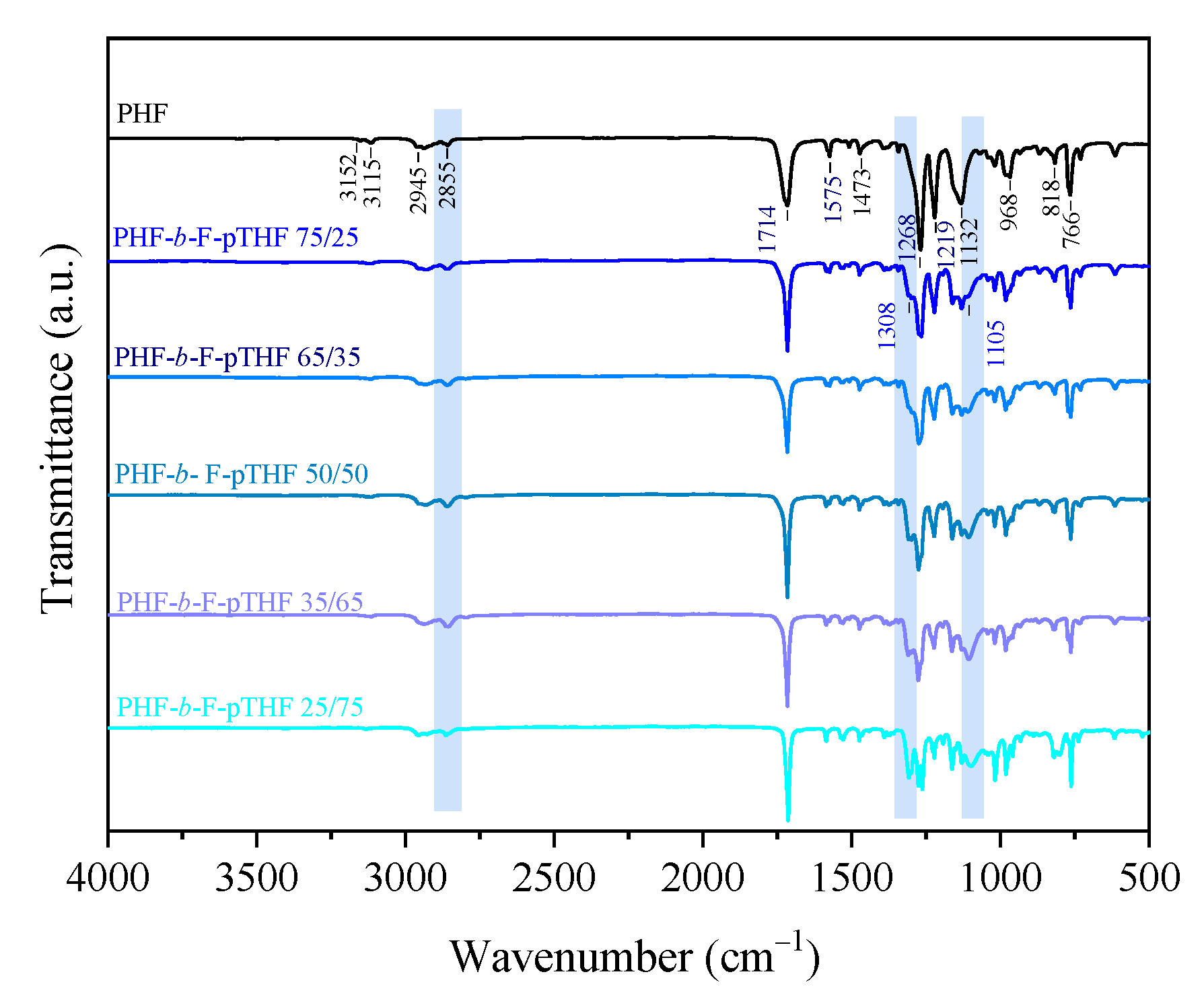
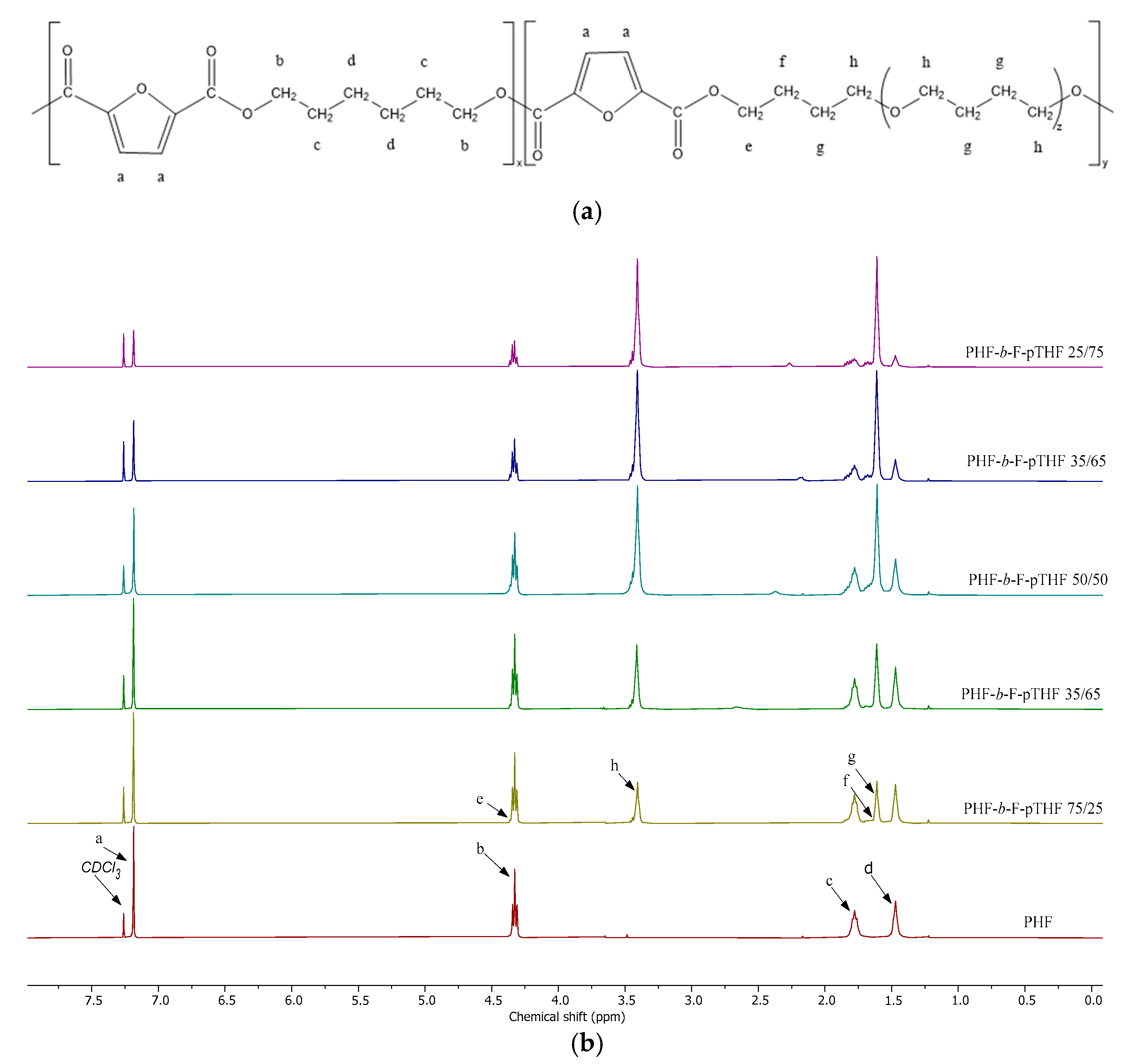
3.1. Analysis of Structure and Composition
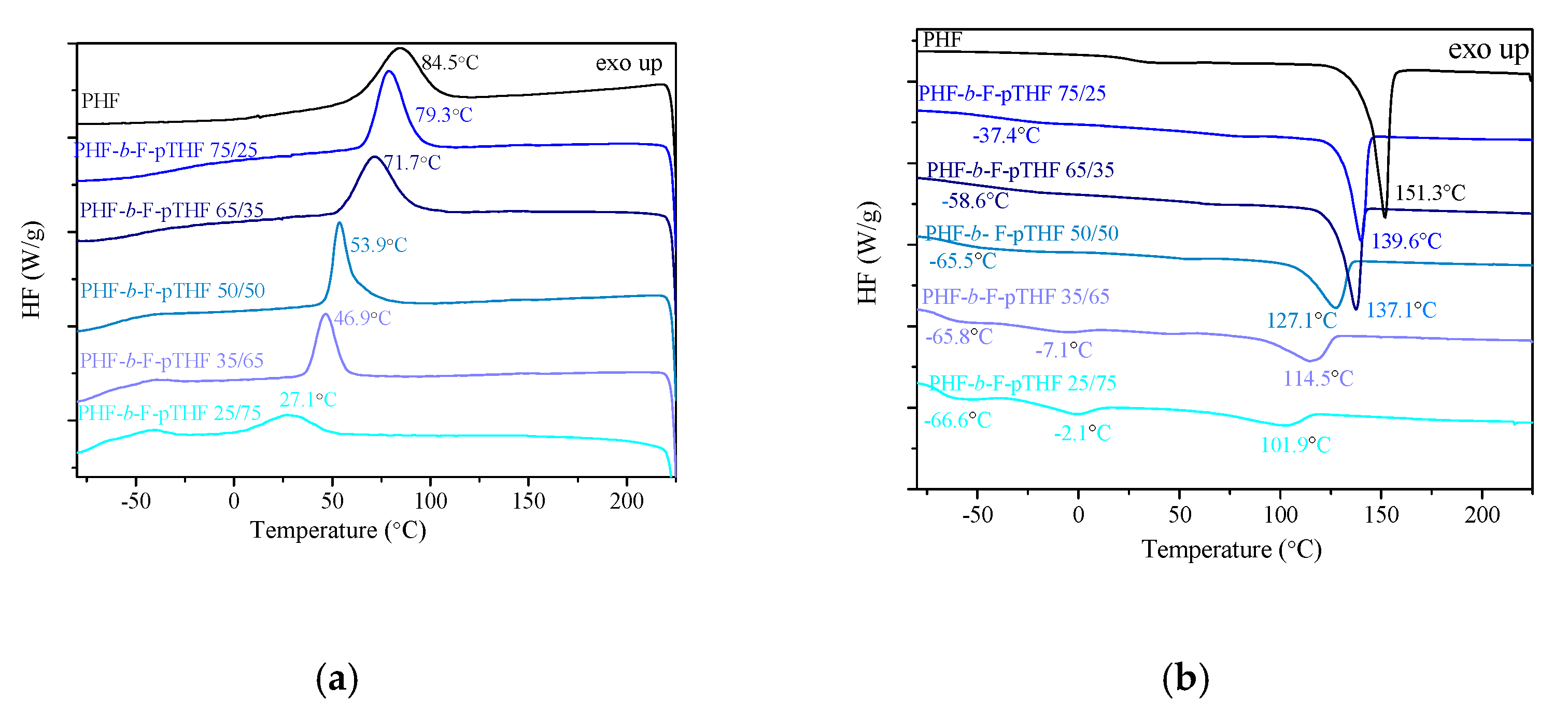
3.2. Phase Structure and Morphology
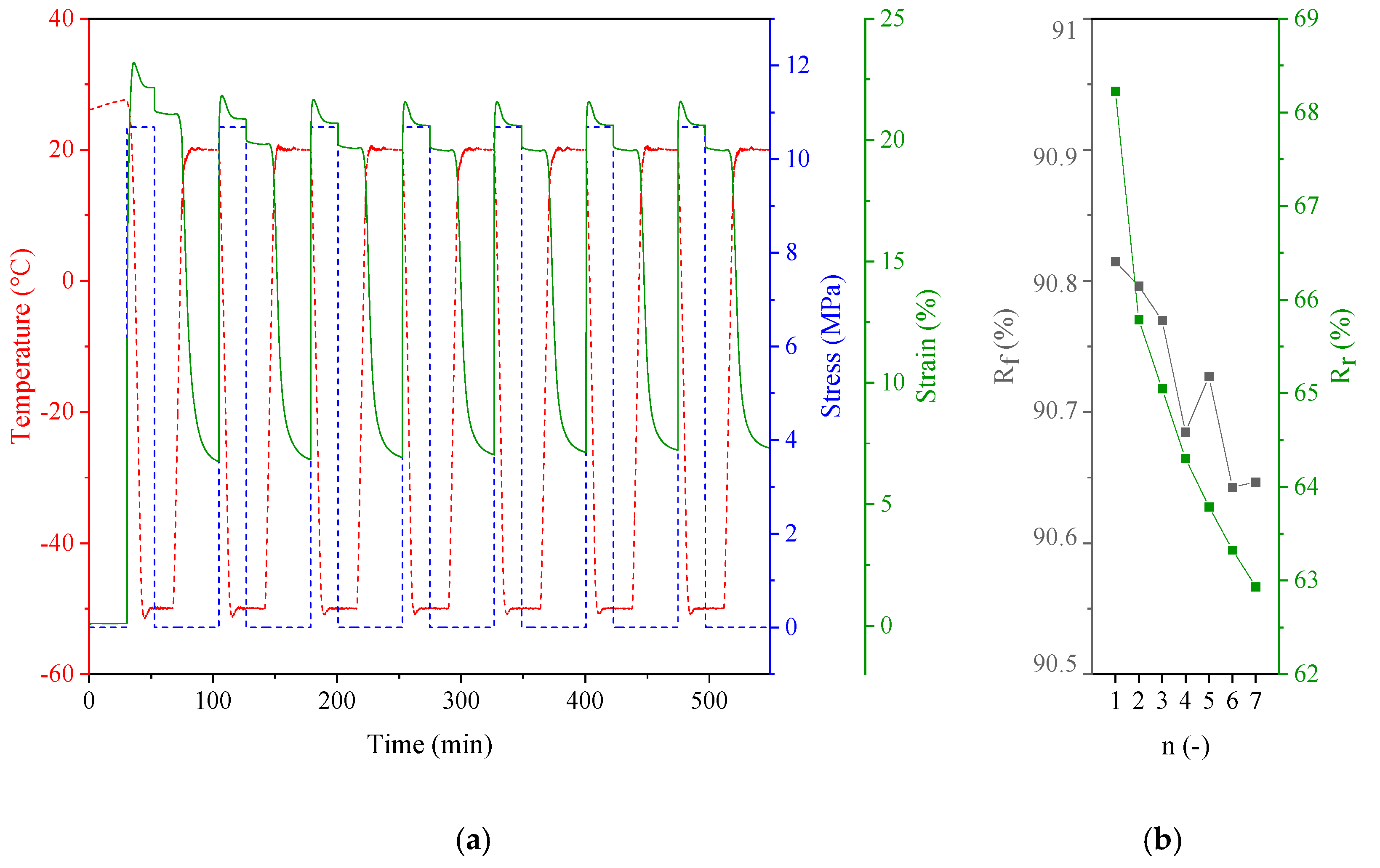
3.3. Shape Memory Behavior
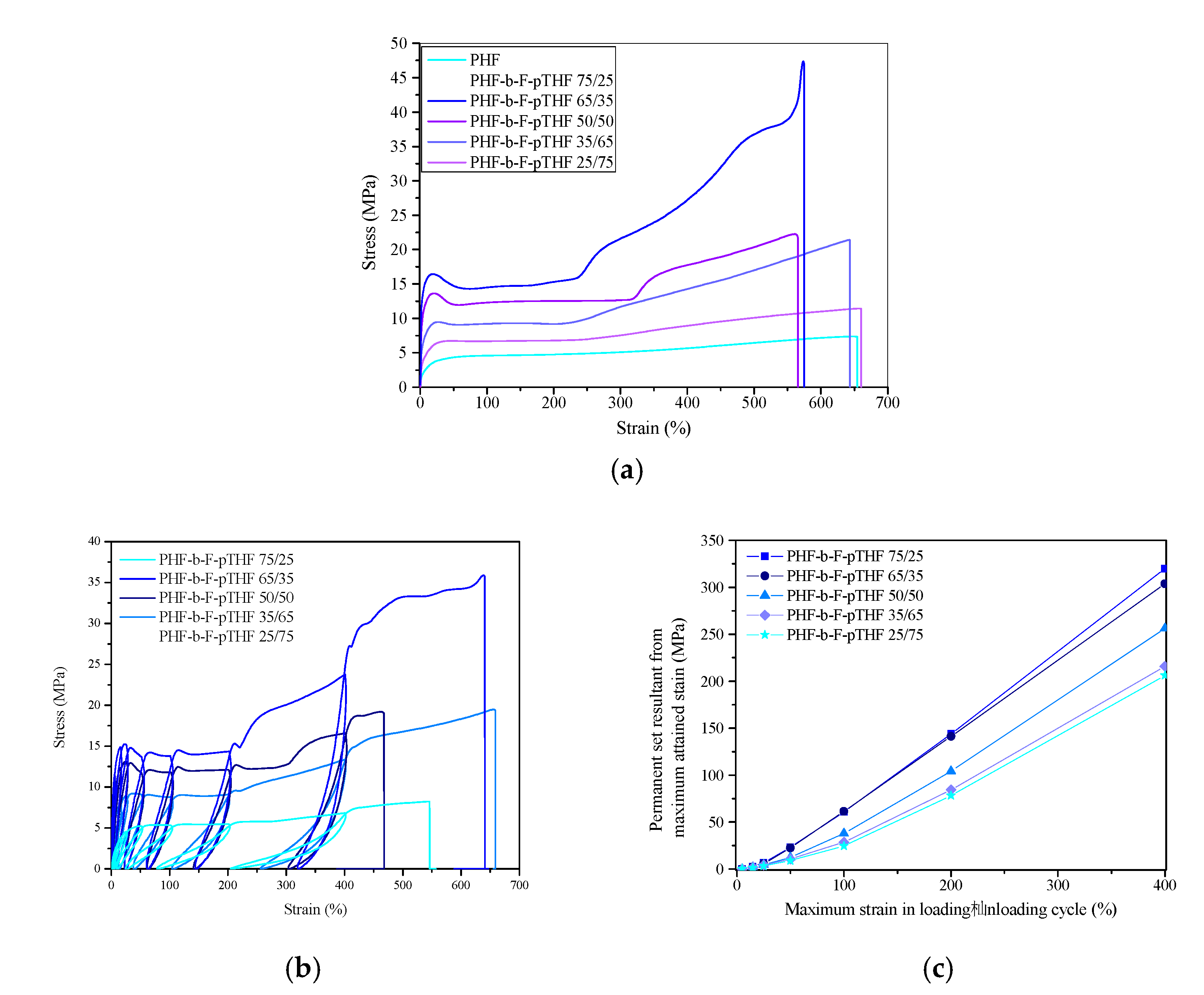
3.4. Mechanical and Elastic Properties of PHF-b-F-pTHF
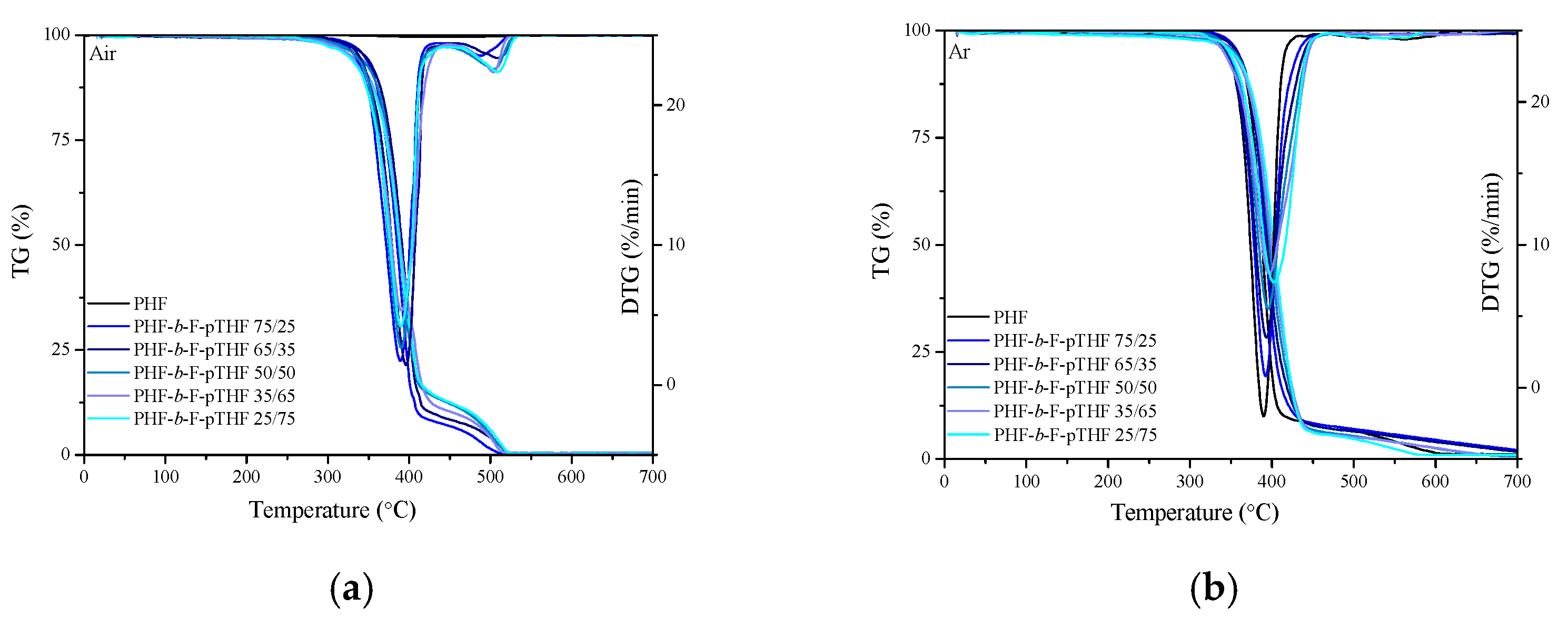
3.5. Thermo-Oxidative and Thermal Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baltá Calleja, F.J.; Roslaniec, Z. Block Copolymers; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Fakirov, S. Handbook of Condensation Thermoplastic Elastomers; Wiley: Hoboken, NJ, USA, 2006; pp. 1–619. [Google Scholar]
- Van Krevelen, D.W. Properties of Polymers; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 9780080548197. [Google Scholar]
- Kobayashi, T.; Matsumura, S. Enzymatic synthesis and properties of novel biodegradable and biobased thermoplastic elastomers. Polym. Degrad. Stab. 2011, 96, 2071–2079. [Google Scholar] [CrossRef]
- BASF Now Offers Bio-Based PolyTHF. Available online: https://www.basf.com/global/en/media/news-releases/2015/03/p-15-163.html (accessed on 27 December 2020).
- Introduction of Bio-Based polyTHF. Available online: http://adbrevio.com/2015/04/04/introduction-of-bio-based-polythf/ (accessed on 27 December 2020).
- Knoop, R.J.I.; Vogelzang, W.; Van Haveren, J.; Van Es, D.S. High molecular weight poly(ethylene-2,5-furanoate); critical aspects in synthesis and mechanical property determination. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4191–4199. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Jia, Z.; Sun, L.; Zhu, J. Highly crystalline polyesters synthesized from furandicarboxylic acid (FDCA): Po-tential bio-based engineering plastic. Eur. Polym. J. 2018, 109, 379–390. [Google Scholar] [CrossRef]
- Wilsens, C.H.R.M. Exploring the Application of 2,5-Furandicarboxylic Acid as a Monomer in High Performance Polymers: Synthesis, Characterization, and Properties. Ph.D. Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2015. [Google Scholar]
- Iwata, T. Biodegradable and Bio-Based Polymers: Future Prospects of Eco-Friendly Plastics. Angew. Chem. Int. Ed. 2015, 54, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.; Gandini, A.; Silvestre, A.J.D.; Reis, B. Synthesis and characterization of poly(2,5-furan dicarboxylate)s based on a variety of diols. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 3759–3768. [Google Scholar] [CrossRef]
- Vannini, M.; Marchese, P.; Celli, A.; Lorenzetti, C. Fully biobased poly(propylene 2,5-furandicarboxylate) for packaging applications: Excellent barrier properties as a function of crystallinity. Green Chem. 2015, 17, 4162–4166. [Google Scholar] [CrossRef]
- Ma, J.; Yu, X.; Xu, J.; Pang, Y. Synthesis and crystallinity of poly(butylene 2,5-furandicarboxylate). Polymer 2012, 53, 4145–4151. [Google Scholar] [CrossRef]
- Moore, J.A.; Kelly, J.E. Thermally initiated crosslinking of an unsaturated heterocyclic polyester. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 2407–2409. [Google Scholar] [CrossRef]
- Gandini, A. The behaviour of furan derivatives in polymerization reactions. Adv. Polym. Sci. 1977, 25, 47–96. [Google Scholar] [CrossRef]
- Gandini, A.; Armando, J.D.; Silvestre, C.P.N.; Souza, A.F.; Gomes, M. The Furan Counterpart of Poly(ethylene terephthalate): An Alternative Material Based on Renewable Resources. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 295–298. [Google Scholar] [CrossRef]
- Sajid, M.; Zhao, X.; Liu, D. Production of 2,5-furandicarboxylic acid (FDCA) from 5-hydroxymethylfurfural (HMF): Recent progress focusing on the chemical-catalytic routes. Green Chem. 2018, 20, 5427–5453. [Google Scholar] [CrossRef]
- Gopalakrishnan, P.; Narayan-Sarathy, S.; Ghosh, T.; Mahajan, K.; Belgacem, N. Synthesis and characterization of bio-based furanic polyesters. J. Polym. Res. 2013, 21, 1–9. [Google Scholar] [CrossRef]
- Wu, L.; Mincheva, R.; Xu, Y.; Raquez, J.M.; Dubois, P. High molecular weight poly(butylene succinate-co-butylene furandi-carboxylate) copolyesters: From catalyzed polycondensation reaction to thermomechanical properties. Biomacromolecules 2012, 13, 2973–2981. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Cai, J.; Xie, W.; Chen, P.H.; Gazzano, M.; Scandola, M.; Gross, R.A. Poly(butylene 2,5-furan dicarboxylate), a bi-obased alternative to PBT: Synthesis, physical properties, and crystal structure. Macromolecules 2013, 46, 796–804. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Tsanaktsis, V.; Papageorgiou, D.G.; Chrissafis, K.; Exarhopoulos, S.; Bikiaris, D.N. Furan-based polyes-ters from renewable resources: Crystallization and thermal degradation behavior of poly(hexamethylene 2,5-furan-dicarboxylate). Eur. Polym. J. 2015, 67, 383–396. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top value added chemicals from biomass. Volume 1-Results of screening for potential candidates from sugars and synthesis gas. Biomass 2004, 1, 1–76. [Google Scholar]
- Wu, M.C.; Woo, E.M.; Yoshioka, T.; Tsuji, M. Thermal analysis, X-ray and electron diffraction studies on crystalline phase transitions in solvent-treated poly(hexamethylene terephthalate). Polymer 2006, 47, 5523–5530. [Google Scholar] [CrossRef]
- Hall, I.; Ibrahim, B. The structure and properties of poly(hexamethylene terephthalate): 1. The preparation, morphology and unit cells of three allomorphs. Polymer 1982, 23, 805–816. [Google Scholar] [CrossRef]
- González-Vidal, N.; Muñoz-Guerra, S.; De Ilarduya, A.M.; Benali, S.; Peeterbroeck, S.; Dubois, P. Poly(hexamethylene terephthalate)–layered silicate nanocomposites. Eur. Polym. J. 2010, 46, 156–164. [Google Scholar] [CrossRef]
- González-Vidal, N.; De Ilarduya, A.M.; Muñoz-Guerra, S.; Castell, P.; Martinez, M.T. Synthesis and properties of poly(hexamethylene terephthalate)/multiwall carbon nanotubes nanocomposites. Compos. Sci. Technol. 2010, 70, 789–796. [Google Scholar] [CrossRef] [Green Version]
- David, C.; Lefèbvre, X.; Lefèvre, C.; Demarteau, W.; Loutz, J.M. Thermal behaviour of polyesters of hexanediol with ter-ephthalic and isophthalic acids. Prog. Org. Coat. 1999, 35, 45–54. [Google Scholar] [CrossRef]
- Moore, J.A.; Kelly, J.E. Polyesters Derived from Furan and Tetrahydrofuran Nuclei. Macromolecules 1978, 11, 568–573. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, Q.; Zhang, Q.; Ye, C.; Zhou, G. A series of furan-aromatic polyesters synthesized via direct esterification method based on renewable resources. J. Polym. Sci. Part A Polym. Chem. 2011, 50, 1026–1036. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Tsanaktsis, V.; Nerantzaki, M.; Papageorgiou, G.Z.; Bikiaris, D.N. Decomposition mechanism of polyesters based on 2,5-furandicarboxylic acid and aliphatic diols with medium and long chain methylene groups. Polym. Degrad. Stab. 2016, 132, 127–136. [Google Scholar] [CrossRef]
- Haernvall, K.; Zitzenbacher, S.; Amer, H.; Zumstein, M.T.; Sander, M.; McNeill, K.; Yamamoto, M.; Schick, M.B.; Ribitsch, D.; Guebitz, G.M. Polyol Structure Influences Enzymatic Hydrolysis of Bio-Based 2,5-Furandicarboxylic Acid (FDCA) Polyesters. Biotechnol. J. 2017, 12, 1600741. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, Q.; Xie, W.; Peng, L.; He, L.; He, Z.; Chowdhury, S.P.; Christensen, R.; Ni, Y. An Eco-Friendly Method to Get a Bio-Based Dicarboxylic Acid Monomer 2,5-Furandicarboxylic Acid and Its Application in the Synthesis of Poly(hexylene 2,5-furandicarboxylate) (PHF). Polymers 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Y.; Qi, Z.; He, L.; Peng, L. Progress in the Synthesis and Properties of 2,5-Furan Dicarboxylate based Polyesters. BioResources 2020, 15, 4502–4527. [Google Scholar]
- Rennovia Enters Piloting Stage of Its Bio-Based 1,6-hexanediol Process. Available online: https://www.bioplasticsmagazine.com/en/news/meldungen/20170329-Rennovia-takes-step-towards-commercialization-of-biobased-platform.php (accessed on 27 December 2020).
- Imhof, P.; Van Der Waal, J.C. Catalytic Process Development for Renewable Materials; Wiley-VCH: Hoboken, NJ, USA, 2013; ISBN 978-3-527-65665-3. [Google Scholar]
- Wang, G.; Jiang, M.; Zhang, Q.; Wang, R.; Qu, X.; Zhou, G. Biobased multiblock copolymers: Synthesis, properties and shape memory behavior of poly(hexamethylene 2,5-furandicarboxylate)-b-poly(ethylene glycol). Polym. Degrad. Stab. 2018, 153, 292–297. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, M.; Zhang, Q.; Wang, R.; Qu, X.; Zhou, G. Poly(hexamethylene 2,5-furandicarboxylate) copolyesters con-taining phosphorus: Synthesis, crystallization behavior, thermal, mechanical and flame retardant properties. Polym. Degrad. Stab. 2018, 153, 272–280. [Google Scholar] [CrossRef]
- Xie, H.; Wu, L.; Li, B.-G.; Dubois, P. Biobased Poly(ethylene-co-hexamethylene 2,5-furandicarboxylate) (PEHF) Copolyesters with Superior Tensile Properties. Ind. Eng. Chem. Res. 2018, 57, 13094–13102. [Google Scholar] [CrossRef]
- Xie, F.; Huang, C.; Wang, F.; Huang, L.; Weiss, R.A.; Leng, J.; Liu, Y. Carboxyl-Terminated Polybutadi-ene-Poly(styrene-co-4-vinylpyridine) Supramolecular Thermoplastic Elastomers and Their Shape Memory Behavior. Macromolecules 2016, 49, 7322–7330. [Google Scholar] [CrossRef]
- Irska, I.; Paszkiewicz, S.; Goracy, K.; Linares, A.; Ezquerra, T.A.; Jedrzejewski, R.; Roslaniec, Z.; Piesowicz, E. Poly(butylene terephthalate)/polylactic acid based copolyesters and blends: Miscibility-structure-property relationship. Express Polym. Lett. 2020, 14, 26–47. [Google Scholar] [CrossRef]
- Szymczyk, A.; Senderek, E.; Nastalczyk, J.; Roslaniec, Z. New multiblock poly(ether-ester)s based on poly(trimethylene terephthalate) as rigid segments. Eur. Polym. J. 2008, 44, 436–443. [Google Scholar] [CrossRef]
- Szymczyk, A. Structure and properties of new polyester elastomers composed of poly(trimethylene terephthalate) and poly(ethylene oxide). Eur. Polym. J. 2009, 45, 2653–2664. [Google Scholar] [CrossRef]
- Piesowicz, E.; Paszkiewicz, S.; Szymczyk, A. Phase separation and elastic properties of poly(trimethylene tereph-thalate)-blockpoly(ethylene oxide) copolymers. Polymers 2016, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Irska, I.; Linares, A.; Piesowicz, E.; Paszkiewicz, S.; Rosłaniec, Z.; Nogales, A.; Šics, I. Dielectric spectroscopy of novel bio-based aliphatic-aromatic block copolymers: Poly(butylene terephthalate)-b-poly(lactic acid). Eur. Phys. J. E 2019, 42, 107. [Google Scholar] [CrossRef]
- Paszkiewicz, S.; Szymczyk, A.; Pawlikowska, D.; Irska, I.; Taraghi, I.; Pilawka, R.; Gu, Y.; Li, X.; Tu, Y.; Piesowicz, E. Synthe-sis and characterization of poly(ethylene terephthalate-co-1,4-cyclohexanedimethylene terephtlata-te)-block-poly(tetramethylene oxide) copolymers. RSC Adv. 2017, 7, 41745–41754. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Zhou, J.; Cao, F.; Zhang, Q.; Huang, K.; Wei, P. Synthesis, Characterization and Thermal Properties of Bio-Based Poly(Ethylene 2,5-Furan Dicarboxylate). J. Macromol. Sci. Part B 2016, 55, 1135–1145. [Google Scholar] [CrossRef]
- Sousa, A.F.; Guigo, N.; Pożycka, M.; Delgado, M.; Soares, J.; Mendonça, P.V.; Coelho, J.F.J.; Sbirrazzuoli, N.; Silvestre, A.J.D. Tailored design of renewable copolymers based on poly(1,4-butylene 2,5-furandicarboxylate) and poly(ethylene glycol) with refined thermal properties. Polym. Chem. 2018, 9, 722–731. [Google Scholar] [CrossRef]
- Sousa, A.F.; Coelho, J.F.J.; Silvestre, A.J.D. Renewable-based poly((ether)ester)s from 2,5-furandicarboxylic acid. Polymer 2016, 98, 129–135. [Google Scholar] [CrossRef]
- Chi, D.; Liu, F.; Na, H.; Chen, J.; Hao, C.; Zhu, J. Poly(neopentyl glycol 2,5-furandicarboxylate): A Promising Hard Segment for the Development of Bio-based Thermoplastic Poly(ether-ester) Elastomer with High Performance. ACS Sustain. Chem. Eng. 2018, 6, 9893–9902. [Google Scholar] [CrossRef]
- Chen, J.; Chen, D.; Huang, W.; Yang, X.; Li, X.; Tu, Y.; Zhu, X. A one pot facile synthesis of Poly(butylene tereph-thalate)-block-poly(tetramethylene oxide) alternative multiblock copolymers via PROP method. Polymer 2016, 107, 29–36. [Google Scholar] [CrossRef]
- Lempesis, N.; In’T Veld, P.J.; Rutledge, G.C. Atomistic Simulation of the Structure and Mechanics of a Semicrystalline Poly-ether. Macromolecules 2016, 49, 5714–5726. [Google Scholar] [CrossRef]
- Szymczyk, A.; Nastalczyk, J.; Sablong, R.; Roslaniec, Z. The influence of soft segment length on structure and properties of poly(trimethylene terephthalate)-block -poly(tetramethylene oxide) segmented random copolymers. Polym. Adv. Technol. 2010, 22, 72–83. [Google Scholar] [CrossRef]
- Guidotti, G.; Soccio, M.; García-Gutiérrez, M.C.; Ezquerra, T.A.; Siracusa, V.; Gutiérrez-Fernández, E.; Munari, A.; Lotti, N. Fully Biobased Superpolymers of 2,5-Furandicarboxylic Acid with Different Functional Properties: From Rigid to Flexible, High Performant Packaging Materials. ACS Sustain. Chem. Eng. 2020, 8, 9558–9568. [Google Scholar] [CrossRef]
- Rousseau, I.A.; Mather, P.T. Shape Memory Effect Exhibited by Smectic-C Liquid Crystalline Elastomers. J. Am. Chem. Soc. 2003, 125, 15300–15301. [Google Scholar] [CrossRef]
- Kratz, K.; Madbouly, S.A.; Wagermaier, W.; Lendlein, A. Temperature-memory polymer networks with crystallizable con-trolling units. Adv. Mater. 2011, 23, 4058–4062. [Google Scholar] [CrossRef]
- Kratz, K.; Voigt, U.; Lendlein, A. Temperature-Memory Effect of Copolyesterurethanes and their Application Potential in Minimally Invasive Medical Technologies. Adv. Funct. Mater. 2012, 22, 3057–3065. [Google Scholar] [CrossRef]
- Shi, Y.; Weiss, R. Sulfonated Poly(ether ether ketone) Ionomers and Their High Temperature Shape Memory Behavior. Macromolecules 2014, 47, 1732–1740. [Google Scholar] [CrossRef]
- Szymczyk, A.; Rosłaniec, Z. Degradacja i stabilizacja termoplastycznych elastomerów eterowo-estrowych. Polimery/Polymers 2006, 51, 627–642. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | x [mol] | wPHF [wt.%] | wFpTHF [wt.%] | WFpTHF NMR [wt.%] | [η] [dl/g] | Mn [g/mol] | Mw [g/mol] | PDI |
|---|---|---|---|---|---|---|---|---|
| PHF | - | 100 | - | - | 0.532 | 19,200 | 54,100 | 2.72 |
| PHF-b-F-pTHF 75/25 | 14.12 | 75 | 25 | 31.98 | 0.696 | 27,500 | 71,000 | 2.58 |
| PHF-b-F-pTHF 65/35 | 8.73 | 65 | 35 | 40.95 | 0.837 | 26,400 | 69,200 | 2.62 |
| PHF-b-F-pTHF 50/50 | 4.71 | 50 | 50 | 58.51 | 0.865 | 27,500 | 70,900 | 2.58 |
| PHF-b-F-pTHF 35/65 | 2.53 | 35 | 65 | 64.60 | 0.893 | 26,400 | 68,800 | 2.61 |
| PHF-b-F-pTHF 25/75 | 1.57 | 25 | 75 | 72.18 | 0.922 | 26,900 | 74,700 | 2.77 |
| Sample | Tg [°C] | ΔCp [J/g °C] | Tc [°C] | ΔHc [J/g] | Tm [°C] | ΔHm [J/g] | Xc [%] | E’ at 25 °C [MPa] | TB [°C] |
|---|---|---|---|---|---|---|---|---|---|
| PHF | 16.6 | 0.26 | 84 | 47.4 | 146 | 50.3 | 35.2 | 1373.3 | 143–152 |
| PHF-b-F-pTHF 75/25 | −36.4 | 0.28 | 79 | 40.8 | 141 | 35.1 | 24.5 | 310.5 | 134–145 |
| PHF-b-F-pTHF 65/35 | −58.6 | 0.33 | 72 | 39.7 | 137 | 36.6 | 25.6 | 232.2 | 129–143 |
| PHF-b-F-pTHF 50/50 | −65.5 | 0.38 | 54 | 28.8 | 127 | 27.6 | 19.3 | 131.2 | 115–135 |
| PHF-b-F-pTHF 35/65 | −65.8 | 0.43 | 47 | 22.9 | 115 | 20.4 | 14.1 | 77.7 | 98–119 |
| PHF-b-F-pTHF 25/75 | −66.6 | 0.58 | 27 | 16.5 | 102 | 11.4 | 7.9 | 35.2 | 90–105 |
| Sample | Hardness [Sh D] | E [MPa] | σy [MPa | εy [%] | σb [MPa] | εb [%] |
|---|---|---|---|---|---|---|
| PHF | 68 ± 1 | 1134.9 ± 84.8 | - | - | 16.1 ± 4.2 | 1.01 ± 0.01 |
| PHF-b-F-pTHF 75/25 | 51 ± 1 | 344.9 ± 69.1 | 16.4 ± 0.3 | 17.2 ± 1.7 | 43.8 ± 4.9 | 583.8 ± 46.1 |
| PHF-b-F-pTHF 65/35 | 48 ± 1 | 232.7 ± 12.3 | 13.6 ± 0.3 | 18.0 ± 1.9 | 23.2 ± 0.7 | 541.3 ± 49.7 |
| PHF-b-F-pTHF 50/50 | 41 ± 1 | 114.2 ± 13.4 | - | - | 21.8 ± 2.3 | 662.8 ± 65.5 |
| PHF-b-F-pTHF 35/65 | 33 ± 1 | 70.0 ± 10.9 | - | - | 11.5 ± 0.1 | 665.0 ± 30.5 |
| PHF-b-F-pTHF 25/75 | 26 ± 1 | 32.1 ± 7.6 | - | - | 7.2 ± 0.2 | 638.8 ± 33.1 |
| Sample | T5% [°C] | T10% [°C] | T50% [°C] | TDTG1 [°C] | Ea, (R) [kJ/mol] | TDTG2 [°C] |
|---|---|---|---|---|---|---|
| Measurement in an oxidizing atmosphere | ||||||
| PHF | 354 | 365 | 388 | 390 | 320.29 (0.9999) | 500 |
| PHF-b-F-pTHF 75/25 | 346 | 359 | 387 | 389 | 322.64 (0.9972) | 505 |
| PHF-b-F-pTHF 65/35 | 344 | 358 | 389 | 392 | 297.79 (0.9996) | 506 |
| PHF-b-F-pTHF 50/50 | 337 | 356 | 388 | 390 | 294.97 (0.9999) | 504 |
| PHF-b-F-pTHF 35/65 | 329 | 350 | 389 | 390 | 246.08 (0.9985) | 504 |
| PHF-b-F-pTHF 25/75 | 327 | 349 | 388 | 390 | 242.87 (0.9983) | 507 |
| Measurement in an inert atmosphere | ||||||
| PHF | 357 | 367 | 388 | 390 | 357.06 (1.0000) | - |
| PHF-b-F-pTHF 75/25 | 358 | 368 | 392 | 392 | 363.30 (0.9994) | - |
| PHF-b-F-pTHF 65/35 | 358 | 368 | 395 | 394 | 340.46 (0.9997) | - |
| PHF-b-F-pTHF 50/50 | 354 | 368 | 397 | 395 | 279.37 (0.9993) | - |
| PHF-b-F-pTHF 35/65 | 351 | 367 | 399 | 397 | 214.18 (0.9987) | - |
| PHF-b-F-pTHF 25/75 | 351 | 368 | 402 | 402 | 210.99 (0.9993) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paszkiewicz, S.; Irska, I.; Zubkiewicz, A.; Szymczyk, A.; Piesowicz, E.; Rozwadowski, Z.; Goracy, K. Biobased Thermoplastic Elastomers: Structure-Property Relationship of Poly(hexamethylene 2,5-furanodicarboxylate)-Block-Poly(tetrahydrofuran) Copolymers Prepared by Melt Polycondensation. Polymers 2021, 13, 397. https://doi.org/10.3390/polym13030397
Paszkiewicz S, Irska I, Zubkiewicz A, Szymczyk A, Piesowicz E, Rozwadowski Z, Goracy K. Biobased Thermoplastic Elastomers: Structure-Property Relationship of Poly(hexamethylene 2,5-furanodicarboxylate)-Block-Poly(tetrahydrofuran) Copolymers Prepared by Melt Polycondensation. Polymers. 2021; 13(3):397. https://doi.org/10.3390/polym13030397
Chicago/Turabian StylePaszkiewicz, Sandra, Izabela Irska, Agata Zubkiewicz, Anna Szymczyk, Elżbieta Piesowicz, Zbigniew Rozwadowski, and Krzysztof Goracy. 2021. "Biobased Thermoplastic Elastomers: Structure-Property Relationship of Poly(hexamethylene 2,5-furanodicarboxylate)-Block-Poly(tetrahydrofuran) Copolymers Prepared by Melt Polycondensation" Polymers 13, no. 3: 397. https://doi.org/10.3390/polym13030397