
Compression and Stretching of Confined Linear and Ring Polymers by Applying Force

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Simulation Methods
3. Results and Discussion
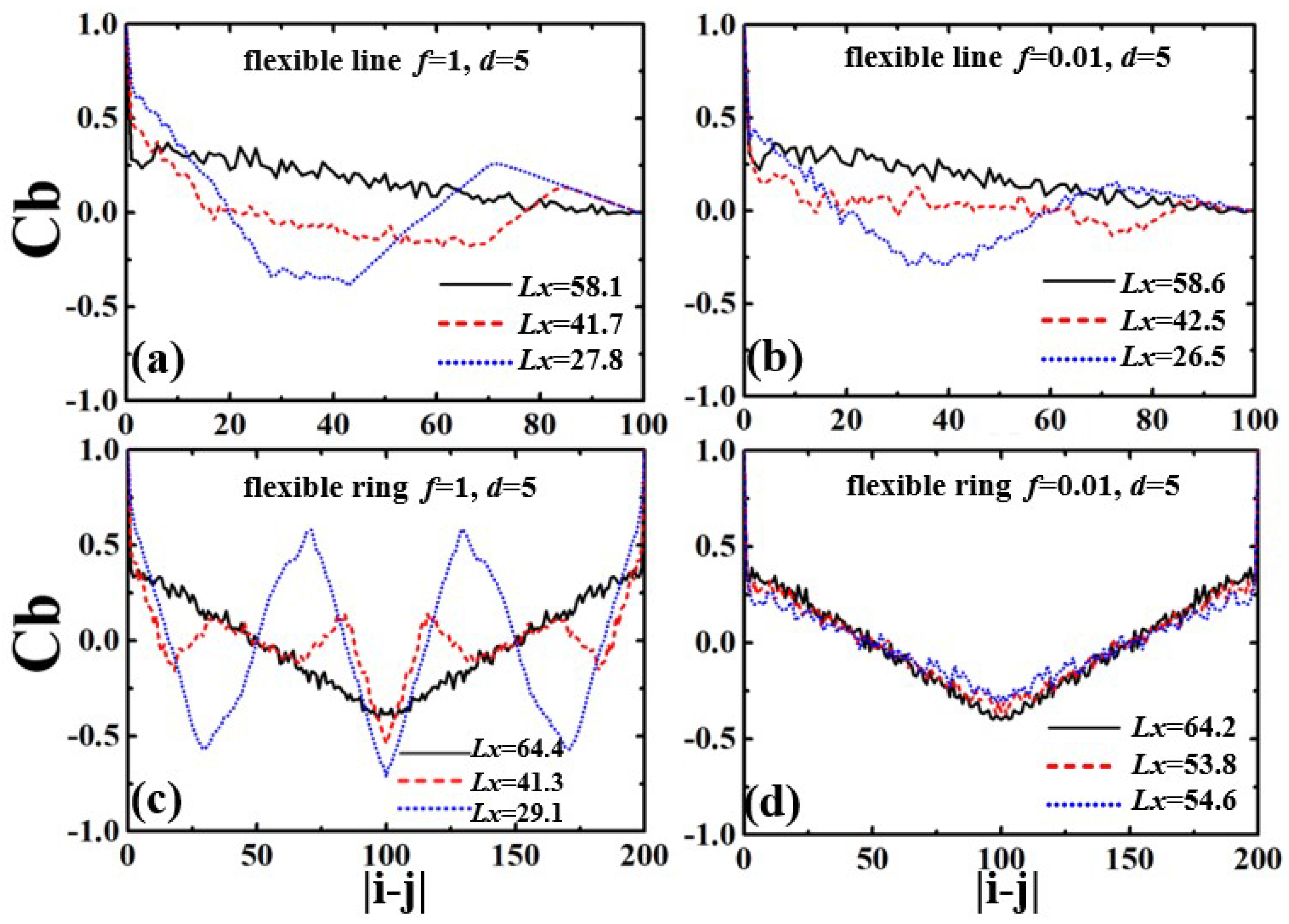
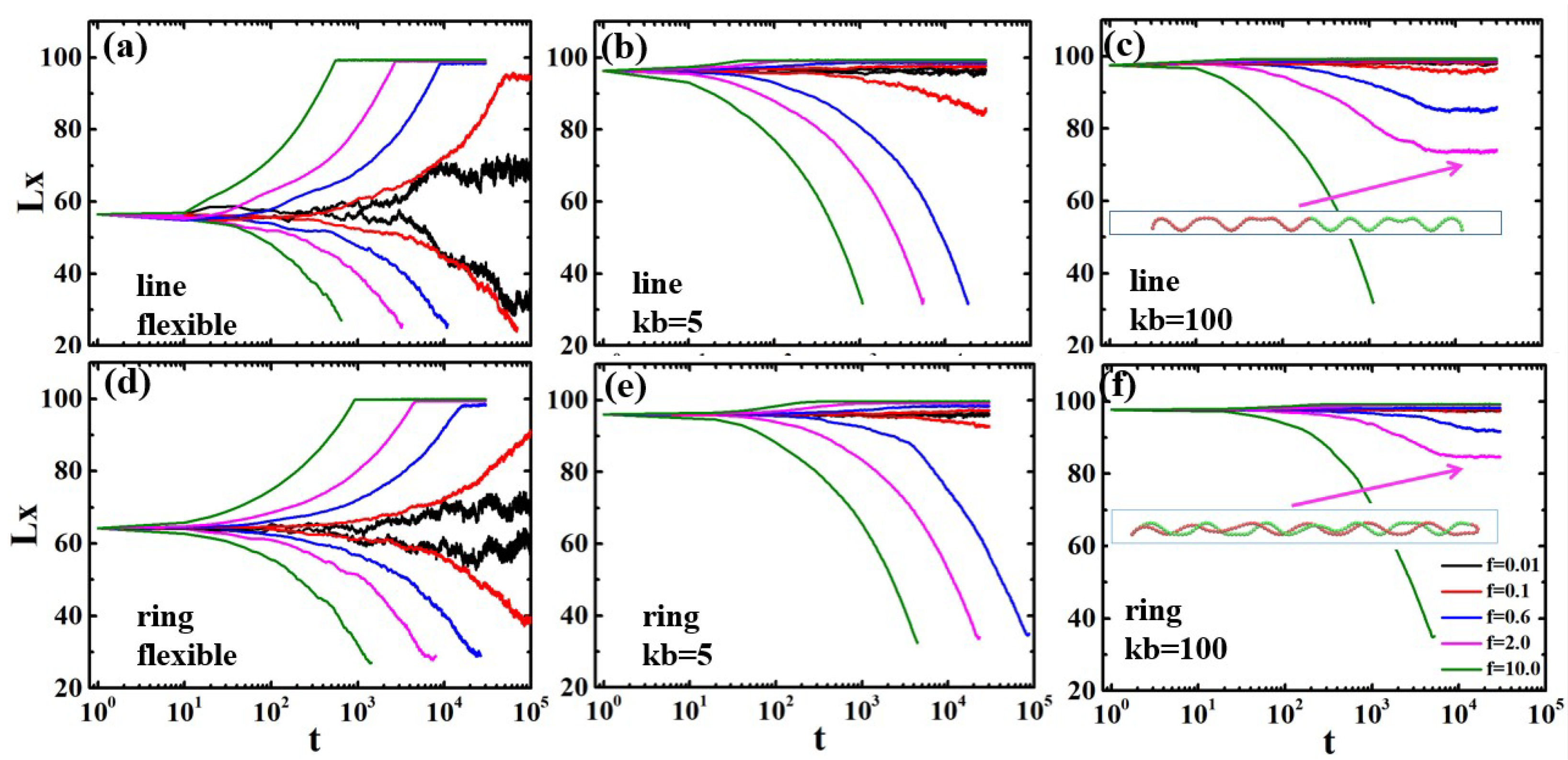
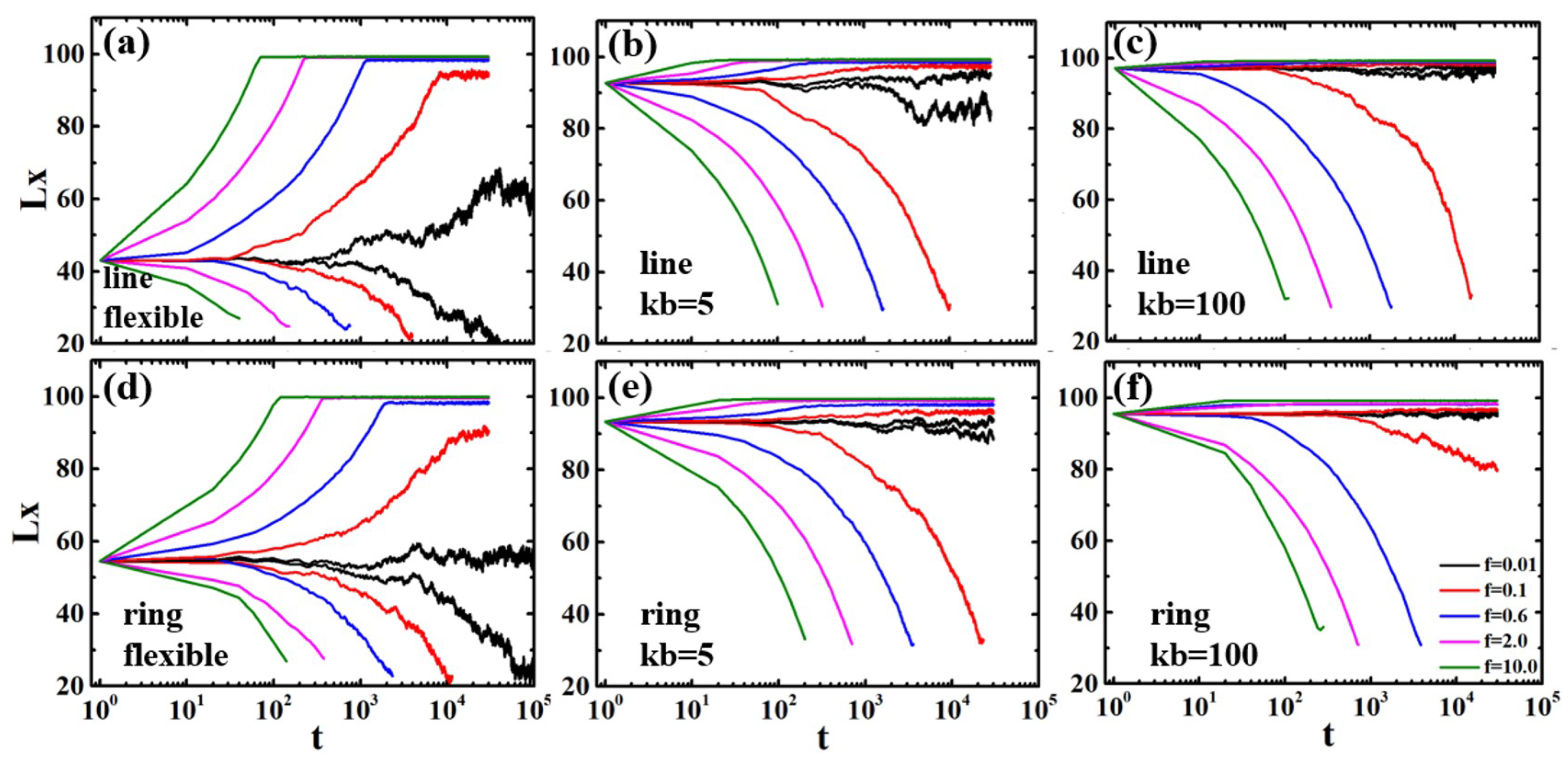
3.1. Semiflexible Polymers in Strong Confinement
3.2. Flexible Polymers in Weak Confinement
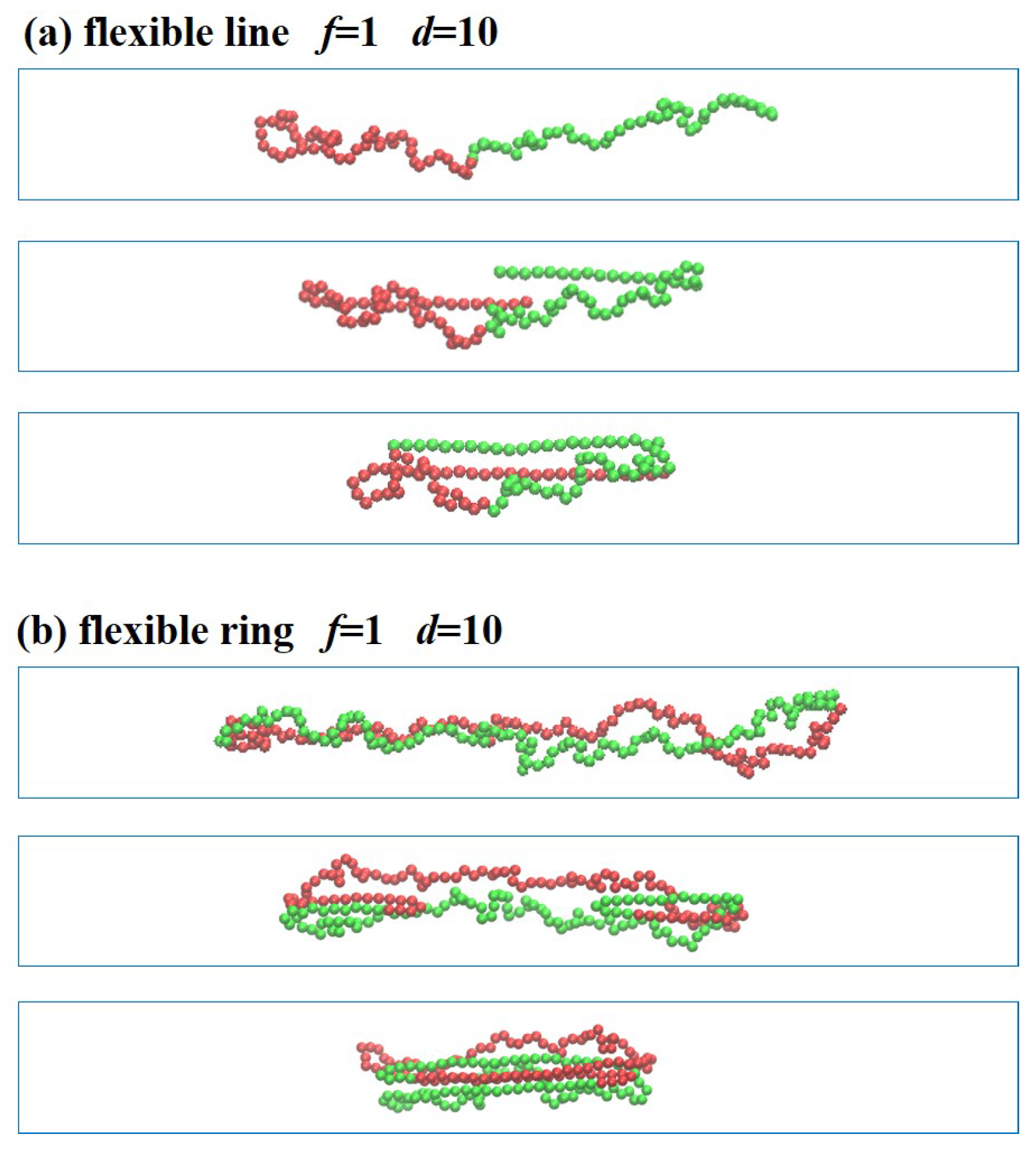
3.3. Trajectories of Polymer Deformation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, L.; Renner, C.B.; Doyle, P.S. The polymer physics of single DNA confined in nanochannels. Adv. Colloid Interface 2016, 232, 80–100. [Google Scholar] [CrossRef]
- Qin, J.; Milner, S.T. Tube Diameter of Oriented and Stretched Polymer Melts. Macromolecules 2013, 46, 1659–1672. [Google Scholar] [CrossRef]
- Steenbakkers, R.J.A.; Peters, G.W.M. A stretch-based model for flow-enhanced nucleation of polymer melts. J. Rheol. 2011, 55, 401–433. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, J.C.M.; Bornschlogla, T.; Rief, M. Full distance-resolved folding energy landscape of one single protein molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 2013–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, K.C.; Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods 2008, 5, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Bouchiat, C.; Wang, M.D.; Allemand, J.F.; Strick, T.; Block, S.M.; Croquette, V. Estimating the persis-tence length of a worm-like chain molecule from force-extension measurements. Biophys. J. 1999, 76, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.; Halvorsen, K.; Ha, B.Y.; Paparcone, R.; Sandler, S.J.; Woldringh, C.L.; Wong, W.P.; Jun, S. Physical manipulation of the Escherichia coli chromosome reveals its soft nature. Proc. Natl. Acad. Sci. USA 2012, 109, E2649–E2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanzio, P.; Mussi, V.; Manneschi, C.; Angeli, E.; Firpo, G.; Repetto, L.; Valbusa, U. DNA detection with a polymeric nanochannel device. Lab Chip 2011, 11, 2961–2966. [Google Scholar] [CrossRef]
- Tessier, F.; Labrie, J.; Slater, G.W. Electrophoretic separation of long polyelectrolytes in submolecu-lar-size constrictions: A Monte Carlo study. Macromolecules 2002, 35, 4791–4800. [Google Scholar] [CrossRef]
- Jun, S.; Thirumalai, D.; Ha, B.Y. Compression and stretching of a self-avoiding chain in cylindrical na-nopores. Phys. Rev. Lett. 2008, 101, 138101. [Google Scholar] [CrossRef] [PubMed]
- Yata, T.; Nianiaris, N.; Songsivilai, S.; Hajitou, A. Bacteriophage: From Bacteria to a Successful Targeted Systemic Gene Delivery for Cancer; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- de Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Egorov, S.A.; Milchev, A.; Binder, K. Semiflexible Polymers in the Bulk and Confined by Planar Walls. Polymers 2016, 8, 296. [Google Scholar] [CrossRef] [Green Version]
- Tree, D.R.; Reinhart, W.F.; Dorfman, K.D. The Odijk Regime in Slits. Macromolecules 2014, 47, 3672–3684. [Google Scholar] [CrossRef]
- Dai, L.; Doyle, P.S. Comparisons of a Polymer in Confinement versus Applied Force. Macromolecules 2013, 46, 6336–6344. [Google Scholar] [CrossRef]
- Pincus, P. Excluded Volume Effects and Stretched Polymer Chains. Macromolecules 1976, 9, 386–388. [Google Scholar] [CrossRef]
- Dittmore, A.; McIntosh, D.B.; Halliday, S.; Saleh, O.A. Single-Molecule Elasticity Measurements of the Onset of Excluded Volume in Poly(Ethylene Glycol). Phys. Rev. Lett. 2011, 107, 149301. [Google Scholar] [CrossRef] [Green Version]
- Khorshid, A.; Zimny, P.; Tetreault-La Roche, D.; Massarelli, G.; Sakaue, T.; Reisner, W.A. Dynamic Compression of Single Nanochannel Confined DNA via a Nanodozer Assay. Phys. Rev. Lett. 2014, 113, 269104. [Google Scholar] [CrossRef] [PubMed]
- Khorshid, A.; Amin, S.; Zhang, Z.Y.; Sakaue, T.; Reisner, W.W. Nonequilibrium Dynamics of Nanochan-nel Confined DNA. Macromolecules 2016, 49, 1933–1940. [Google Scholar] [CrossRef]
- Bleha, T.; Cifra, P. Compression and Stretching of Single DNA Molecules under Channel Confinement. J. Phys. Chem. B 2020, 124, 1691–1702. [Google Scholar] [CrossRef]
- Bleha, T.; Cifra, P. Piston Compression of Semiflexible Ring Polymers in Channels. Macromol. Theory Simul. 2021, 30, 2100027. [Google Scholar]
- Li, Y.C.; Wen, T.C.; Wei, H.H. Electrophoretic stretching of tethered polymer chains by travelling-wave electric fields: Tunable stretching, expedited coil-stretch transition, and a new paradigm of dynamic mo-lecular probing. Soft Matter 2012, 8, 1977–1990. [Google Scholar] [CrossRef]
- Lamura, A.; Winkler, R.G. Tethered Semiflexible Polymer under Large Amplitude Oscillatory Shear. Polymers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayase, Y.; Sakaue, T.; Nakanishi, H. Compressive response and helix formation of a semiflexible poly-mer confined in a nanochannel. Phys. Rev. E 2017, 95, 052502. [Google Scholar] [CrossRef] [Green Version]
- Bleha, T.; Cifra, P. Stretching and compression of DNA by external forces under nanochannel confinement. Soft Matter 2018, 14, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; MacKintosh, F.C.; Kumar, S.; Geisse, N.A.; Talbot, J.; Mahadevan, L.; Parker, K.K.; Ingber, D.E.; Weitz, D.A. Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J. Cell Biol. 2006, 173, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Broedersz, C.P.; MacKintosh, F.C. Modeling semiflexible polymer networks. Rev. Mod. Phys. 2014, 86, 995–1036. [Google Scholar] [CrossRef] [Green Version]
- Chelakkot, R.; Winkler, R.G.; Gompper, G. Flow-Induced Helical Coiling of Semiflexible Polymers in Structured Microchannels. Phys. Rev. Lett. 2012, 109, 178101. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, B.; Liu, Y.A.; LaGrone, J.; Cortez, R.; Fauci, L.; du Roure, O.; Saintillan, D.; Lindner, A. Flexible filaments buckle into helicoidal shapes in strong compressional flows. Nat. Phys. 2020, 16, 689. [Google Scholar] [CrossRef] [Green Version]
- Haque, F.M.; Grayson, S.M. The synthesis, properties and potential applications of cyclic polymers. Nat. Chem. 2020, 12, 433–444. [Google Scholar] [CrossRef]
- Jung, Y.; Jeon, C.; Kim, J.; Jeong, H.; Jun, S.; Ha, B.Y. Ring polymers as model bacterial chromosomes: Confinement, chain topology, single chain statistics, and how they interact. Soft Matter 2012, 8, 2095–2102. [Google Scholar] [CrossRef]
- Witz, G.; Rechendorff, K.; Adamcik, J.; Dietler, G. Conformation of Ring Polymers in 2D Constrained Environments. Phys. Rev. Lett. 2011, 106, 248301. [Google Scholar] [CrossRef] [Green Version]
- Vafabakhsh, R.; Ha, T. Extreme Bendability of DNA Less than 100 Base Pairs Long Revealed by Sin-gle-Molecule Cyclization. Science 2012, 337, 1097–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.D.; Chen, J.Z.; An, L.J. Tumbling and tank-treading dynamics of individual ring polymers in shear flow. Soft Matter 2013, 9, 4312–4318. [Google Scholar] [CrossRef]
- Chen, W.D.; Chen, J.Z.; Liu, L.J.; Xu, X.L.; An, L.J. Effects of Chain Stiffness on Conformational and Dynamical Properties of Individual Ring Polymers in Shear Flow. Macromolecules 2013, 46, 7542–7549. [Google Scholar] [CrossRef]
- Jeong, S.H.; Cho, S.; Ha, T.Y.; Roh, E.J.; Baig, C. Structural and Dynamical Characteristics of Short-Chain Branched Ring Polymer Melts at Interface under Shear Flow. Polymers 2020, 12, 3068. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Chen, W.D.; Zhao, H.C.; Liu, L.J.; Chen, J.Z.; Li, Y.Q.; An, L.J. Effects of excluded volume and hydrody-namic interaction on the deformation, orientation and motion of ring polymers in shear flow. Soft Matter 2016, 11, 5265–5273. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Winkler, R.G.; Sutmann, G.; Gompper, G. Semidilute Polymer Solutions at Equilibrium and under Shear Flow. Macromolecules 2010, 43, 10107–10116. [Google Scholar] [CrossRef] [Green Version]
- Pilyugina, E.; Krajina, B.; Spakowitz, A.J.; Schieber, J.D. Buckling a Semiflexible Polymer Chain under Compression. Polymers 2017, 9, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgärtner, A. Statistics of self-avoiding ring polymers. J. Chem. Phys. 1982, 76, 4275. [Google Scholar] [CrossRef]
- Pedersen, J.N.; Marie, R.; Kristensen, A.; Flyvbjerg, H. How to determine local stretching and tension in a flow-stretched DNA molecule. Phys. Rev. E 2016, 93, 042405. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Kong, X.; Wei, Q.; Chen, H.; Liu, J.; Jiang, D. Compression and Stretching of Confined Linear and Ring Polymers by Applying Force. Polymers 2021, 13, 4193. https://doi.org/10.3390/polym13234193
Chen W, Kong X, Wei Q, Chen H, Liu J, Jiang D. Compression and Stretching of Confined Linear and Ring Polymers by Applying Force. Polymers. 2021; 13(23):4193. https://doi.org/10.3390/polym13234193
Chicago/Turabian StyleChen, Wenduo, Xiangxin Kong, Qianqian Wei, Huaiyu Chen, Jiayin Liu, and Dazhi Jiang. 2021. "Compression and Stretching of Confined Linear and Ring Polymers by Applying Force" Polymers 13, no. 23: 4193. https://doi.org/10.3390/polym13234193