1. Introduction
Understanding the relation between viscosity and structure and its implications on macroscopic flow is of prime importance, particularly for semiflexible polymers, which are omnipresent in nature (DNA, actin filaments, microtubules), and synthetic polymers (polyelectrolytes, dendronised polymers). Technological applications are manifold and include, e.g., purifying DNA in microfluidic devices [
1,
2,
3] or separation of polymers [
4]. Computer simulations at multiple scales nowadays provide powerful tools to probe and understand fundamental structure-flow relations as well as their applications.
At the microscopic level, non-equilibrium molecular dynamics (NEMD) simulations have been applied for forty years [
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16] to study how macroscopic flow properties, such as shear-thinning, emerge from dynamics of microscopic structures. From the beginning, fundamental issues were approached from two sides. On the one hand, generic bead-spring models (similar to the ones applied in this work) were used to probe the general behaviour of polymer melts under shear, such as shear viscosity at zero shear rate [
5], progressive alignment and elongation with shear and corresponding correlations with stress [
7], the increase in viscosity when approaching the glass-transition in polymer melts and movement modes of individual chains [
10]. On the other hand, research in this direction was also driven by modelling and comparing simulations with actual experiments involving specific polymers [
6,
8,
9]. The underlying models tend to be more involved in these cases and include bending and even torsion terms absent in the early studies mentioned above. It therefore comes as a bit of a surprise that the specific influence of stiffness on shear-thinning in polymers has only recently come into the focus of attention. Particularly, Reference [
13] provides a comprehensive study and already anticipates some of the effects also discussed in this work, while [
14] focuses on the influence of chain stiffness on individual movement modes in single chain simulations in a MPCD solvent. We will argue, amongst others, that the emergence and decline of entanglements (e.g., as considered for melts of long flexible chains under shear in [
17] and for polyethylene in [
18,
19]) may also play an important role for short oligomers in this context.
For complex multiscale systems, bridging over a large range of dynamically coupled scales is a challenging problem. In previous decades, this question has led to the development of new mathematical algorithms and hybrid multiscale methods. One possibility to build a multiscale algorithm relies on the Lagrangian–Eulerian decomposition, where the Lagrangian-type particles are embedded in the Eulerian description of fluid; see, e.g., [
20,
21,
22]. Another approach is based on the domain decomposition. Hereby, a small accurate atomistic region is embedded into a coarser macroscopic model, see, e.g., [
23]. Several hybrid models combining particle dynamics with the macroscopic continuum model can be found in the literature. In this context, we should mention, e.g., the hybrid heterogeneous multiscale methods [
21,
22,
24,
25,
26,
27,
28], the seamless multiscale methods [
29,
30], the equation-free multiscale methods [
31,
32], the triple-decker atomistic-mesoscopic-continuum method [
23], and the internal-flow multiscale method [
33,
34]. A nice overview of multiscale flow simulations using particles is presented in [
35].
In this paper, we apply a hybrid multiscale method that couples atomistic details obtained by molecular dynamics with a continuum model approximated by the discontinuous Galerkin method. In order to extract mean flow field information from the molecular dynamics, averaging needs to be performed. Specifically, the required rheological information for the complex stress tensor is calculated by means of the Irving–Kirkwood formula [
36]. Consequently, the averaged stress tensor is passed to the macroscopic continuum model. Thus, our method belongs to the class of hybrid particle-continuum methods under the statistical influence of microscale effects. We note in passing that the degree of scale separation of a physical system influences the sensitivity of the accuracy of a solution and the computational speed-up over a full molecular simulation [
34].
The present paper is organised in the following way. First, we review and explore with NEMD simulations the molecular foundation of shear-thinning in low molecular weight polymers as a function of chain stiffness in the framework of a microscopic bead-spring model (
Section 2 and
Section 3). In particular, we will show how shear-thinning emerges from an intricate interplay of molecular alignment, stretching and tumbling modes [
12,
14,
37]. The comparison of our high-density melt with simulations of single chains will also allow us to provide educated guesses for the density dependence of individual effects. In
Section 4 and
Section 5, we use microscopic data obtained from simulations in
Section 3 as input for the study of various macroscopic channel flows using a hybrid multiscale method and investigate differences from Newtonian flow behaviour arising due to shear-thinning effects.
2. Microscopic Model and Simulation Techniques
For our microscopic model, we use standard bead-spring chains to represent the oligomers, as formulated by Kremer and Grest [
38]. In this model, all beads interact with each other via a repulsive Weeks–Chandler–Andersen potential [
39]:
with
and
. Adjacent beads are connected with an additional FENE interaction [
40,
41]:
with
and
. Semiflexibility is implemented with a bending potential:
with
being the angle between the three involved consecutive atoms and
being the coefficient of stiffness. A cosine type bending potential such as Equation (
3) originates from the well-known Kratky–Porod model [
42,
43,
44] and is a common choice for modelling semiflexibility in polymers [
45]. Note that even though our short flexible chains are essentially unentangled, Reference [
46] suggests for a very similar model that the entanglement length drops steeply with increasing stiffness for semiflexible chains, implying that chains of length
are already entangled for our intermediate range of stiffnesses
.
Non-equilibrium molecular dynamics simulations of a sheared melt at density
were performed using the LAMMPS simulation package [
47]. System sizes were set to
if not mentioned otherwise. Shear along the x-direction was introduced by superimposing a velocity gradient on thermal velocities using the SLLOD equations [
48,
49,
50,
51] and coupling the latter to the Nose–Hoover thermostat [
50,
52]. Temperature T = 1 was maintained throughout our simulations, and the Velocity Verlet algorithm was used to integrate the equations of motion. Note that LAMMPS implements a non-orthogonal simulation box with periodic boundary conditions that deforms continuously in accordance with the applied shear rate [
53,
54]—an approach that has been shown to be equivalent to the commonly used Lees–Edwards boundary conditions [
51,
55].
Shear viscosity
was calculated using the relation
where
is the applied shear rate and
is a non-diagonal component of the stress tensor as determined by the Irving–Kirkwood formula [
36,
56]:
Here,
is the mass of the
ith particle,
the peculiar velocity of the
ith particle, and
and
are the distance and force vectors between the
ith and the
jth particle, respectively. For comparison, we have also calculated shear viscosity via the Green–Kubo relation:
where
V is the volume of the system, and
is the Boltzmann constant.
is measured under equilibrium conditions (
) and serves as a reference value for
.
Note that forces arising from the thermostat and its coupling to the SLLOD conditions are not explicitly considered in Equation (
5) and may potentially result in a small systematic error. For a detailed discussion of this effect in the context of dissipative thermostats, the reader is referred to Reference [
57].
In addition, we have carried out Brownian dynamics simulations of single chains (same potentials as above) in an external shear flow profile. The equation of motion of monomers
i is given by
where
is the monomeric friction,
the intermolecular force acting on
i, and
an uncorrelated Gaussian white noise with mean zero obeying the fluctuation–dissipation theorem, i.e.,
. In the simulations, we used an Euler forward algorithm with a time step
. The time scales of the two models were adjusted by mapping the Rouse times of fully flexible chains (
) at equilibrium (
). To determine the Rouse time, we determined the autocorrelation function of the squared end-to-end distance,
For ideal Rouse chains, it can be calculated analytically, giving
where
p sums over the Rouse modes of the chain. At late times, the behaviour is dominated by the first Rouse mode with
. We thus fitted the late time behaviour of
to the function
for chains of length
in a melt and for the corresponding single Brownian chains. The fit parameters for the prefactor
A were in rough agreement with the theoretical value
in both cases (
for melt chains, and
for single oligomers). The fitted Rouse time of melt chains was
, and that of single oligomers was
. Hence, the time scales match when choosing
. We have also carried out a more intricate mapping (discussed in
Appendix A), which matches Rouse times for each value of
, but does not change our results qualitatively.
3. Shear-Thinning in Oligomer Melts—A Molecular Analysis
In the following section, we would like to investigate and review the molecular foundation of shear-thinning in low molecular weight polymer melts. We will show and highlight that macroscopic flow properties of polymers are governed by an intricate interplay of stretching, alignment and tumbling of individual molecules as well as collective modes at the molecular level.
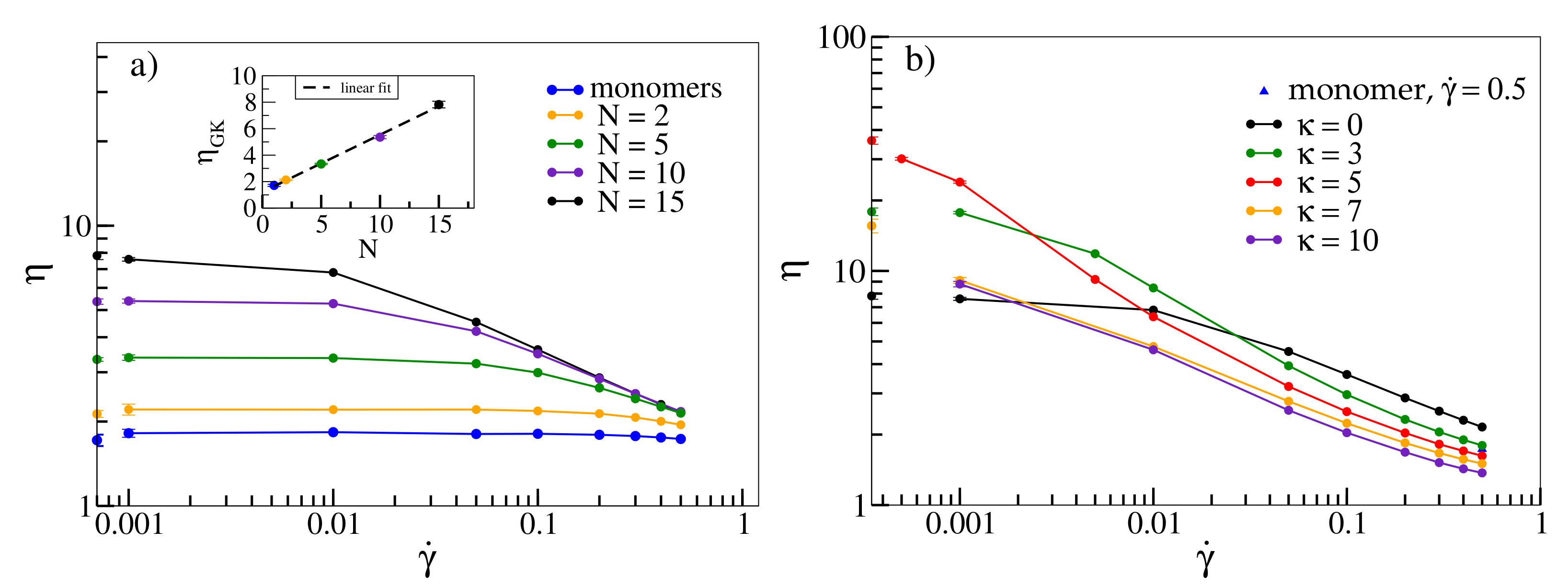
Figure 1a displays viscosity
as a function of shear rate
for flexible oligomers with different chain lengths
N. Flexible oligomers exhibit shear-thinning [
10], i.e., decrease of viscosity with increasing shear rate. For consistency, we also compare viscosities as derived from Equation (
5) with those obtained from the Green–Kubo relation, Equation (
6) (points on the
y-axis). The latter agree with the values for
within the error bars. The overall shape of
, namely a plateau at low shear rates followed by a shear-thinning regime, which becomes more pronounced with increasing molecular weight, has also been observed for various polymers experimentally [
9,
58,
59]. The inset shows that
increases linearly with
N for small chain lengths in agreement with simulations from [
5].
In
Figure 1b, we investigate the dependence of viscosity on shear rate as a function of stiffness for an oligomer melt with chain length
. While for large shear rates, viscosity decreases with increasing stiffness and intriguingly even drops below the value determined for monomers (for
),
becomes non-monotonic for low shear rates. For
, flexible chains with
have the lowest viscosity, while viscosity increases for semiflexible chains and drops down again for rigid chains, an effect described in [
13] for a similar model. A similar non-monotonic behaviour is exhibited by
(shown on the margins of
Figure 1b as a function of
). While the rise of viscosity can already be explained with the emergence of entanglements for intermediate stiffnesses, at the end of this section we will associate the following decline with a collective alignment of chains (and associated disentanglements), which are amplified by shear (Figure 4).
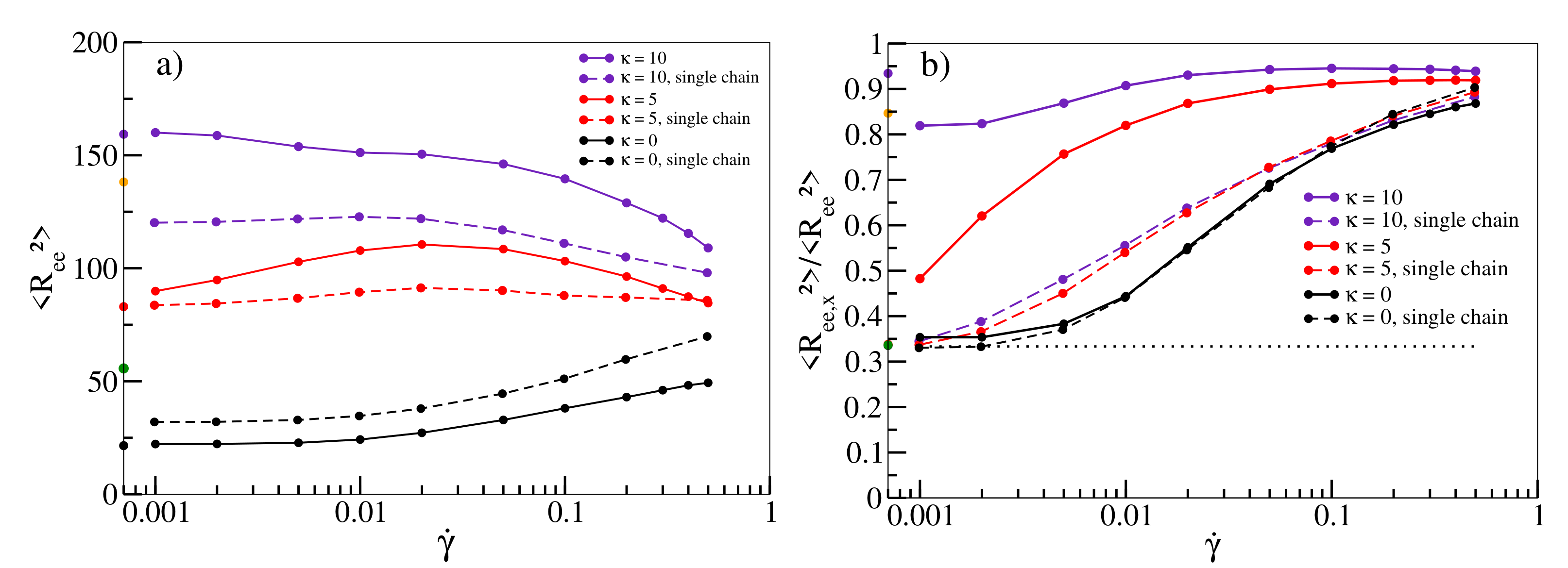
In
Figure 2a, we quantify the stretching of individual chains with shear. The size of a flexible chain as measured by the mean square end-to-end distance
increases continuously as a function of shear rate. For
, the average size only increases slightly towards moderate shear rates before decreasing again at high rates similar to [
14], ruling out stretching as a main driving force for shear-thinning in this regime for melts of semiflexible chains.
Figure 2b visualises the alignment of chains along the shear direction by plotting the ratio of the x-component to the total
. Without shear, each component contributes equally, yielding a ratio of 1/3 (dotted line in
Figure 2b). While this holds for flexible chains at low shear rates, deviations become more pronounced for shear rates exceeding
. This is also roughly the rate at which noticeable deviations from
start to occur in
Figure 1a and shear-thinning sets in. This behaviour becomes even more pronounced for semiflexible chains. At
, chains are already partially aligned, and the viscosity in
Figure 1b already deviates significantly from the value obtained from the Green–Kubo relation. Shear-thinning sets in at even lower shear rates and is reinforced with progressive alignment of chains. This relation provides a clear indication that chain alignment is strongly correlated with the occurrence of shear-thinning in agreement with previous observations [
13,
14]. Note that for
, chains are already stretched and aligned in equilibrium simulations without shear (values on
y-axis of
Figure 2a,b) indicating the emergence of nematic behaviour in agreement with previous observations in a similar model [
46,
60]. For
, there is even an initial drop from the bulk ratio once shear sets in. Interestingly, stretching and alignment of chains can already be observed qualitatively in simulations of single chains in shear flow (dashed lines in
Figure 2a,b), indicating that these phenomena should in principle be observable for melts of all densities. However, while the alignment of flexible chains is well-reproduced, the increase is less pronounced for higher stiffnesses, indicating that collective alignment contributions due to stiffness are not captured by single chain simulations.
In the following, we turn to movement modes of individual chains.
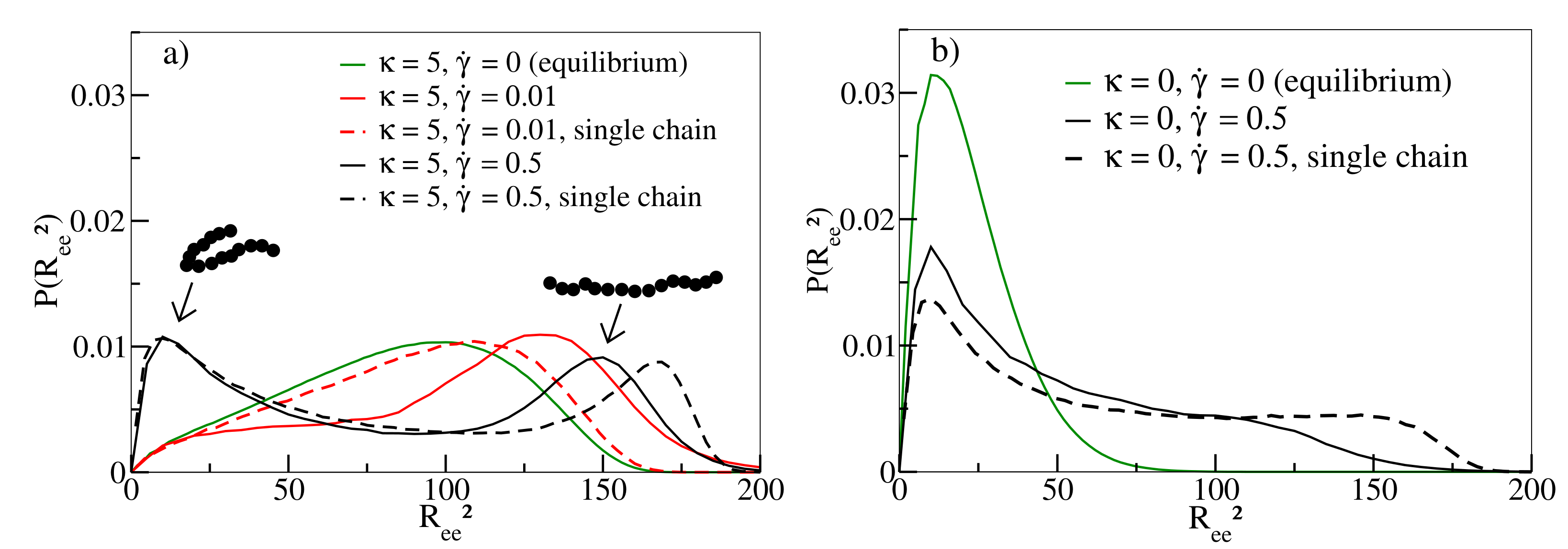
Figure 3 shows the distribution of
of individual chains in melts. While equilibrium simulations (
) have a broad distribution of end-to-end distances for
(solid green line in
Figure 3a), conformations develop a preference for stretched chains at moderate shear rates (
, solid red line). At about this rate,
displays a maximum in
Figure 2a. For the highest shear rate
(solid black line), U-shaped tumbling conformations coexist with stretched conformations explaining the decrease of
in
Figure 2a for
[
14]. For flexible chains, compact conformations dominate the behaviour at small and large shear rates (
Figure 3b), as already noted in [
10], even though the latter also exhibit some degree of stretched conformations. This also explains why
is significantly smaller in comparison to semiflexible chains (
Figure 2a). It is worth noting that the occurrence of compact conformations does not impede the continuous alignment of chains along the shear direction with increasing shear rate, as demonstrated in
Figure 2b. The intricate interplay between stretching and tumbling as a function of shear rate and stiffness can also be observed for our single chain simulations (as noted for flexible chains already in [
10] and studied in [
11]), indicating that these movement modes are not a collective phenomenon and should occur in melts of all densities [
15].
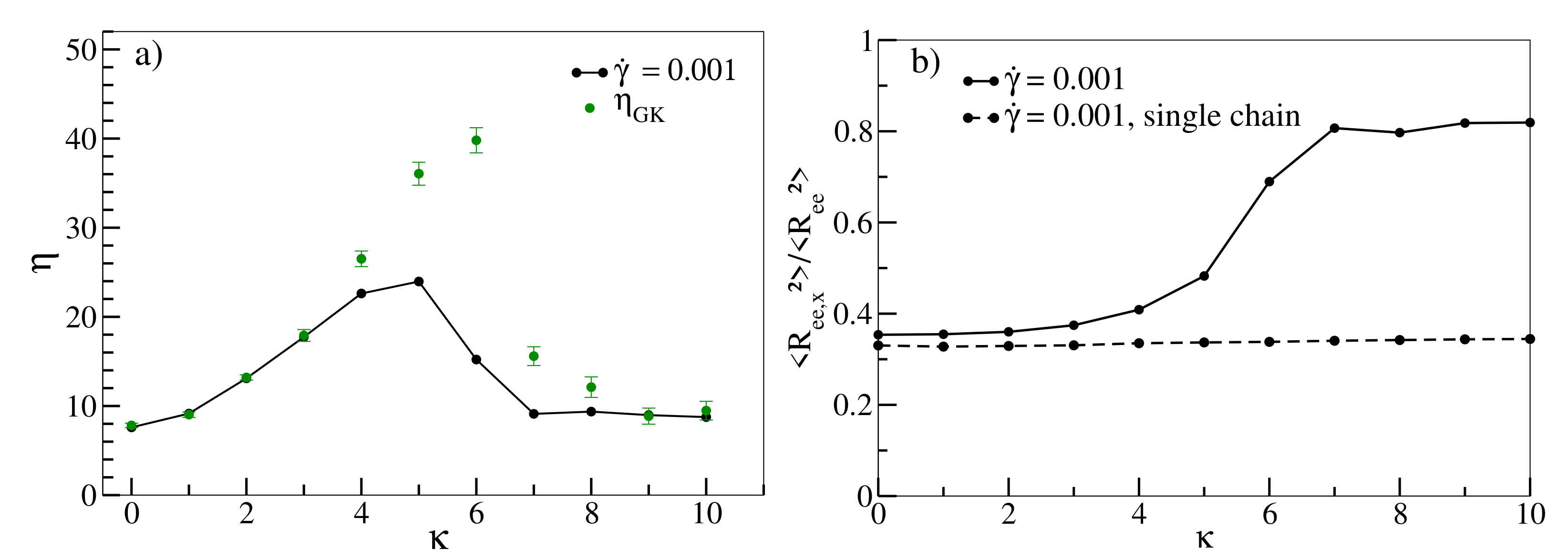
In
Figure 4, we finally investigate the non-monotonous behaviour of the Green–Kubo viscosity
and viscosity at low shear rates (
) as functions of
.
increases with increasing chain stiffness, reaches a maximum at about
and undergoes a sharp decrease after that. Furthermore,
increases up to
and decreases subsequently. Intriguingly,
matches with
up to
, but, between
and
,
is significantly larger than
. As already pointed out, Reference [
46] estimates that the entanglement length decreases with increasing persistence length,
, to a point at which the entanglement length becomes smaller than the chain size. It estimates that at
, the entanglement lengths are approximately equal to 15, 8 and 6, respectively. Reference [
61] estimates that the numerical values of
are quite close to the numerical values of
. For example,
and 5 correspond to
2.5 and ≈5, respectively. Therefore, chains become entangled, and this effect increases with increasing
. It should be noted, however, that for both Reference [
46] and [
61], the number density was 0.85, which is a bit higher than the number density of our system (0.8). Bond and angular potentials forms also differ slightly in [
46]. Incidentally, Reference [
62] also obtains an analytic expression that estimates entanglement length as a function of chain stiffness for isotropic polymer chains. Unaligned and entangled chains impede collective motion under equilibrium conditions, and as a result,
increases up to
. Following
, however, there is a sharp drop in
. This is consistent with our observation from
Figure 2b that for
and
, chains are already stretched and aligned under equilibrium conditions, indicating that our system indeed undergoes an isotropic-nematic transition following
. Entanglements decrease as chains align and conformations become more susceptible to the applied shear, resulting in a decrease in
.
as a function of
also exhibits a similar non-monotonous behaviour. While the initial increase can also be attributed to progressive entanglements, for
, the applied shear already begins to align the chains towards the shear direction, counteracting entanglement effects, as is evident in
Figure 4b. As a result,
are lower than the corresponding
values. For
, chains are more strongly aligned along the shear direction, thus
as a function of
decreases beyond
[
13]. This behaviour is, in contrast to stretching and alignment with increasing shear rate, a collective phenomenon that cannot be observed in corresponding simulations of single chains and should therefore vanish gradually at smaller densities. Our finding that rheological properties in rather short oligomeric systems are dominated by the emergence and decline of entanglements may come as a surprise. However, it should be noted that some prior studies have also associated shear-thinning with decreasing entanglements, albeit for much longer chains. Reference [
18] shows that in a linear polyethylene melt (
) system comprising polymers with a finite stiffness, the number of entanglement strands per chain decrease with increasing shear rate. Reference [
19] argues along similar lines for a melt comprising of even longer linear polyethylene chains (
), and Reference [
17] shows a similar decrease in entanglements with increasing shear rate for flexible polymer chains (
and 400). In
Appendix B,
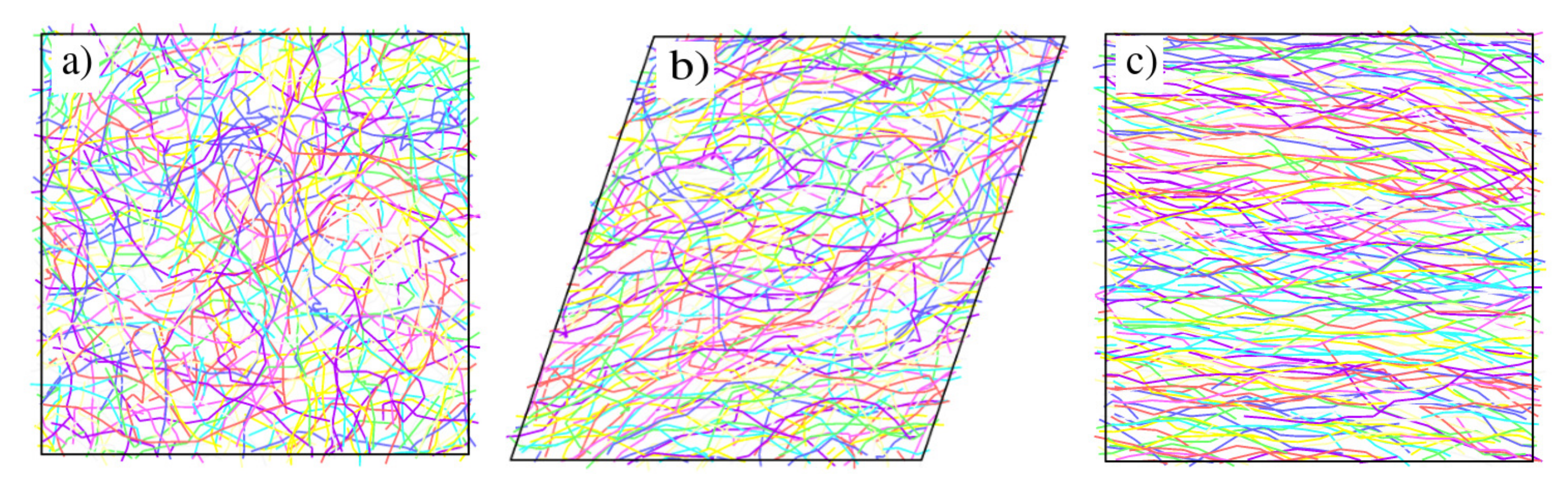
Figure A2 displays various configuration snapshots of oligomer melts, which further visualise the interplay between disentanglement, alignment and shear.
4. Hybrid Multiscale Method
After studying the molecular origin of non-Newtonian behaviour of short polymer melts, we now turn our attention to macroscopic simulations combining them with the results of molecular dynamics.
The motion of an incompressible fluid flow at the macroscopic level is governed by the continuity and the momentum equations
where
is the velocity vector,
the Cauchy stress tensor, and
an external body force. The boundary of the computational domain
consists of the Dirichlet, Neumann and periodic boundary, i.e.,
.
The Cauchy stress tensor can be split into two parts with p being an isotropic hydrostatic pressure and a viscous stress tensor. For the Navier–Stokes equations, we have with being a constant viscosity. This relation is more complex when non-Newtonian polymer fluids are considered.
In this work, we apply the hybrid multiscale method that couples the molecular dynamics simulations with the macroscopic model (10a–e). As explained in
Section 2, the macroscopic stress tensor can be derived from the Irving–Kirkwood formula (
5).
Our extensive molecular dynamics simulations imply that the stress tensor can actually be expressed in the following simple way
Finally, the viscosity–shear rate dependence leads to a well-known Carreau–Yasuda rheological model [
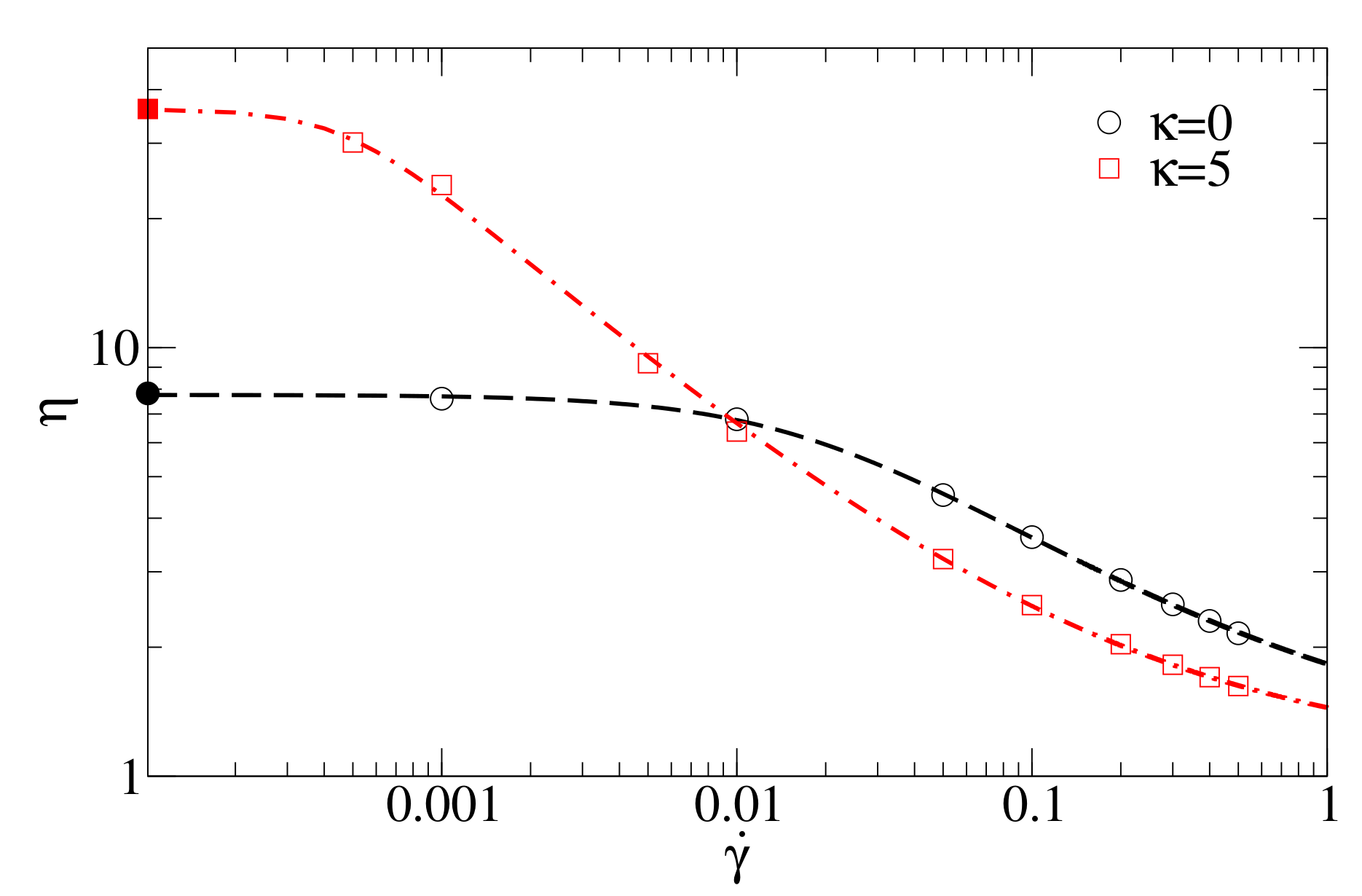
63]
with the following coefficients: for flexible polymers (stiffness
)
,
,
,
,
; and for semiflexible polymers with
= 5
,
,
,
,
.
We note in passing that for a particular situation considered in this paper, our hybrid multiscale method can be seen as a parameter passing sequential coupling multiscale method, see, e.g., ref. [
29] for a detailed description of the concurrent and sequential coupling strategies.
The oligomer chain length in both cases is
.
Figure 5 compares the MD data and the fitting with the Carreau–Yasuda model.
Our next goal is to calculate the shear-dependent viscosity
In what follows, we consider for simplicity the situation of two-dimensional shear flows and use the notation
. Applying (
12), we need the value of the shear rate
of the polymer flow. It can be obtained from the strain-rate tensor
by rotating it with respect to the streamlines to the anti-diagonal matrix
by an angle
, which for incompressible flows is
. Here,
are components of the strain-rate tensor
and
the rotation matrix [
64,
65]. The new strain-rate tensor
corresponds to a pure-shear deformation (i.e., in absence of normal stresses). The shear rate can therefore be calculated from the components of the original strain-rate tensor
and the angle
The stress tensor in the shear flow can be calculated in the normal-stress free basis and transformed back to the original basis according to (
11)
As shown in (
16), we consider in the present work shear dependent flows, where the stress tensor or, more precisely, the viscosity are nonlinear functions of the local shear rate. In order to consider more general flow conditions, such as the extensional flow, rigid rotation and mixed flows, one needs to take into account not only the shear rate dependence but a complete decomposition of a three-dimensional symmetric tensor (stress tensor) into a six-dimensional basis. In such a way, not only the viscosity but also additional response coefficients will be computed from microscopic simulations in order to determine the local stress tensor. We refer the reader to our recent work [
66] where complex flows in general geometries were studied, see also [
67].
We proceed by describing a numerical method applied to (10a–e). For
time integration, we apply the implicit BDF2 scheme, which leads to the following system
Discretisation in space is realised by the discontinuous Galerkin (dG) method [
68,
69,
70]. Domain
is discretised by a quadrilateral mesh
with a meshsize
h. Mesh faces
can be either the inner interfaces between adjacent elements
or boundary faces
. The face normal
points from an arbitrarily chosen (but fixed) element
towards
. The face normal points outward of the domain
on boundary faces.
We consider broken Sobolev spaces
that consist of piecewise quadratic and piecewise linear polynomials, respectively,
The usual average and jump operators of a scalar-valued function
on interfaces between adjacent elements
and
are defined as
Vector-valued functions are treated componentwisely. For boundary faces, we set , when not mentioned otherwise.
The Discontinuous Galerkin (dG) method for an oligomer fluid flow (10a–e) is formulated as follows. Given the initial data
, we look for a sequence of numerical solutions
for
such that
The numerical shear rate is computed locally, i.e., in each quadrature point from the broken gradients . Hereby, we compute the first approximation , e.g., by the Euler implicit method. The scalar product is denoted by In what follows, we define the discrete forms a, b, and
The
convective term is rewritten in the conservative form and the interface integrals are approximated by means of the Lax–Friedrichs numerical flux
where
, and
is the absolute eigenvalue of the Jacobian matrix
. The average and jump operators on the Dirichlet boundaries are defined as
The divergence of the velocity
as well as the discrete gradient of the pressure
are both approximated using the form
To discretize the viscous, terms we employ an almost-standard symmetric interior penalty method (SIP), first introduced in [
71] and extensively analysed in [
72]
On the Dirichlet boundaries
, we set
Coefficient
is a penalty parameter that we choose for a quadraliteral mesh in the following way [
73]
where
is a user-defined small coefficient. Area and volume are denoted by
A and
V, respectively. We conclude this section by mentioning that the resulting nonlinear system is approximated by the Newton method employing the Dogleg-globalisation and a direct sparse solver for the linearised system; see [
74].
5. Two Channel Flows of a Non-Newtonian Oligomer Fluid
In this section, we illustrate the consequences of shear-thinning, whose microscopic origins have been discussed in previous sections, on two examples of macroscopic channel flows. Numerical simulations have been realised within BoSSS code, for more details see also [
20,
68,
69,
74,
75].
In the first test, the so-called Poiseuille flow [
76] is simulated. Here, an oligomer melt flows through a narrow channel of length
, driven by a pressure difference between the outlet and inlet of the channel. The intensity of the flow is controlled by the pressure parameter related to the external pressure gradient
. Since the viscosity is a function of the shear rate, we compute the Reynolds number using the averaged viscosity
i.e.,
where
U is the characteristic velocity (maximal inflow velocity) and
L the characteristic length, i.e., the channel diameter
. In order to also take into account the effects of asymptotic viscosity values, we define
,
and introduce them in
Table 1.
For the macroscopic model of our hybrid multiscale method, we have used a grid with
mesh cells, i.e., the mesh parameter
. Numerical simulations on a coarser grid confirm that the steady-state results presented here are independent on the mesh resolution.
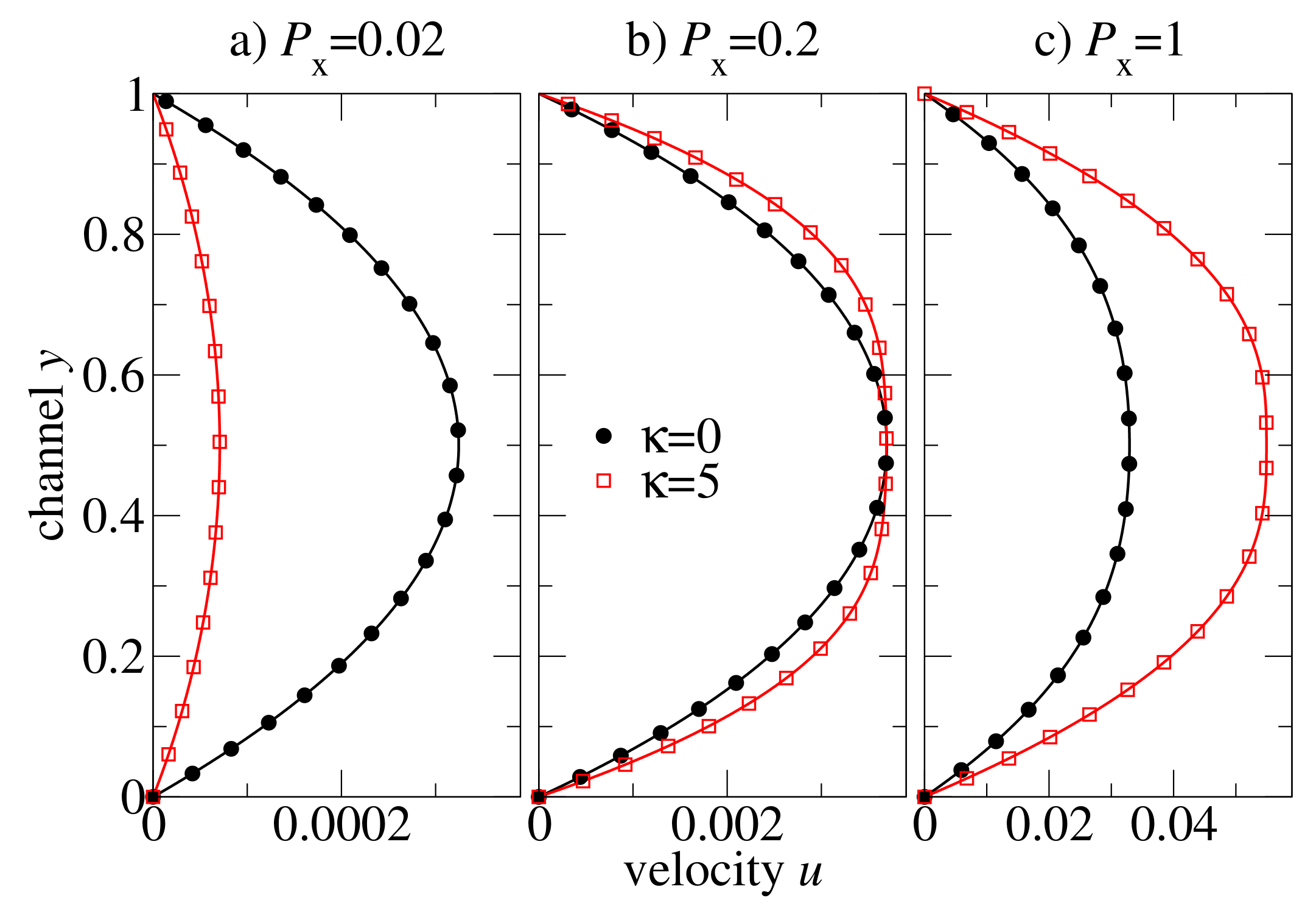
Figure 6 shows the steady-state velocity profiles across the channel for flexible (
) and semiflexible (
) polymer chains of the length
. The external pressure drop is chosen at values of
in order to study the influence of the different regimes of the shear rate-dependent viscosity on an oligomer flow. At low-pressure drop (
in
Figure 6a), the flow is very slow and the shear rates are low, too. The viscosity of both polymeric systems is at the Newtonian plateau (cf.
Figure 5). The viscosity of flexible chains is lower than that of the semiflexible chains (ca. 8 vs. 36), and therefore, the velocity of the flexible chains is higher than that of the semiflexible chain melt. At moderate pressure drop (
in
Figure 6b), the viscosity of the flexible chains is still nearly a constant (the Newtonian regime), whereas the semiflexible chain melt is already in the shear-thinning regime. By coincidence, the amplitude of the two profiles at the centre of the channel is approximately the same, and one can easily recognise that the shapes of the velocity profiles are very different: the semiflexible chains exhibit a broader distribution (
Figure 6b). Further increase in the pressure drop leads to an inverse situation: the melt of semiflexible chain flows faster through the channel than the flexible chains melt (cf.
in
Figure 6c).
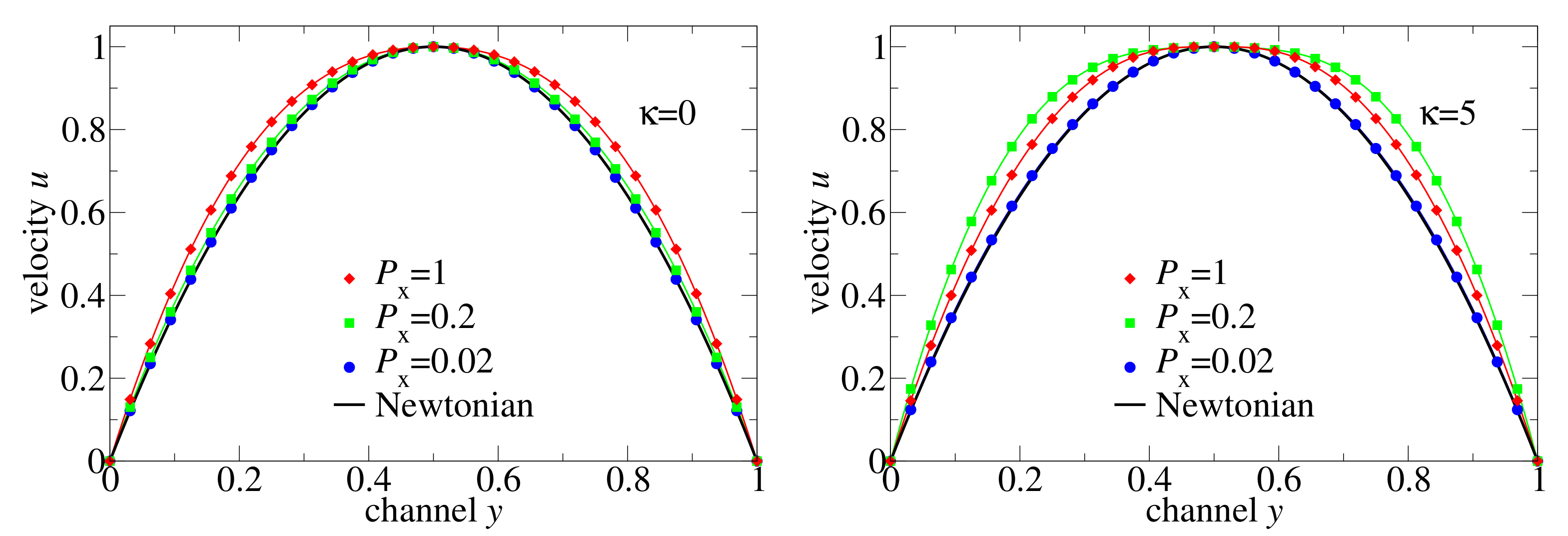
To compare different non-Newtonian and Newtonian flows, we plotted in
Figure 7 the velocity distributions normalised by the maximal velocity together with the Newtonian fluid solution. (i) At low-pressure drop, the flow of both flexible and semiflexible melts is Newtonian, and it deviates from the Newtonian regime as the external pressure difference increases. (ii) The non-Newtonian effect increases progressively in the flexible chain melt, but it is non-monotone for the semiflexible chains:
causes a larger non-Newtonian effect on the velocity distribution than
. The reason for this retrograde non-Newtonian behaviour is the following: The semiflexible chain melt is coming into the regime of a second plateau of viscosity at high shear rates where the melt behaves like at low shear rates, but the flow velocity is larger due to lower viscosity. The conjecture on the existence of the second plateau at high shear rates is based in the existence of a positive curvature of
for
in
Figure 5. In contrast, the microscopic data for a flexible chain does not indicate positive curvature within the measured shear rates.
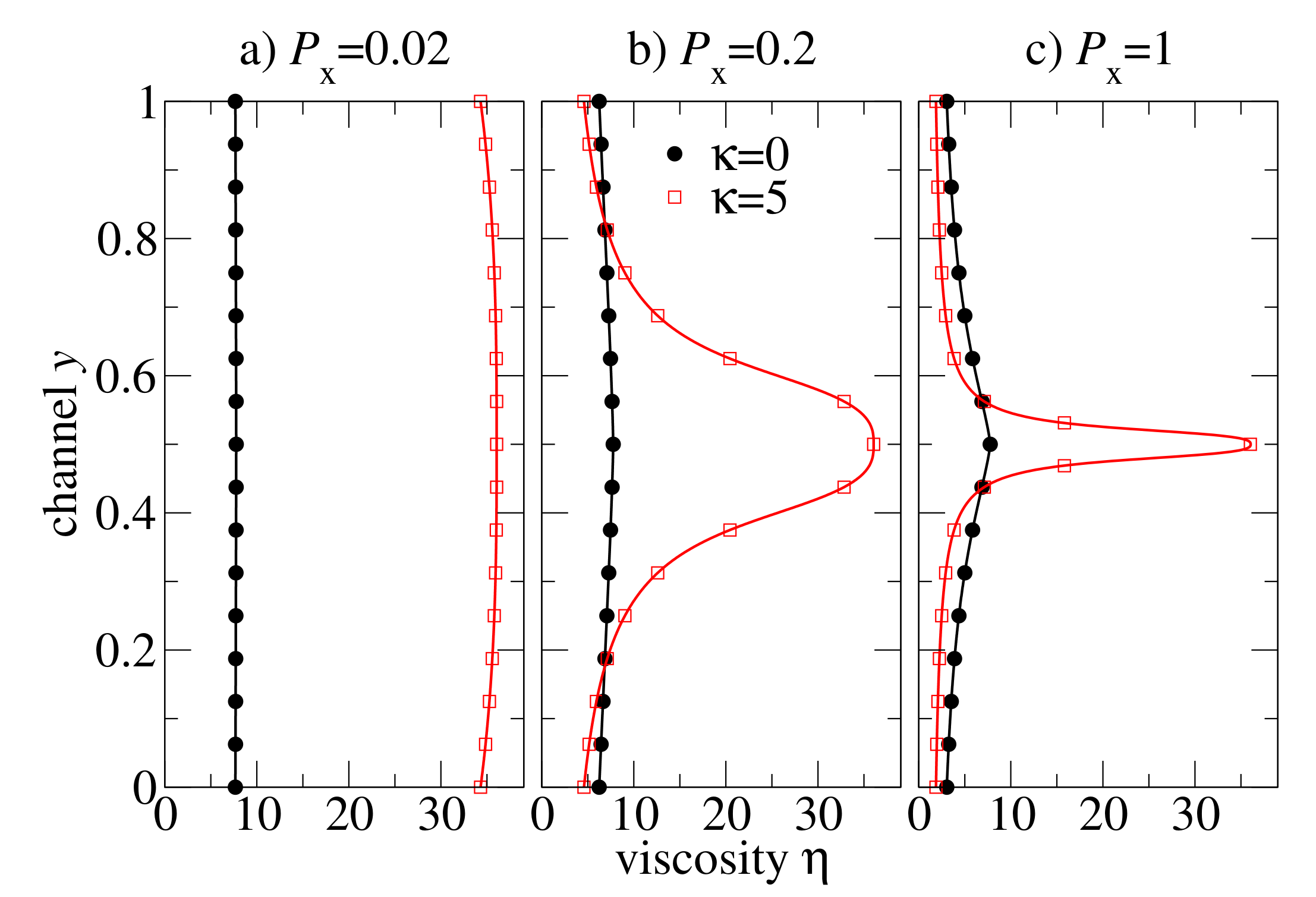
We proceed with the analysis of viscosity distributions
, see
Figure 8. For slowly flowing melts (
in
Figure 8a) the viscosity is almost constant in flexible and semiflexible chain melts. At moderate pressure drop
, the viscosity is almost constant in the flexible chain melt, but it is
-shaped with a large amplitude variation between the centre of the channel and near the walls for the semiflexible chain melt. At large pressure drop (
), the semiflexible chains develop a kind of viscosity spike localised at the centre of the channel due to
. Except for this singular region, the viscosity distribution of semiflexible chains is very flat, and the viscosity value is smaller than in the flexible chain melt.
The second test is devoted to the study of complex geometry effects. Here we investigate non-Newtonian flows in a channel with a backward-facing step, see Armaly et al. [
77]. Newtonian flows in such a channel have been studied in, e.g., [
78]. Depending on the inflow velocity, this flow can be laminar, transitional or turbulent. In the laminar regime at low Reynolds number, one or more recirculation zones of the secondary flow after the expansion arise. One zone is located directly behind the backward-facing step, and it can be observed already for a very low Reynolds number. Further zones randomly appear and disappear in the case of high Reynolds numbers. In this test, there is a broad distribution of the shear rates since the intensity of the flow and the shape of streamlines are very different in the main and the secondary flows.
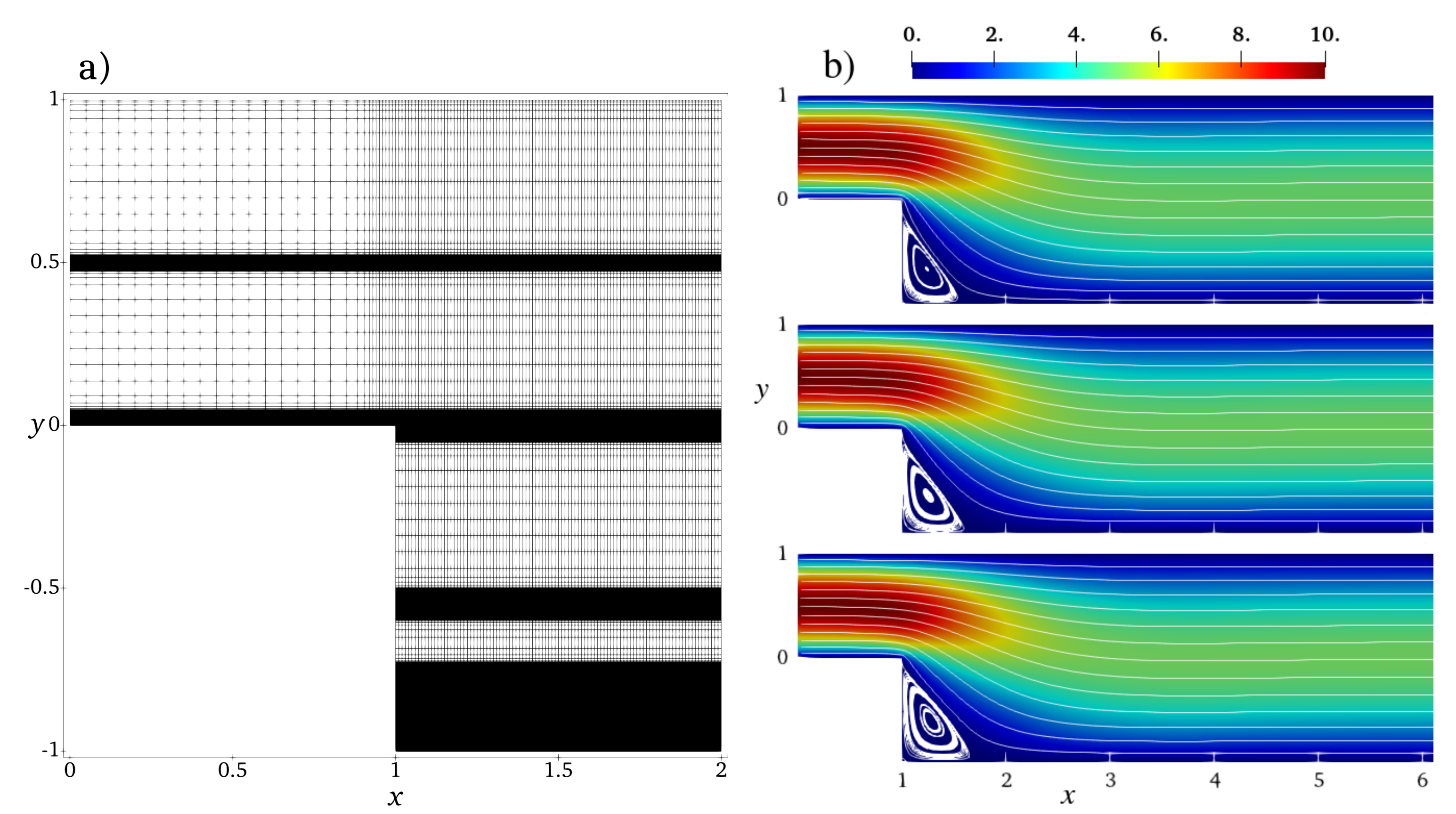
We study the flow of chain molecules in the channel of the length
and the height at the inlet
and at the outlet
. The inlet is located at
. The computation domain is a structured quadrilateral mesh shown in
Figure 9a. In the
x-direction, the mesh is homogeneous with the mesh step
except for the region near the backward-facing step
where the grid density has been smoothly increased in order to better resolve the transit region. The fine mesh step is
. In the
y-direction, the mesh has several high-resolution regions: near the walls at
, in the middle of the inlet part (
) and of the main part (
), and one zone at
. The fine mesh resolution in these zones is
, and the coarse resolution is
elsewhere with a smooth transition in-between.
At the inlet at , the Dirichlet boundary condition is applied for the inflow velocity , where is the amplitude of the inflow velocity used as a control parameter for simulations. The outlet boundary conditions at impose zero stress (Neumann boundary conditions), i.e., .
Figure 9b compares the velocity and the flow streamlines for flexible, semiflexible chain molecules and the Newtonian fluid for inflow velocity
. Although the overall pictures look similar, one can clearly observe variations in the recirculation zone in the corner behind the step. To analyse the solution in details, we plot in
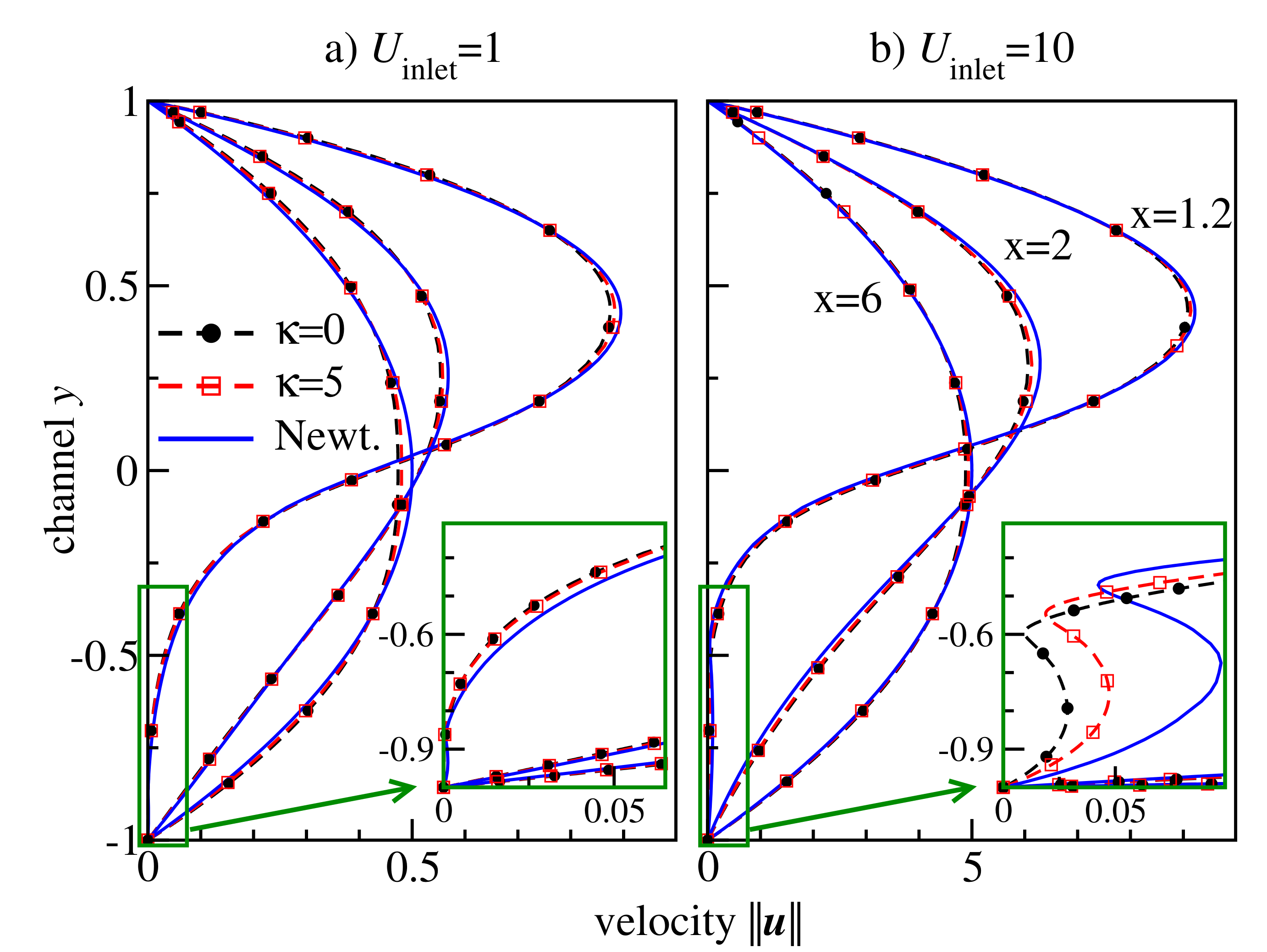
Figure 10 velocity cross-sections at
for different inflow velocities. These positions represent three characteristic regions of the flow behind the backward-facing step: the secondary flow vortex at the bottom corner directly behind the backward-facing step (
), flow directly behind the secondary flow (
), and flow far behind the secondary flow (
). The velocity profiles change between
and
from strongly asymmetric in the transit region at the backward-facing step to a symmetric one in the region far behind the step, where the characteristic non-Newtonian shape is observed. The curve for semiflexible chains is located between the curves for flexible chains and Newtonian fluid, indicating the retrograde non-Newtonian effect discussed also for the Poisseuile flow (cf.
Figure 6c). This effect becomes more visible in the case of high inflow velocity, cf. the recirculation region shown in the insets of
Figure 10.
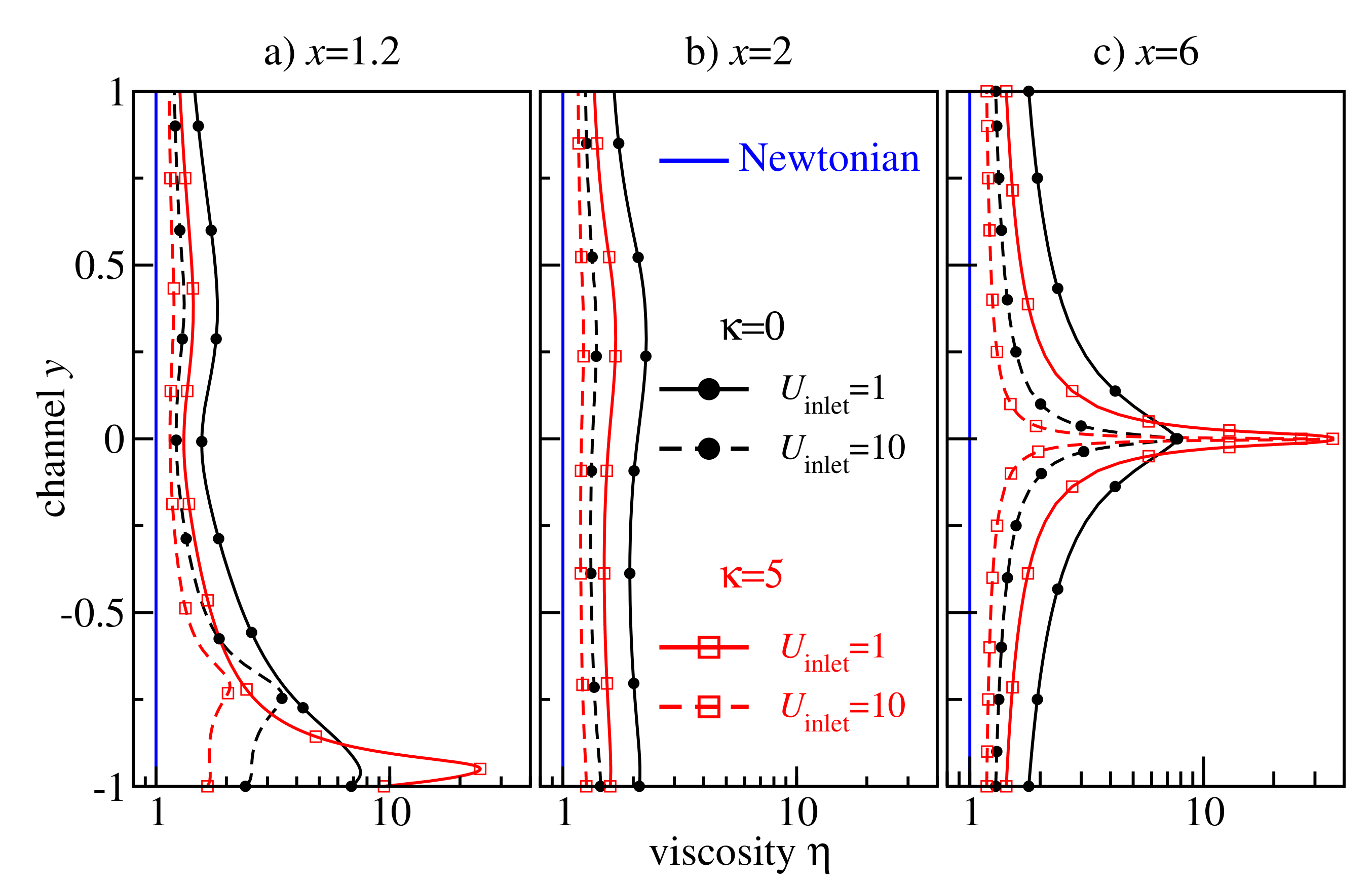
Figure 11 shows a detailed analysis of the viscosity profiles at the cross-sections
. One sharp viscosity peak is located at the centre of the main part of the channel (
) far behind the backward-facing step (cf.
Figure 11c). An analogous peak has already been observed for the non-Newtonian Poiseuille flow in
Figure 8. Another peak arises at
due to the secondary flow at the corner (cf.
Figure 11a), indicating that the non-Newtonian effect plays an important role in this region, too.
Our extensive simulations explain that (i) the retrograde non-Newtonian behaviour is a result of a nearly homogeneous distribution of the viscosity
at high shear rates in the flow regime when
does not vary significantly (post shear-thinning plateau). (ii) By tuning the flow parameters, it is possible to control the strength of the shear-thinning effect in a melt by varying the area under the
-shaped viscosity curve in
Figure 8. (iii) A higher flow intensity leads to a wider shear rate distribution and to a narrower
-shaped viscosity spike. Note that the height of this spike is limited by the shear viscosity in the non-sheared system, as the flat region of the velocity profile corresponds to shear rates close to zero.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}