
Ionic Polymer Nanocomposites Subjected to Uniaxial Extension: A Nonequilibrium Molecular Dynamics Study



Abstract
:
1. Introduction
2. Methods
2.1. Model Description and Setup
2.2. Choice of Systems
2.3. Uniaxial Extension
2.4. Quantities
- nonequilibrium contribution to the stress tensor, : its potential and kinetic contributions are calculated via the tensorial virial theorem, using coordinates and momenta.
- number of temporary crosslinks, : between a single polymer and all NPs, averaged over all polymers. Such a temporary crosslink is formed when one (or more) monomer(s) of a chain comes into the neighborhood of an NP surface [42].
- NP–NP radial pair correlation function, : calculated from the center coordinates of the NPs, , where now denotes the position of an NP center, and the average is taken over all pairs of NPs.
- non-affine NP displacement, : quantifies the extent of non-affine motion of NPs at given strain. If denotes the th component of the time-dependent position vector of an NP center and the box size in -direction, then is the mean length of the vectors (one for each NP), whose components are given by , where is the time between snapshots. We take 500 snapshots during each uniaxial extension experiment up to strain , hence .
- mean pore radius: calculated from the pore radius histogram , which offers the probability that the largest sphere that can be inserted into the polymer matrix without any overlap with existing NPs, and which contains point and has radius r, provided that the insertion point is chosen randomly from the points residing in the space available to the polymers. See for example, Ref. [7] for more details on the pore size definition.
- disorder parameter S: defined, based on the NP–NP pair correlation function, in Equation (5).
- segment orientation tensor : obtained with from all normalized bond vectors along the polymer chains, as a function of strain.
3. Results and Discussion
3.1. Stress–Strain Behavior
3.2. NP Structure during Uniaxial Extension
3.3. Polymer Conformations
3.4. Polymer–Polymer Entanglements
3.5. Polymer-NP Temporary Crosslinks
3.6. Pore Size Analysis
3.7. Stretching of the Entangled IPNC Systems at Low Strain Rates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Odent, J.; Raquez, J.M.; Dubois, P.; Giannelis, E.P. Ultra-stretchable ionic nanocomposites: From dynamic bonding to multi-responsive behaviors. J. Mater. Chem. A 2017, 5, 13357–13363. [Google Scholar] [CrossRef]
- Potaufeux, J.E.; Odent, J.; Notta-Cuvier, D.; Delille, R.; Barrau, S.; Giannelis, E.P.; Lauro, F.; Raquez, J.M. Mechanistic insights on ultra-tough polylactide-based ionic nanocomposites. Compos. Sci. Techn. 2020, 191, 108075. [Google Scholar] [CrossRef]
- Rodriguez, R.; Herrera, R.; Archer, L.A.; Giannelis, E.P. Nanoscale Ionic Materials. Adv. Mater. 2008, 20, 4353–4358. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, N.J.; Wallin, T.J.; Vaia, R.A.; Koerner, H.; Giannelis, E.P. Nanoscale ionic materials. Chem. Mater. 2014, 26, 84–96. [Google Scholar] [CrossRef]
- Fernandes, N.J.; Akbarzadeh, J.; Peterlik, H.; Giannelis, E.P. Synthesis and properties of highly dispersed ionic silica-poly(ethylene oxide) nanohybrids. ACS Nano 2013, 7, 1265–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersen, M.L.; Mirau, P.A.; von Meerwall, E.D.; Koerner, H.; Vaia, R.A.; Fernandes, N.J.; Giannelis, E.P. Hierarchical Canopy Dynamics of Electrolyte-Doped Nanoscale Ionic Materials. Macromolecules 2013, 46, 9669–9675. [Google Scholar] [CrossRef]
- Moghimikheirabadi, A.; Kröger, M.; Karatrantos, A.V. Insights from modeling into structure, entanglements, and dynamics in attractive polymer nanocomposites. Soft Matter 2021, 17, 6362–6373. [Google Scholar] [CrossRef]
- Karatrantos, A.; Koutsawa, Y.; Dubois, P.; Clarke, N.; Kröger, M. Miscibility and diffusion in ionic nanocomposites. Polymers 2018, 10, 1010. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zeng, X.; Pidaparti, R.; Wang, X. Tough and strong bioinspired nanocomposites with interfacial cross-links. Nanoscale 2016, 8, 18531–18540. [Google Scholar] [CrossRef]
- Shah, D.; Maiti, P.; Jiang, D.D.; Batt, C.A.; Giannelis, E.P. Effect of nanoparticle mobility on toughness of polymer nanocomposites. Adv. Mater. 2005, 17, 525. [Google Scholar] [CrossRef]
- Gersappe, D. Molecular Mechanisms of Failure in Polymer Nanocomposites. Phys. Rev. Lett. 2002, 89, 058301. [Google Scholar] [CrossRef] [PubMed]
- Karatrantos, A.; Clarke, N.; Kröger, M. Modeling of polymer structure and conformations in polymer nanocomposites from atomistic to mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Borodin, O.; Bedrov, D.; Smith, G.D.; Nairn, J.; Bardenhagen, S. Multiscale Modeling of Viscoelastic Properties of Polymer Nanocomposites. J. Polym. Sci. B 2005, 43, 1005–1013. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, L.; Cao, D.; Wang, W. Static, rheological and mechanical properties of polymer nanocomposites studied by computer modeling and simulation. Phys. Chem. Chem. Phys. 2009, 11, 11365–11384. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzis, G.G.; Theodorou, D.N. Multiscale Molecular Simulations of Polymer-Matrix Nanocomposites. Arch. Comput. Meth. Eng. 2017, 25, 591–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y. Effect of nanoinclusions on the structural and physical properties of polythelene polymer matrix. Polymer 2011, 52, 2310–2318. [Google Scholar] [CrossRef]
- Fu, Y.; Song, J.H. Large deformation mechanism of glassy polyethylene polymer nanocomposites: Coarse grain molecular dynamics study. Comput. Mater. Sci. 2015, 96, 485–494. [Google Scholar] [CrossRef]
- Kutvonen, A.; Rossi, G.; Ala-Nissila, T. Correlations between mechanical, structural, and dynamical properties of polymer nanocomposites. Phys. Rev. E 2012, 85, 041803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggleman, R.A.; Toepperwein, G.N.; Papakonstantopoulos, G.J.; de Pablo, J.J. Dynamics of a Glassy Polymer Nanocomposite during Active Deformation. Macromolecules 2009, 42, 3632–3640. [Google Scholar] [CrossRef]
- Vorselaars, B.; Lyulin, A.V.; Michels, M.A.J. Development of Heterogeneity near the Glass Transition: Phenyl-Ring-Flip Motions in Polystyrene. Macromolecules 2007, 40, 6001–6011. [Google Scholar] [CrossRef]
- Riggleman, R.A.; Toepperwein, G.; Papakonstantopoulos, G.J.; Barrat, J.L.; de Pablo, J.J. Entanglement network in nanoparticle reinforced polymers. J. Chem. Phys. 2009, 130, 244903. [Google Scholar] [CrossRef]
- Lin, E.Y.; Frischknecht, A.L.; Riggleman, R.A. Origin of Mechanical Enhancement in Polymer Nanoparticle (NP) Composites with Ultrahigh NP Loading. Macromolecules 2020, 53, 2976–2982. [Google Scholar] [CrossRef]
- Hagita, K.; Morita, H.; Takano, H. Molecular dynamics simulation study of a fracture of filler-filled polymer nanocomposites. Polymer 2016, 99, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Hagita, K.; Morita, H.; Doi, M.; Takano, H. Coarse-Grained Molecular Dynamics Simulation of Filled Polymer Nanocomposites under Uniaxial Elongation. Macromolecules 2016, 49, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Lin, X.; Liu, J.; Li, X. Revisiting stress–strain behavior and mechanical reinforcement of polymer nanocomposites from molecular dynamics simulations. Phys. Chem. Chem. Phys. 2020, 22, 16760–16771. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantopoulos, G.J.; Yoshimoto, K.; Doxastakis, M.; Nealey, P.F.; de Pablo, J.J. Local mechanical properties of polymeric nanocomposites. Phys. Rev. E 2005, 72, 031801. [Google Scholar] [CrossRef]
- Molinari, N.; Sutton, A.P.; Mostofi, A.A. Mechanisms of reinforcement in polymer nanocomposites. Phys. Chem. Chem. Phys. 2018, 20, 23085–23094. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Liu, J.; Gao, Y.; Cao, D.; Zhang, L. Molecular dynamics simulations of the structural, mechanical and visco-elastic properties of polymer nanocomposites filled with grafted nanoparticles. Phys. Chem. Chem. Phys. 2015, 17, 7196. [Google Scholar] [CrossRef]
- Gao, N.; Hou, G.; Liu, J.; Shen, J.; Gao, Y.; Lyulin, A.V.; Zhang, L. Tailoring the mechanical properties of polymer nanocomposites via interfacial engineering. Phys. Chem. Chem. Phys. 2019, 21, 18714–18726. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Zeng, F.; Peng, C.; Wang, Y. Coarse-grained molecular dynamics simulation of cis-1,4-polyisoprene with silica nanoparticles under extreme uniaxial tension. Model. Simul. Mater. Sci. Eng. 2021, 29, 055013. [Google Scholar] [CrossRef]
- Chao, H.; Riggleman, R.A. Effect of particle size and grafting density on the mechanical properties of polymer nanocomposites. Polymer 2013, 54, 5222–5229. [Google Scholar] [CrossRef]
- Frankland, S. The stress–strain behavior of polymer–nanotube composites from molecular dynamics simulation. Compos. Sci. Technol. 2003, 63, 1655–1661. [Google Scholar] [CrossRef]
- Toepperwein, G.N.; Riggleman, R.A.; de Pablo, J.J. Dynamics and deformation response of rod-containing nanocomposites. Macromolecules 2011, 45, 543–554. [Google Scholar] [CrossRef]
- Skountzos, E.N.; Anastassiou, A.; Mavrantzas, V.G.; Theodorou, D.N. Determination of the Mechanical Properties of a Poly(methyl methacrylate) Nanocomposite with Functionalized Graphene Sheets through Detailed Atomistic Simulations. Macromolecules 2014, 47, 8073–8088. [Google Scholar] [CrossRef]
- Yang, S.; Choi, J.; Cho, M. Elastic stiffness and filler size effect of covalently grafted nanosilica polyimide composites: Molecular dynamics study. ACS Appl. Mater. Interfaces 2012, 4, 4792–4799. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, M.; Iriarte-Carretero, I.; Ghanbari, A.; Bohm, M.C.; Müller-Plathe, F. Mechanical behavior and interphase structure in a silica-polystyrene nanocomposite under uniaxial deformation. Nanotechnology 2012, 23, 305702. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, A.S.; Khalatur, P.G. Fully atomistic molecular dynamics simulation of nanosilica-filled crosslinked polybutadiene. Chem. Phys. Lett. 2016, 653, 90–95. [Google Scholar] [CrossRef]
- Kutvonen, A.; Rossi, G.; Puisto, S.R.; Rostedt, N.K.J.; Ala-Nissila, T. Influence of nanoparticle size, loading, and shape on the mechanical properties of polymer nanocomposites. J. Chem. Phys. 2012, 137, 214901. [Google Scholar] [CrossRef] [Green Version]
- Papakonstantopoulos, G.J.; Doxastakis, M.; Nealey, P.F.; Barrat, J.L.; de Pablo, J.J. Calculation of local mechanical properties of filled polymers. Phys. Rev. E 2007, 75, 031803. [Google Scholar] [CrossRef]
- Choudhary, S.; Sengwa, R.J. Dielectric spectroscopy and confirmation of ion conduction mechanism in direct melt compounded hot-press polymer nanocomposite electrolytes. Ionics 2011, 17, 811–819. [Google Scholar] [CrossRef]
- Odent, J.; Raquez, J.M.; Samuel, C.; Barrau, S.; Enotiadis, A.; Dubois, P.; Giannelis, E.P. Shape-memory behavior of polylactide/silica ionic hybrids. Macromolecules 2017, 50, 2896. [Google Scholar] [CrossRef]
- Moghimikheirabadi, A.; Mugemana, C.; Kröger, M.; Karatrantos, A.V. Polymer conformations, entanglements and dynamics in ionic nanocomposites: A molecular dynamics study. Polymers 2020, 12, 2591. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Wang, J.; Shen, Y.; Gu, L.; Lin, Y.; Nan, C.W. Hierarchical interfaces induce high dielectric permittivity in nanocomposites containing TiO2@BaTiO3 nanofibers. Nanoscale 2014, 6, 6701–6709. [Google Scholar] [CrossRef] [PubMed]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057. [Google Scholar] [CrossRef]
- Cao, X.Z.; Merlitz, H.; Wu, C.X.; Ungar, G.; Sommer, J.U. A theoretical study of dispersion-to-aggregation of nanoparticles in adsorbing polymers using molecular dynamics simulations. Nanoscale 2016, 8, 6964–6968. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Olson, E.; Jiang, S.; Yong, X. Nanoparticle assembly modulated by polymer chain conformation in composite materials. Nanoscale 2020, 12, 14560–14572. [Google Scholar] [CrossRef]
- Rubinstein, M.; Panyukov, S. Elasticity of Polymer Networks. Macromolecules 2002, 35, 6670. [Google Scholar] [CrossRef]
- Kröger, M.; Hess, S. Rheological evidence for a dynamical crossover in polymer melts via nonequilibrium molecular dynamics. Phys. Rev. Lett. 2000, 85, 1128–1131. [Google Scholar] [CrossRef]
- Miwatani, R.; Takahashi, K.Z.; Arai, N. Performance of coarse graining in estimating polymer properties: Comparison with the atomistic model. Polymers 2020, 12, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, H.R. Kinetic theory and rheology of dilute suspensions of finitely extendible dumbbells. Ind. Eng. Chem. Fund. 1972, 11, 379–387. [Google Scholar] [CrossRef]
- Kröger, M.; Loose, W.; Hess, S. Structural changes and rheology of polymer melts via nonequilibrium molecular dynamics. J. Rheol. 1993, 37, 1057–1079. [Google Scholar] [CrossRef]
- Hagita, K.; Murashima, T.; Iwaoka, N. Thinning approximation for calculating two-dimensional scattering patterns in dissipative particle dynamics simulations under shear flow. Polymers 2018, 10, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguchi, T.; Uehara, E. Statistical and dynamical properties of topological polymers with graphs and ring polymers with knots. Polymers 2017, 9, 252. [Google Scholar] [CrossRef]
- Kröger, M. Simple, admissible, and accurate approximants of the inverse Langevin and Brillouin functions, relevant for strong polymer deformations and flows. J. Non-Newton. Fluid Mech. 2015, 223, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Moghimikheirabadi, A.; Sagis, L.M.C.; Kröger, M.; Ilg, P. Gas–liquid phase equilibrium of a model Langmuir monolayer captured by a multiscale approach. Phys. Chem. Chem. Phys. 2019, 21, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghimikheirabadi, A.; Ilg, P.; Sagis, L.M.C.; Kröger, M. Surface rheology and structure of model triblock copolymers at a liquid–vapor interface: A molecular dynamics study. Macromolecules 2020, 53, 1245–1257. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Hou, J.X.; Svaneborg, C.; Everaers, R.; Grest, G.S. Stress relaxation in entangled polymer melts. Phys. Rev. Lett. 2010, 105, 068301. [Google Scholar] [CrossRef] [Green Version]
- Hoy, R.S.; Foteinopoulou, K.; Kröger, M. Topological analysis of polymeric melts: Chain-length effects and fast-converging estimators for entanglement length. Phys. Rev. E 2009, 80, 031803. [Google Scholar] [CrossRef] [Green Version]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Entanglements in polymer nanocomposites containing spherical nanoparticles. Soft Matter 2016, 12, 2567. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ting, C.L.; Composto, R.J.; Frischknecht, A.L. Nonequilibrium simulations of model ionomers in an oscillating electric field. J. Chem. Phys. 2016, 145, 044902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Archer, L.A. Poly(ethylene oxide)/silica nanocomposites: Structure and rheology. Langmuir 2002, 18, 10435–10442. [Google Scholar] [CrossRef]
- Koizumi, N.; Hanai, T. Dielectric properties of polyethylene glycols: Dielectric relaxation in solid state. Bull. Inst. Chem. Res. 1964, 42, 115–127. [Google Scholar]
- Porter, C.; Boyd, R. A dielectric study of the effects of melting on molecular relaxation in poly(ethylene oxide) and polyoxymethylene. Macromolecules 1971, 4, 589–594. [Google Scholar] [CrossRef]
- Gkolfi, E.; Bacova, P.; Harmandaris, V. Size and shape characteristics of polystyrene and poly(ethylene oxide) star polymer melts studied by atomistic simulations. Macromol. Theory Simul. 2020, 30, 2000067. [Google Scholar] [CrossRef]
- Kröger, M. Shortest multiple disconnected path for the analysis of entanglements in two- and three-dimensional polymeric systems. Comput. Phys. Commun. 2005, 168, 209–232. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Kröger, M. Combined Molecular Algorithms for the Generation, Equilibration and Topological Analysis of Entangled Polymers: Methodology and Performance. Int. J. Molec. Sci. 2009, 10, 5054–5089. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1989. [Google Scholar]
- Kröger, M. Models for Polymeric and Anisotropic Liquids; Springer: New York, NY, USA, 2005. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kremer, K.; Sukumaran, S.K.; Everaers, R.; Grest, G.S. Entangled polymer systems. Comput. Phys. Commun. 2005, 169, 75–81. [Google Scholar] [CrossRef]
- Hoy, R.S.; Robbins, M.O. Strain Hardening of Polymer Glasses: Effect of Entanglement Density, Temperature, and Rate. J. Polym. Sci. B 2006, 44, 3487–3500. [Google Scholar] [CrossRef]
- Wang, D.; An, X.; Gou, D.; Zhao, H.; Wang, L.; Huang, F. DEM construction of binary hard sphere crystals and radical tessellation. AIP Adv. 2018, 8, 105203. [Google Scholar] [CrossRef] [Green Version]
- Salib, I.G.; Kolmakov, G.V.; Bucior, B.J.; Peleg, O.; Kröger, M.; Savin, T.; Vogel, V.; Matyjaszewski, K.; Balazs, A.C. Using Mesoscopic Models to Design Strong and Tough Biomimetic Polymer Networks. Langmuir 2011, 27, 13796–13805. [Google Scholar] [CrossRef] [PubMed]
- Peleg, O.; Savin, T.; Kolmakov, G.V.; Salib, I.G.; Balazs, A.C.; Kröger, M.; Vogel, V. Fibers with Integrated Mechanochemical Switches: Minimalistic Design Principles Derived from Fibronectin. Biophys. J. 2012, 103, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.J.; Wang, H.; Du, K.; Zhang, W.; Sui, M.L.; Mao, S.X. Deformation-induced structural transition in body-centred cubic molybdenum. Nat. Commun. 2014, 5, 3433. [Google Scholar] [CrossRef]
- Xu, H.; Lv, Y.; Qiu, D.; Zhou, Y.; Zeng, H.; Chu, Y. An ultra-stretchable, highly sensitive and biocompatible capacitive strain sensor from an ionic nanocomposite for on-skin monitoring. Nanoscale 2019, 11, 1570–1578. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | N | n | |||||
|---|---|---|---|---|---|---|---|
| L1 | 200 | 11.6% | 24 | 256 | 2304 | 603 | 54.2 × 108.3 × 108.3 |
| L1 | 200 | 11.6% | 50 | 256 | 2304 | 603 | 54.2 × 108.3 × 108.3 |
| L2 | 200 | 20.8% | 24 | 512 | 2304 | 306 | 56.2 × 112.4 × 112.4 |
| L2 | 200 | 20.8% | 50 | 512 | 2304 | 306 | 56.2 × 112.4 × 112.4 |
| S1 | 40 | 11.7% | 24 | 192 | 8640 | 630 | 49.3 × 98.5 × 98.5 |
| S1 | 40 | 11.6% | 50 | 192 | 8640 | 630 | 49.3 × 98.5 × 98.5 |
| S2 | 40 | 20.8% | 24 | 384 | 8640 | 315 | 51.1 × 102.3 × 102.3 |
| S2 | 40 | 20.7% | 50 | 384 | 8640 | 315 | 51.1 × 102.3 × 102.3 |
| Figure | Label | Content | Figure | Label | Content | ||||
|---|---|---|---|---|---|---|---|---|---|
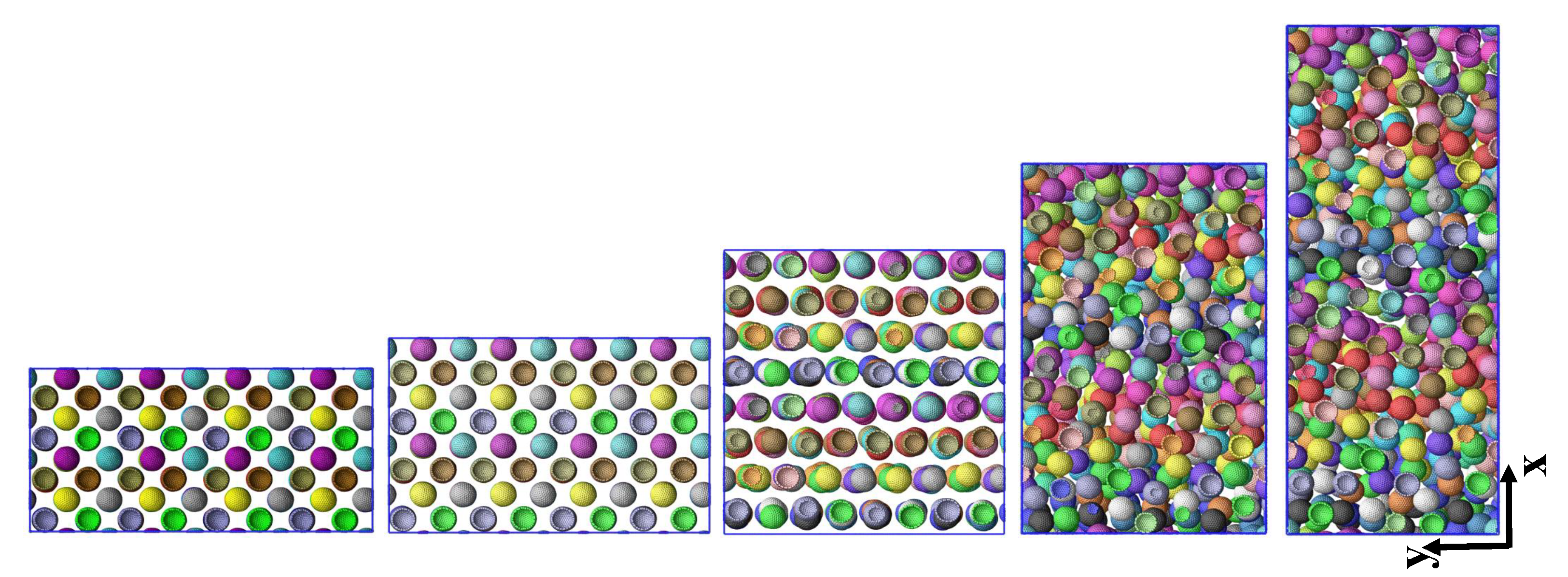
| Figure 1 | L2 | 0 | 0 | snapshot | Figure S1a,b | L2, S2 | varied | 0–8 | stress–strain |
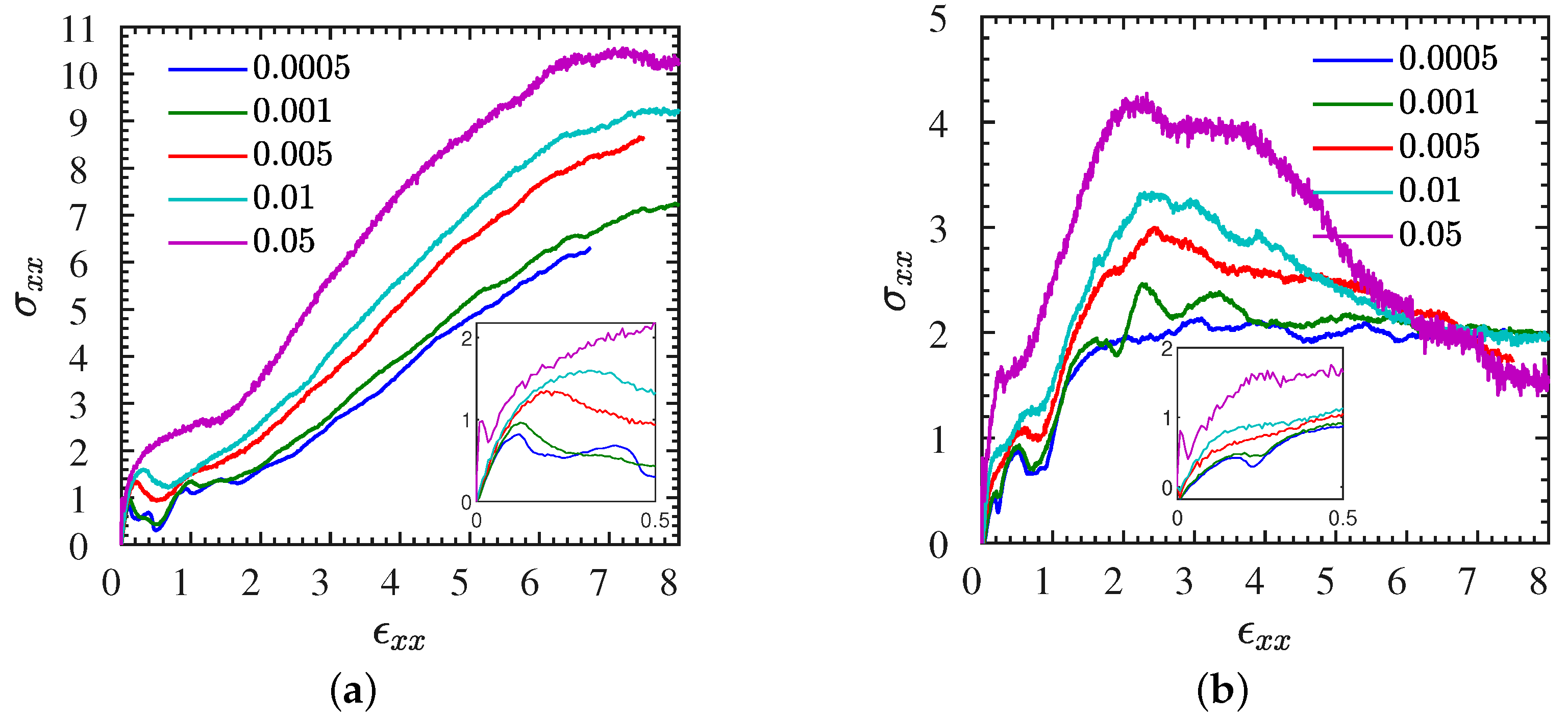
| Figure 2a,b | L1, S1 | varied | 0–8 | stress–strain | Figure S2 | L2 | varied | 0–5 | correlation |
| Figure 3 | S1 | 0.05 | 7.5 | snapshots | Figure S3 | L1 | varied | 0–3 | correlation |
| Figure 4a,b | L, S | 0.01 | 0–3 | stress–strain | Figure S4 | L1 | varied | 0–8 | gyration |
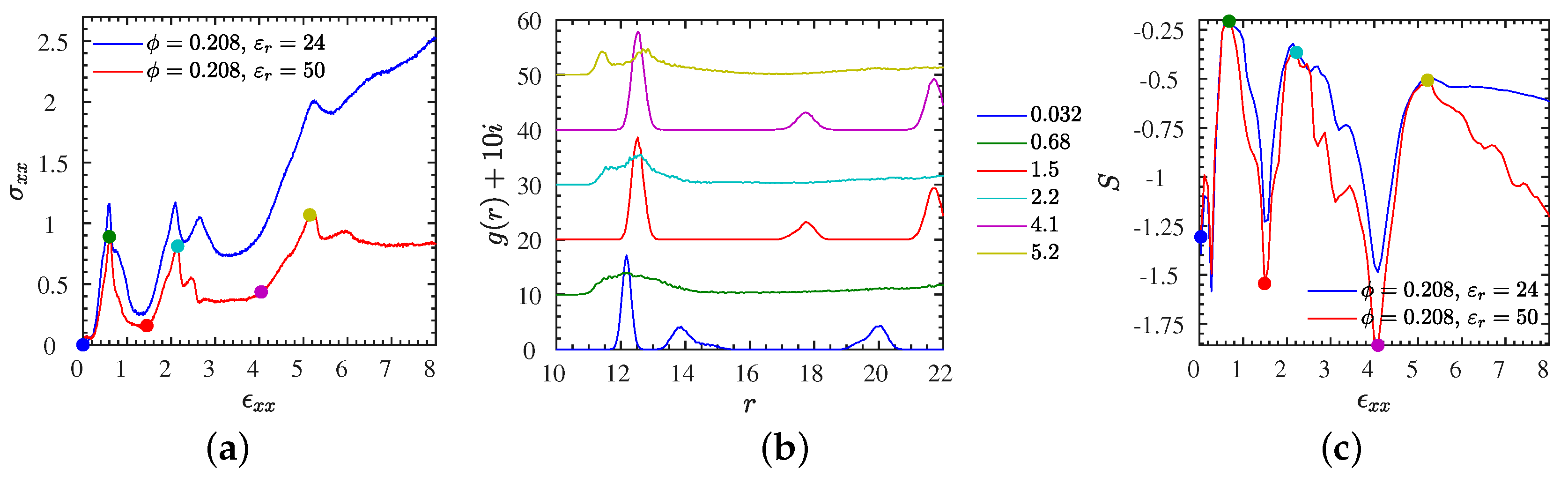
| Figure 5 | L2 | 0.005 | 0–3 | stress–strain, , S | Figure S5 | S1 | varied | 0–8 | gyration |
| Figure 6 | L2 | 0.005 | 0–2 | snapshots | Figure S6 | L, S | 0.01 | 0–8 | stress-optic |
| Figure 7 | L, S | 0.01 | 0–3 | gyration | Figure S7a | S1 | varied | 0–8 | entanglements |
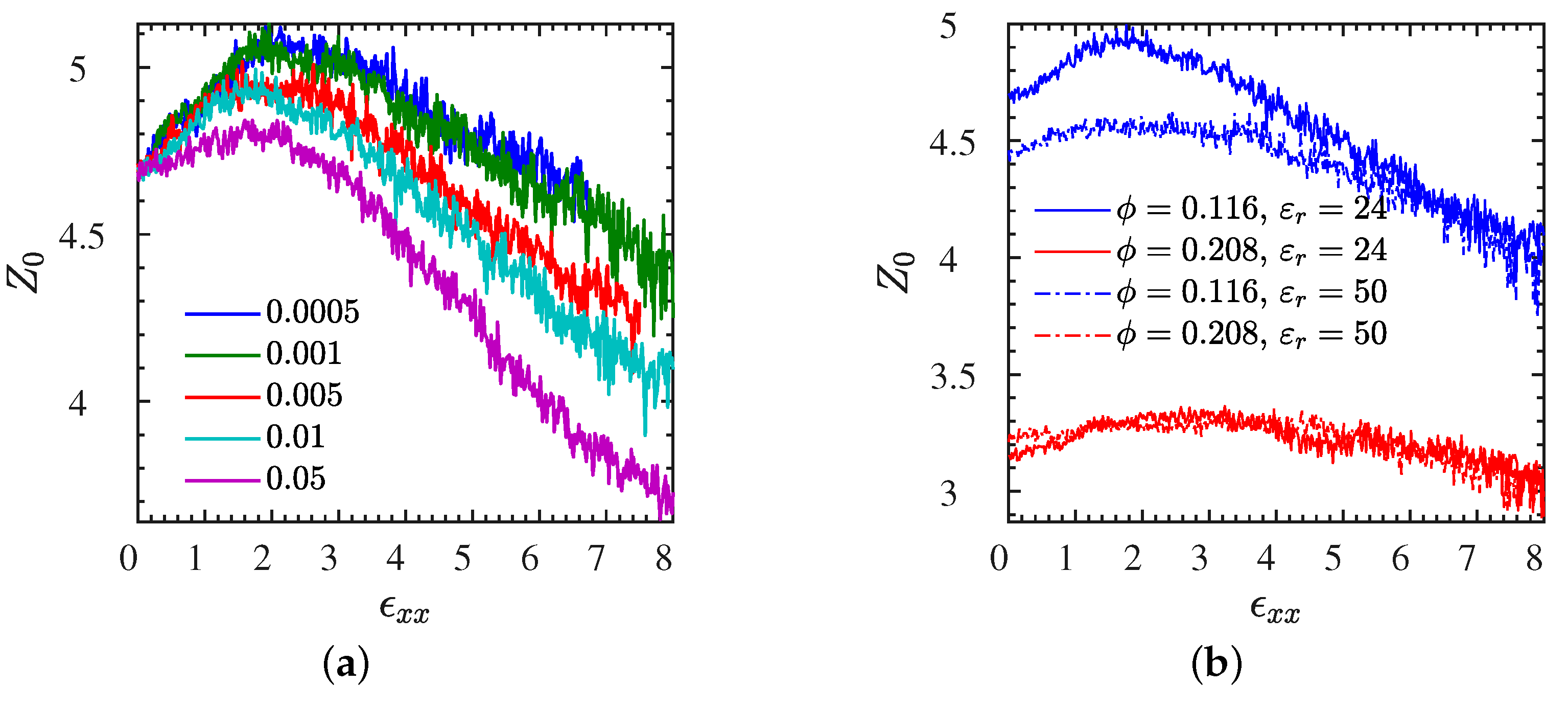
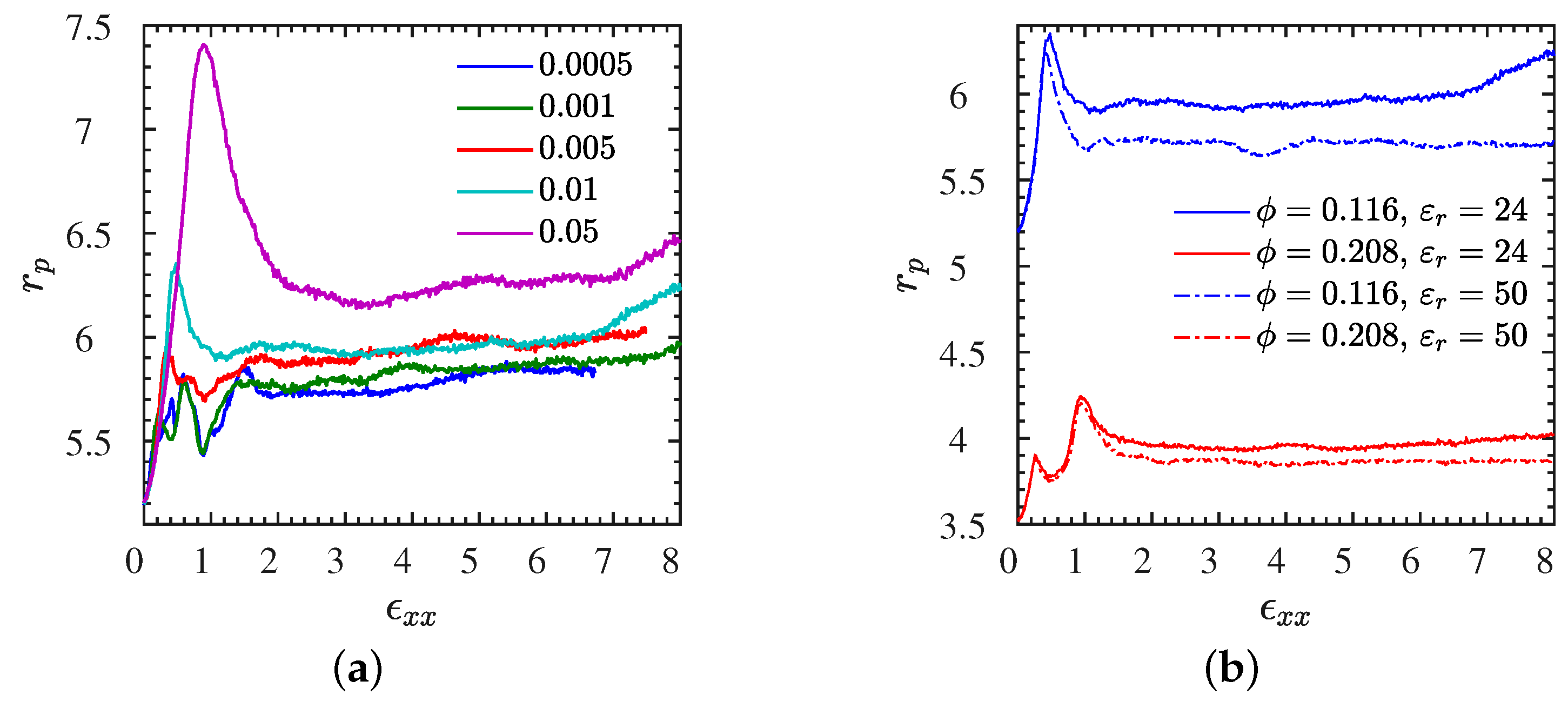
| Figure 8>a | L1 | varied | 0–8 | entanglements | Figure S7b | S | 0.01 | 0–8 | entanglements |
| Figure 8b | L | 0.01 | 0–8 | entanglements | Figure S8a | S2 | varied | 0–8 | crosslinks |
| Figure 9a | L2 | varied | 0–8 | crosslinks | Figure S8b | S | 0.01 | 0–8 | crosslinks |
| Figure 9b | L | 0.01 | 0–8 | crosslinks | Figure S9a | S1 | varied | 0–8 | pore |
| Figure 10a | L1 | varied | 0–8 | pore | Figure S9b | S | 0.01 | 0–8 | pore |
| Figure 10b | L | 0.01 | 0–8 | pore | Figure S10 | L2 | 0.001 | 0–8 | , , |
| Figure 11 | L2 | 0.001 | 0–8 | stress–strain, , S | Figure S11 | L, S | 0.01 | 0–3 | non-affine |
| Figure 12 | L2 | 0.001 | 0–5 | snapshots | Figure S12 | L2 | 0.001 | 0–8 | non-affine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moghimikheirabadi, A.; Karatrantos, A.V.; Kröger, M. Ionic Polymer Nanocomposites Subjected to Uniaxial Extension: A Nonequilibrium Molecular Dynamics Study. Polymers 2021, 13, 4001. https://doi.org/10.3390/polym13224001
Moghimikheirabadi A, Karatrantos AV, Kröger M. Ionic Polymer Nanocomposites Subjected to Uniaxial Extension: A Nonequilibrium Molecular Dynamics Study. Polymers. 2021; 13(22):4001. https://doi.org/10.3390/polym13224001
Chicago/Turabian StyleMoghimikheirabadi, Ahmad, Argyrios V. Karatrantos, and Martin Kröger. 2021. "Ionic Polymer Nanocomposites Subjected to Uniaxial Extension: A Nonequilibrium Molecular Dynamics Study" Polymers 13, no. 22: 4001. https://doi.org/10.3390/polym13224001