Advances in the Multi-Orthogonal Folding of Single Polymer Chains into Single-Chain Nanoparticles


 ,
, 
Abstract
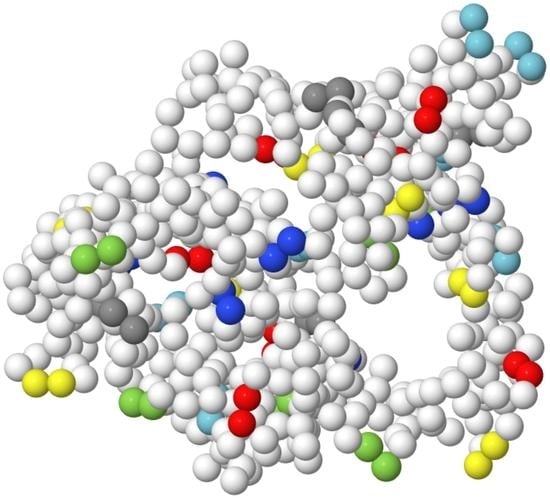
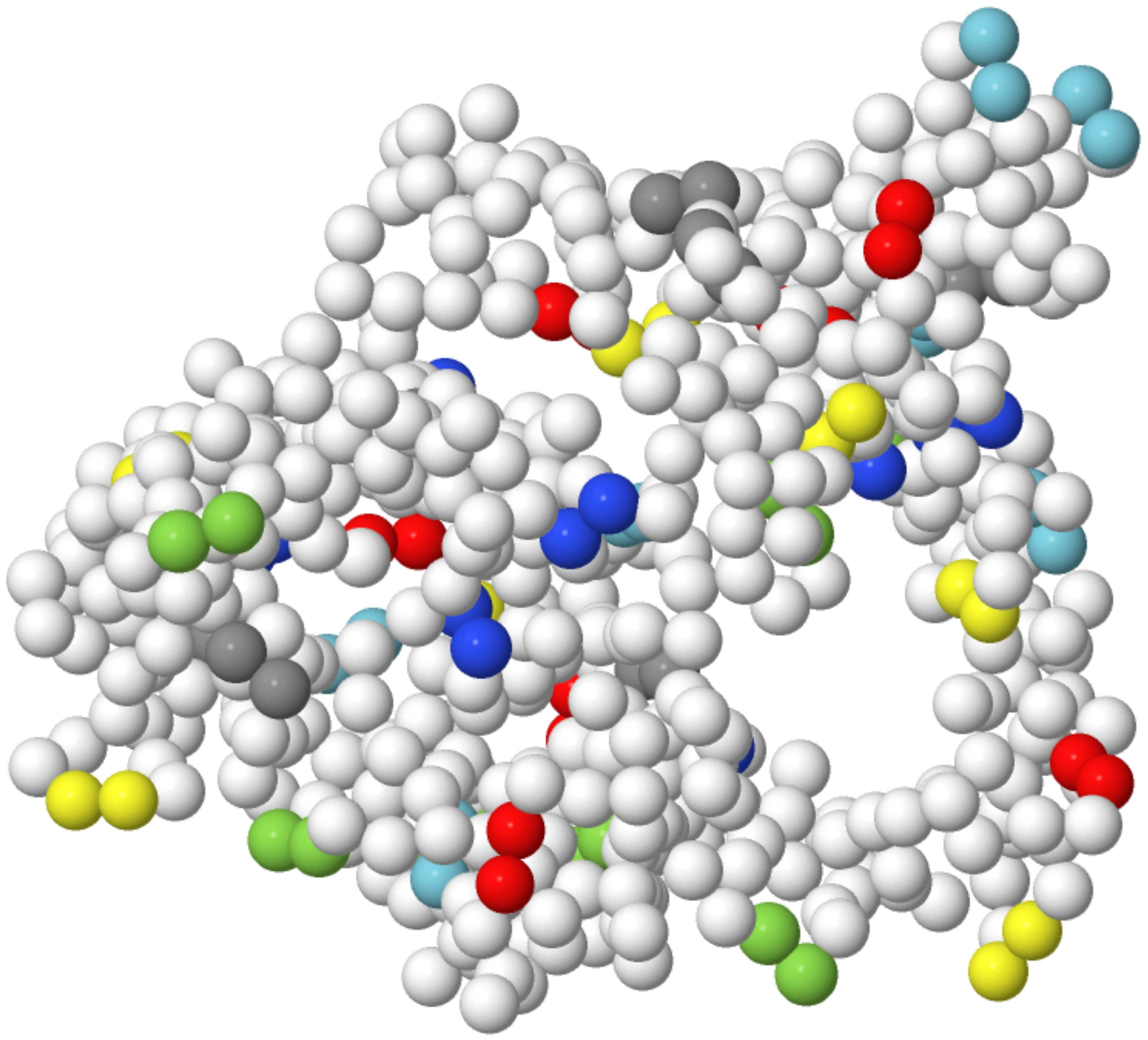
:
1. Introduction
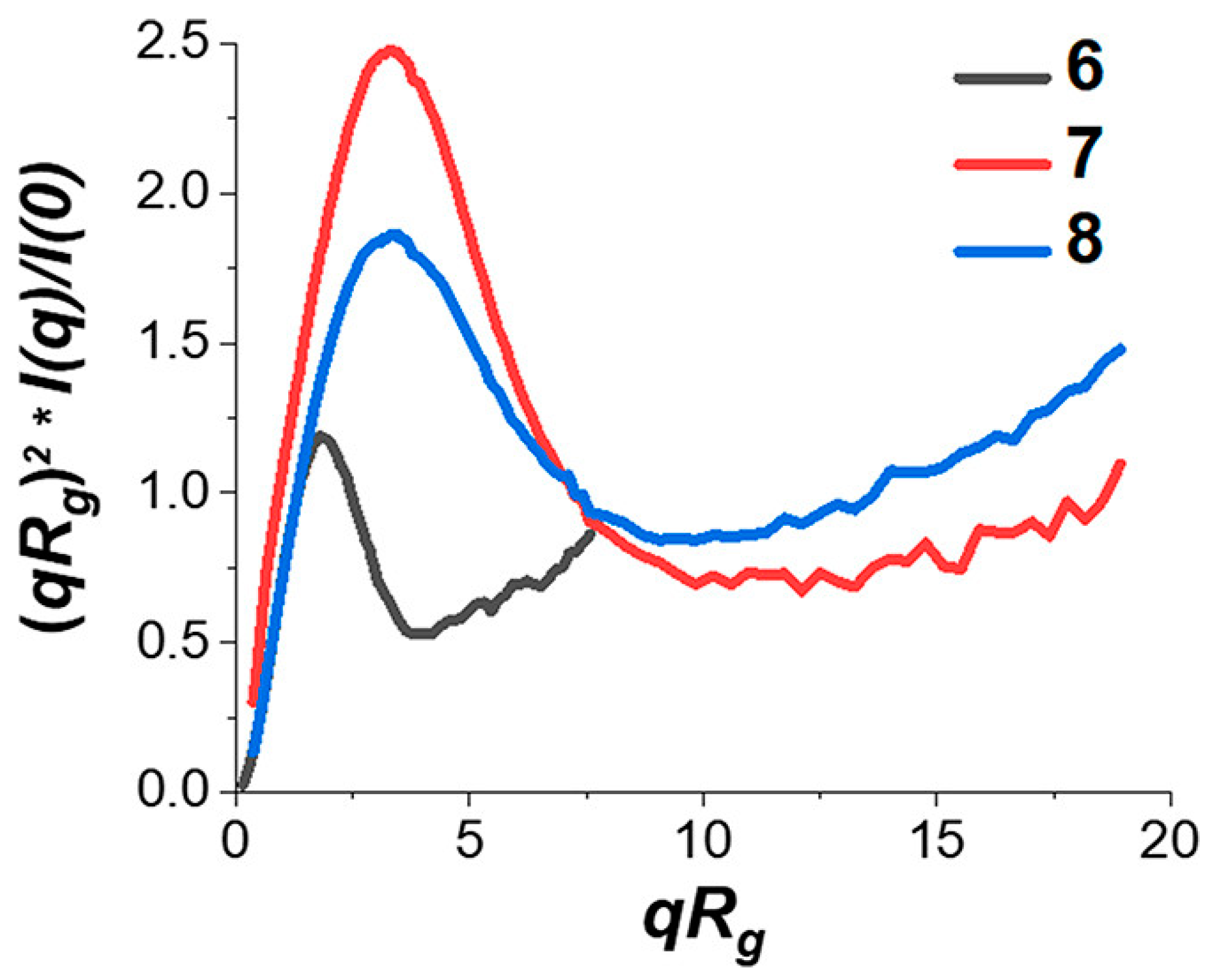
2. Simulations of Multi-Folded Single-Chain Nanoparticles
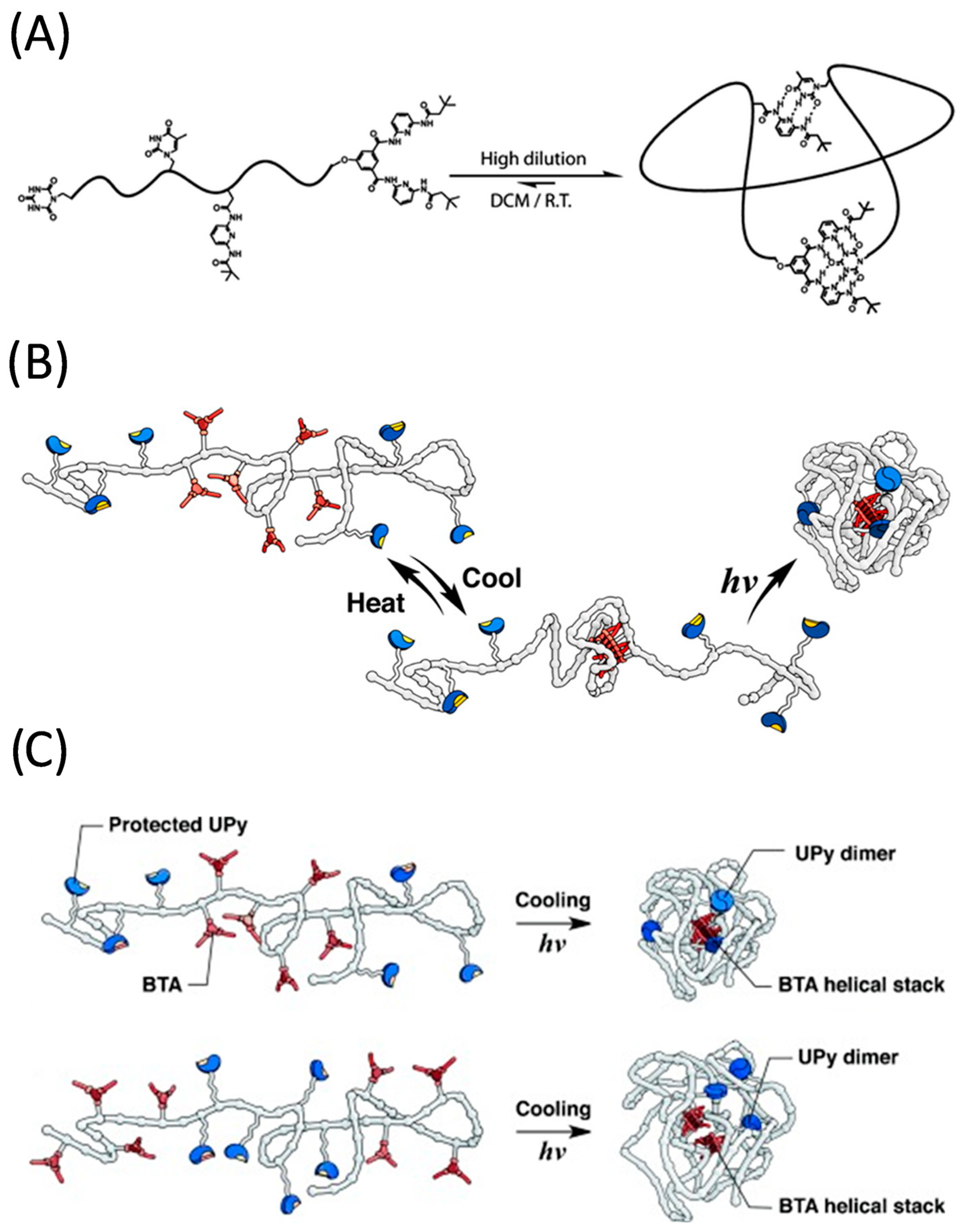
3. Multi-Folded Single-Chain Nanoparticles via Non-Covalent Bonds
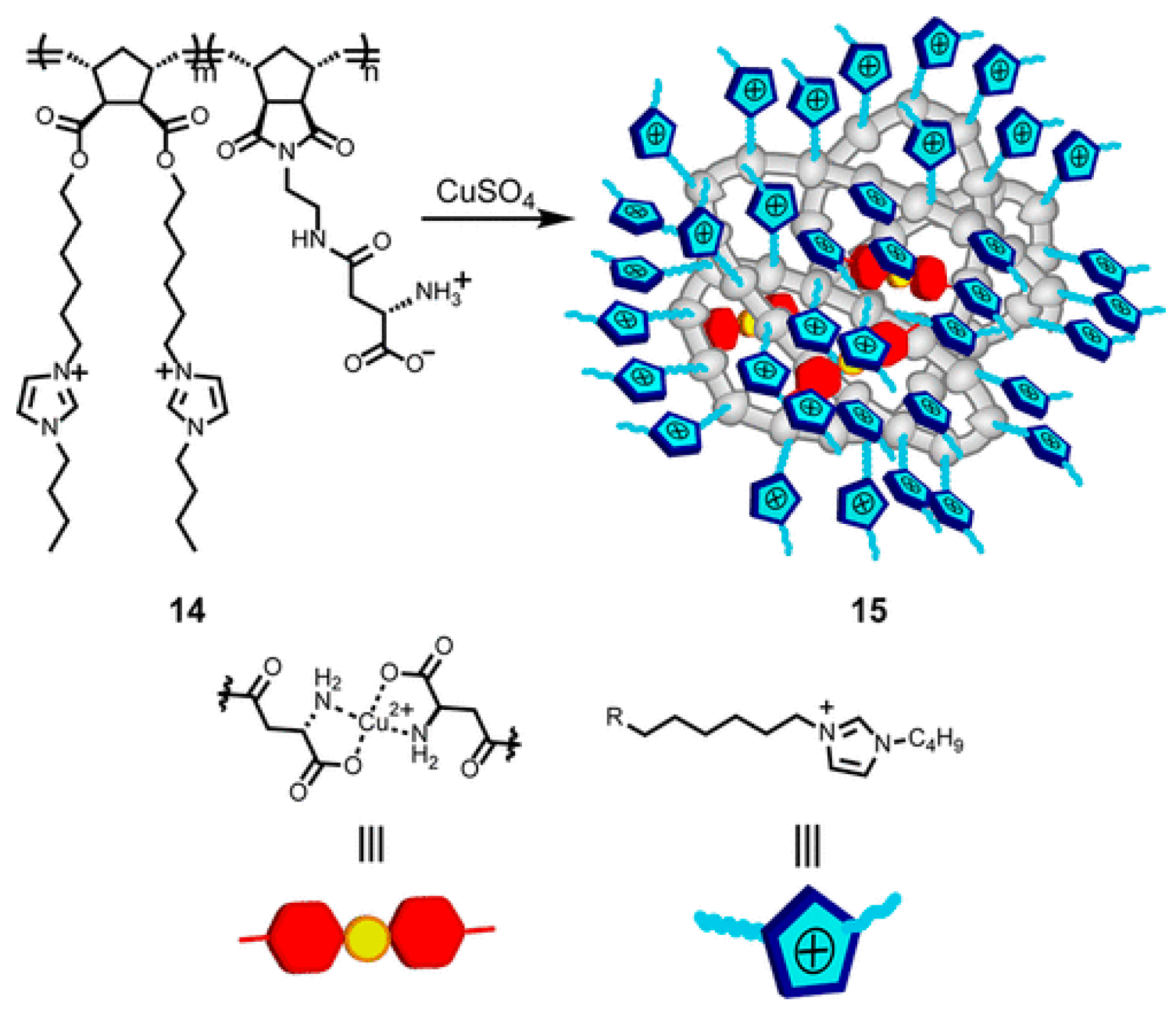
4. Multi-Folded Single-Chain Nanoparticles via Covalent Bonds
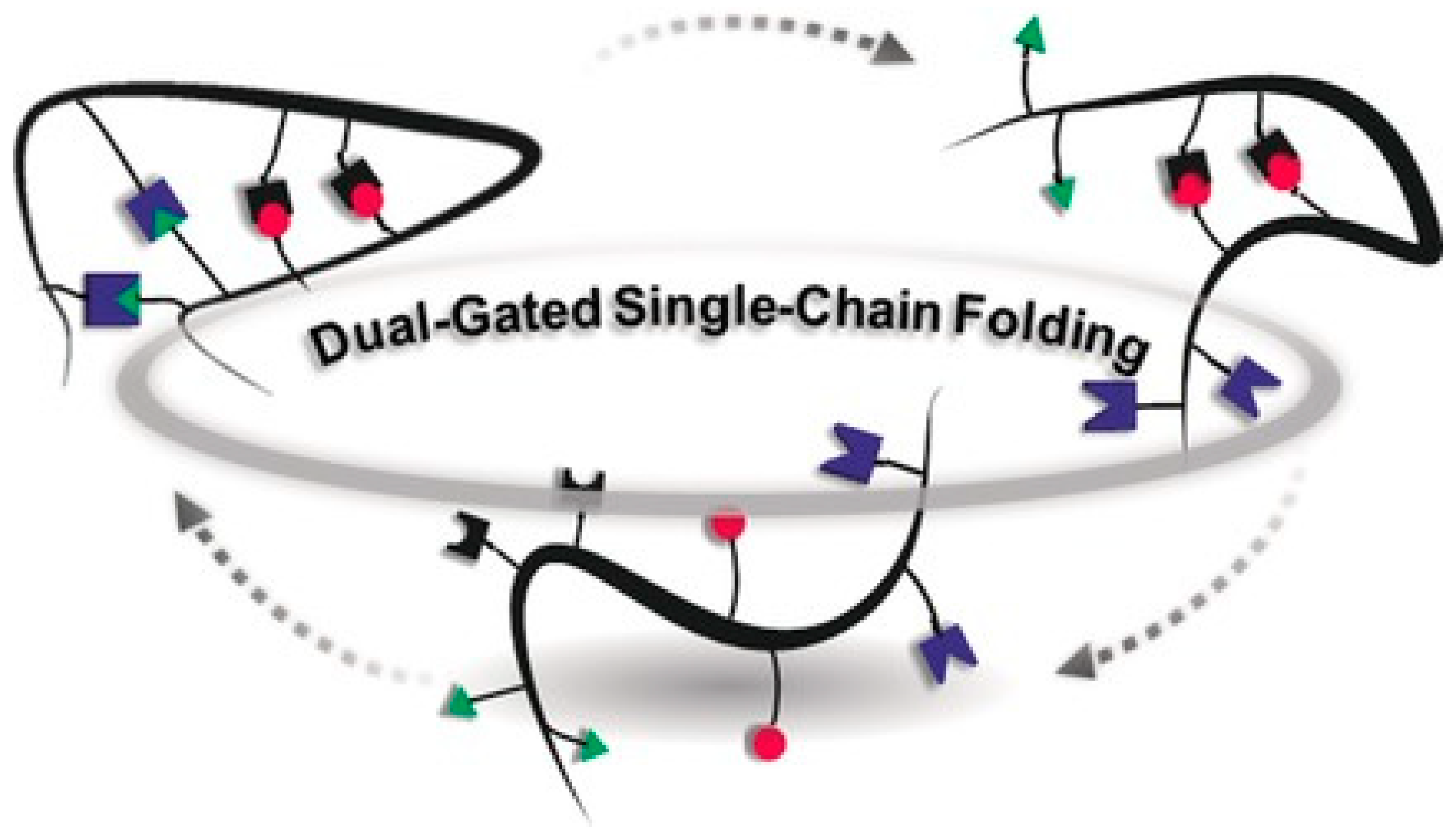
5. Multi-Folded Single-Chain Nanoparticles via Covalent and Non-Covalent Bonds
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
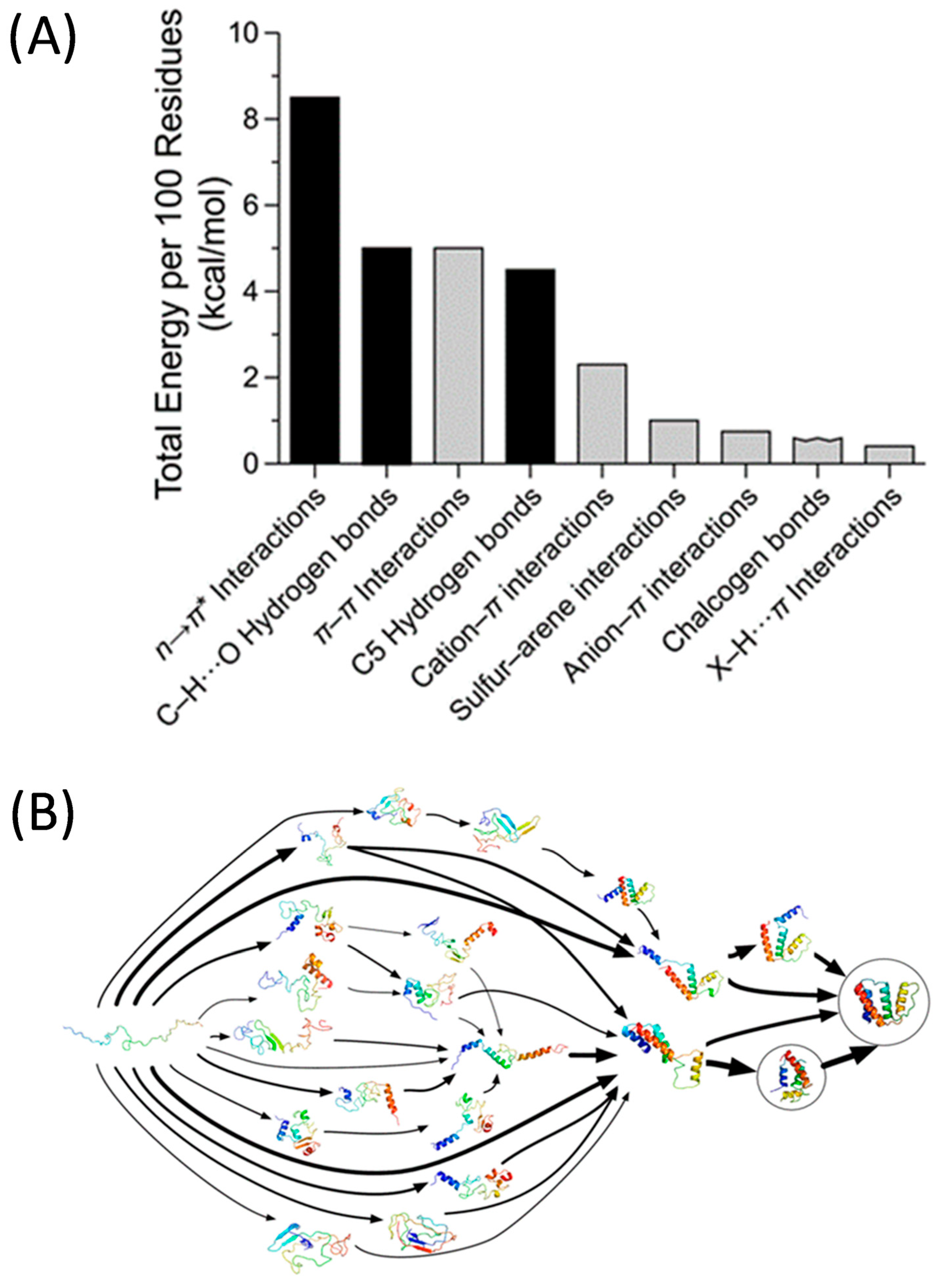
- Newberry, R.W.; Raines, R.T. Secondary Forces in Protein Folding. ACS Chem. Biol. 2019, 14, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- An Orthogonal Interaction Occurs When There Are Two Pairs of Functional Groups and Each Group can Interact with Their Respective Partner, but Does not Interact with Either Functional Group of the other Pair. Available online: https://onlinelibrary.wiley.com/doi/book/10.1002/9783527806386 (accessed on 18 January 2021).
- Voelz, V.A.; Jager, M.; Yao, S.; Chen, Y.; Zhu, L.; Waldauer, S.A.; Bowman, G.R.; Friedrichs, M.; Bakajin, O.; Lapidus, L.J.; et al. Slow Unfolded-State Structuring in Acyl-CoA Binding Protein Folding Revealed by Simulation and Experiment. J. Am. Chem. Soc. 2012, 134, 12565–12577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomposo, J. (Ed.) Single-Chain Polymer Nanoparticles: Synthesis, Characterization, Simulations and Applications; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Frisch, H.; Tuten, B.T.; Barner-Kowollik, C. Macromolecular Superstructures: A Future Beyond Single Chain Nanoparticles. Isr. J. Chem. 2020, 60, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Verde-Sesto, E.; Arbe, A.; Moreno, A.J.; Cangialosi, D.; Alegría, A.; Colmenero, J.; Pomposo, J.A. Single-chain nanoparticles: Opportunities provided by internal and external confinement. Mater. Horiz. 2020, 7, 2292–2313. [Google Scholar] [CrossRef]
- ter Huurne, G.M.; Palmans, A.R.A.; Meijer, E.W. Supramolecular Single-Chain Polymeric Nanoparticles. CCS Chem. 2019, 1, 64–82. [Google Scholar] [CrossRef] [Green Version]
- De-La-Cuesta, J.; González, E.; Pomposo, J.A. Advances in Fluorescent Single-Chain Nanoparticles. Molecules 2017, 22, 1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altintas, O.; Barner-Kowollik, C. Single-chain folding of synthetic polymers: A critical update. Macromol. Rapid Commun. 2016, 37, 29–46. [Google Scholar] [CrossRef]
- Hanlon, A.M.; Lyon, C.K.; Berda, E.B. What is next in single-chain nanoparticles? Macromolecules 2016, 49, 2–14. [Google Scholar] [CrossRef]
- Latorre-Sanchez, A.; Pomposo, J.A. Recent bioinspired applications of single-chain nanoparticles. Polym. Int. 2016, 65, 855–860. [Google Scholar] [CrossRef]
- Mavila, S.; Eivgi, O.; Berkovich, I.; Lemcoff, N.G. Intramolecular cross-linking methodologies for the synthesis of polymer nanoparticles. Chem. Rev. 2016, 116, 878–961. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, M.; Latorre-Sanchez, A.; Pomposo, J.A. Advances in single chain technology. Chem. Soc. Rev. 2015, 44, 6122–6142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, C.K.; Prasher, A.; Hanlon, A.M.; Tuten, B.T.; Tooley, C.A.; Frank, P.G.; Berda, E.B. A brief user’s guide to single-chain nanoparticles. Polym. Chem. 2015, 6, 181–197. [Google Scholar] [CrossRef]
- Müge, A.; Elisa, H.; Meijer, E.W.; Anja, R.A.P. Dynamic single chain polymeric nanoparticles: From structure to function. In Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties; American Chemical Society: Washington, DC, USA, 2014; Volume 1170, pp. 313–325. [Google Scholar]
- Sanchez-Sanchez, A.; Pomposo, J.A. Single-chain polymer nanoparticles via non-covalent and dynamic covalent bonds. Part. Part. Syst. Charact. 2014, 31, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Sanchez, A.; Perez-Baena, I.; Pomposo, J.A. Advances in click chemistry for single-chain nanoparticle construction. Molecules 2013, 18, 3339–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altintas, O.; Barner-Kowollik, C. Single chain folding of synthetic polymers by covalent and non-covalent interactions: Current status and future perspectives. Macromol. Rapid Commun. 2012, 33, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Aiertza, M.; Odriozola, I.; Cabañero, G.; Grande, H.-J.; Loinaz, I. Single-chain polymer nanoparticles. Cell. Mol. Life Sci. 2012, 69, 337–346. [Google Scholar] [CrossRef]
- Seo, M.; Beck, B.J.; Paulusse, J.M.J.; Hawker, C.J.; Kim, S.Y. Polymeric Nanoparticles via Noncovalent Cross-Linking of Linear Chains. Macromolecules 2008, 41, 6413–6418. [Google Scholar] [CrossRef]
- Foster, E.J.; Berda, E.B.; Meijer, E.W. Metastable Supramolecular Polymer Nanoparticles via Intramolecular Collapse of Single Polymer Chains. J. Am. Chem. Soc. 2009, 131, 6964–6966. [Google Scholar] [CrossRef]
- Hosono, N.; Kushner, A.M.; Chung, J.; Palmans, A.R.A.; Guan, Z.; Meijer, E.W. Forced Unfolding of Single-Chain Polymeric Nanoparticles. J. Am. Chem. Soc. 2015, 137, 6880–6888. [Google Scholar] [CrossRef]
- Terashima, T.; Mes, T.; De Greef, T.F.A.; Gillissen, M.A.J.; Besenius, P.; Palmans, A.R.A.; Guan, Z.; Meijer, E.W. Single-Chain Folding of Polymers for Catalytic Systems in Water. J. Am. Chem. Soc. 2011, 133, 4742–4745. [Google Scholar] [CrossRef]
- Appel, E.A.; Dyson, J.; del Barrio, J.; Walsh, Z.; Scherman, O.A. Formation of Single-Chain Polymer Nanoparticles in Water through Host–Guest Interactions. Angew. Chem. Int. Ed. 2012, 51, 4185–4189. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Piyapakorn, P.; Akashi, M. Formation of Unimer Nanoparticles by Controlling the Self-Association of Hydrophobically Modified Poly(amino acid)s. Langmuir 2012, 28, 5249–5256. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and Single-Chain Folding of Amphiphilic Random Copolymers in Water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Altintas, O.; Artar, M.; Huurne, G.; Voets, I.; Palmans, A.R.A.; Barner-Kowollik, C.; Meijer, E.W. Design and Synthesis of Triblock Copolymers for Creating Complex Secondary Structures by Orthogonal Self-Assembly. Macromolecules 2015, 48, 8921–8932. [Google Scholar] [CrossRef]
- Wang, F.; Pu, H.; Che, X. Voltage-responsive single-chain polymer nanoparticles via host–guest interaction. Chem. Commun. 2016, 52, 3516–3519. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pu, H.; Ding, Y.; Lin, R.; Pan, H.; Chang, Z.; Jin, M. Single-chain folding of amphiphilic copolymers in water via intramolecular hydrophobic interaction and unfolding triggered by cyclodextrin. Polymer 2018, 141, 86–92. [Google Scholar] [CrossRef]
- Fischer, T.S.; Schulze-Sinninghausen, D.; Luy, B.; Altintas, O.; Barner-Kowollik, C. Stepwise Unfolding of Single-Chain Nanoparticles by Chemically Triggered Gates. Angew. Chem. Int. Ed. 2016, 55, 11276–11280. [Google Scholar] [CrossRef]
- Ji, X.; Guo, C.; Ke, X.-S.; Xiaodong, X.; Sessler, J.L. Using anion recognition to control the folding and unfolding of a single chain phosphorescent polymer. Chem. Commun. 2017, 53, 8774–8777. [Google Scholar] [CrossRef]
- Yilmaz, G.; Uzunova, V.; Napier, R.; Becer, C.R. Single-Chain Glycopolymer Folding via Host−Guest Interactions and Its Unprecedented Effect on DC-SIGN Binding. Biomacromolecules 2018, 19, 3040–3047. [Google Scholar] [CrossRef]
- Murray, B.S.; Fulton, D.A. Dynamic Covalent Single-Chain Polymer Nanoparticles. Macromolecules 2011, 44, 7242–7252. [Google Scholar] [CrossRef]
- Tuten, B.T.; Chao, D.; Lyon, C.K.; Berda, E.B. Single-chain polymer nanoparticles via reversible disulfide bridges. Polym. Chem. 2012, 3, 3068–3071. [Google Scholar] [CrossRef]
- Song, C.; Li, L.; Dai, L.; Thayumanavan, S. Responsive single-chain polymer nanoparticles with host–guest features. Polym. Chem. 2015, 6, 4828–4834. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Fulton, D.A.; Pomposo, J.A. pH-responsive single-chain polymer nanoparticles utilising dynamic covalent enamine bonds. Chem. Commun. 2014, 50, 1871–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaoglu, S.; Balta, D.K.; Temel, G. Synthesis of Photoactive Single-Chain Folded Polymeric Nanoparticles via Combination of Radical Polymerization Techniques and Menschutkin Click Chemistry. J. Polym. Sci. Part A: Polym. Chem. 2017, 55, 1998–2003. [Google Scholar] [CrossRef]
- Zhang, J.; Tanaka, J.; Gurnani, P.; Wilson, P.; Hartlieb, M.; Perrier, S. Self-assembly and disassembly of stimuli responsive tadpole-like single chain nanoparticles using a switchable hydrophilic/hydrophobic boronic acid cross-linker. Polym. Chem. 2017, 8, 4079–4087. [Google Scholar] [CrossRef] [Green Version]
- Wedler-Jasinski, N.; Lueckerath, T.; Mutlu, H.; Goldmann, A.S.; Walther, A.; Stenzel, M.H.; Barner-Kowollik, C. Dynamic covalent single chain nanoparticles based on hetero Diels–Alder chemistry. Chem. Commun. 2017, 53, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.S.; Spann, S.; An, Q.; Luy, B.; Tsotsalas, M.; Blinco, J.P.; Mutlu, H.; Barner-Kowollik, C. Self-reporting and refoldable profluorescent single-chain nanoparticles. Chem. Sci. 2018, 9, 4696–4702. [Google Scholar] [CrossRef] [Green Version]
- Thirumalai, D.; O’Brien, E.P.; Morrison, G.; Hyeon, C. Theoretical Perspectives on Protein Folding. Annu. Rev. Biophys. 2010, 39, 159–183. [Google Scholar] [CrossRef] [Green Version]
- Pomposo, J.A.; Perez-Baena, I.; Lo Verso, F.; Moreno, A.J.; Arbe, A.; Colmenero, J. How Far Are Single-Chain Polymer Nanoparticles in Solution from the Globular State? ACS Macro Lett. 2014, 3, 767–772. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Bernadó, P.; Svergun, D.I. Structural Analysis of Intrinsically Disordered Proteins by Small-Angle X-ray Scattering. Mol. Bio Syst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
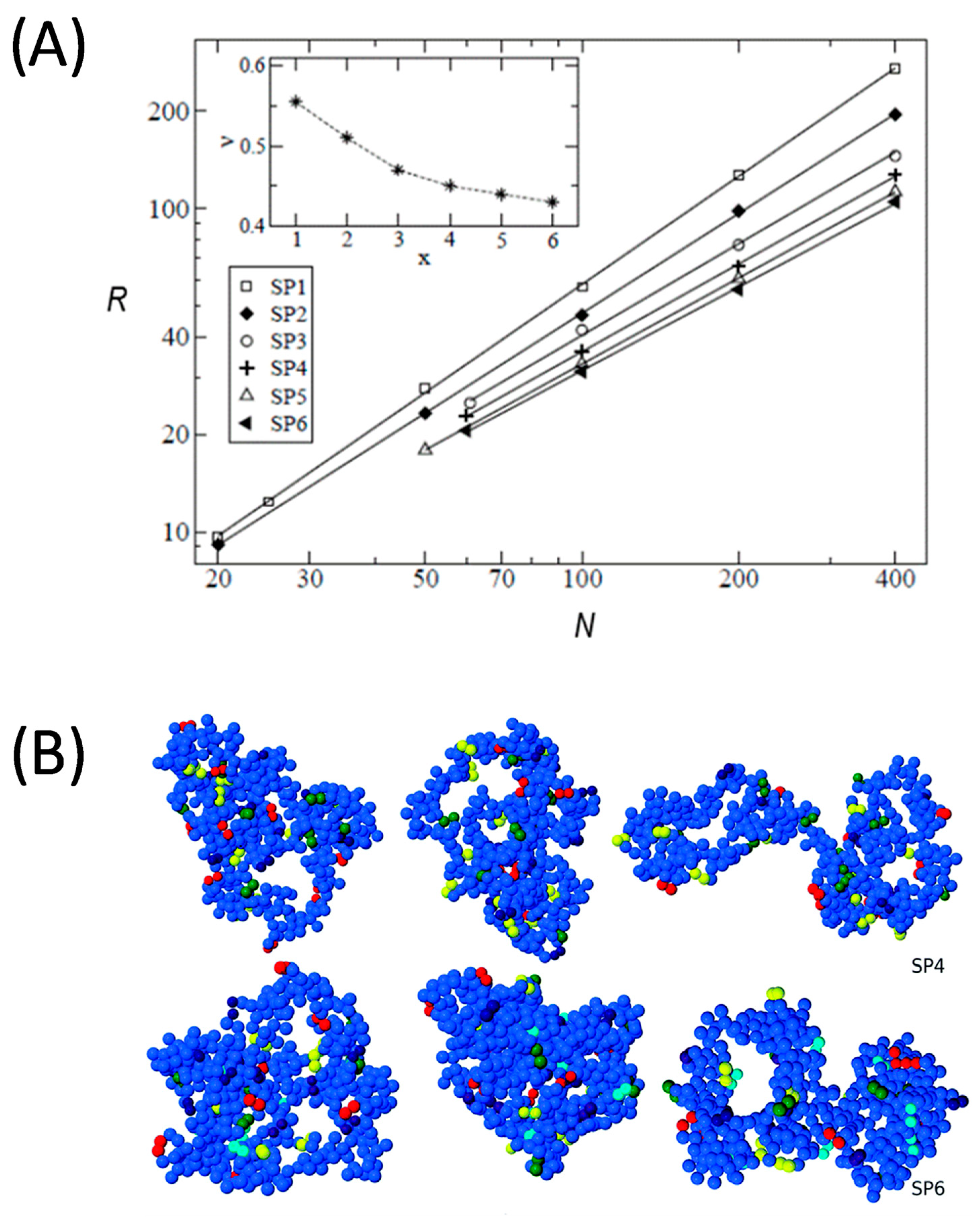
- Moreno, A.J.; Lo Verso, F.; Sanchez-Sanchez, A.; Arbe, A.; Colmenero, J.; Pomposo, J.A. Advantages of Orthogonal Folding of Single Polymer Chains to Soft Nanoparticles. Macromolecules 2013, 46, 9748–9759. [Google Scholar] [CrossRef]
- Lo Verso, F.; Pomposo, J.A.; Colmenero, J.; Moreno, A.J. Multi-orthogonal folding of single polymer chains into soft nanoparticles. Soft Matter 2014, 10, 4813–4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Verso, F.; Pomposo, J.A.; Colmenero, J.; Moreno, A.J. Simulation Guided Design of Globular Single-Chain Nanoparticles by Tuning the Solvent Quality. Soft Matter 2015, 11, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbel, H.; Breier, P.; Sommer, J.-U. Swelling Behavior of Single-Chain Polymer Nanoparticles: Theory and Simulation. Macromolecules 2017, 50, 7410–7418. [Google Scholar] [CrossRef]
- Moreno, A.J.; Bacova, P.; Lo Verso, F.; Arbe, A.; Colmenero, J.; Pomposo, J.A. Effect of Chain Stiffness on the Structure of Single-Chain Polymer Nanoparticles. J. Phys. Condens. Matter. 2018, 30, 034001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-Y.; Jia, X.-M.; Shi, R.; Li, S.-J.; Zhao, H.; Qian, H.-J.; Lu, Z.-Y. Synthesis of Polymer Single-Chain Nanoparticle with High Compactness in Cosolvent Condition: A Computer Simulation Study. Macromol. Rapid Commun. 2020, 41, 1900655. [Google Scholar] [CrossRef]
- González-Burgos, M.; Arbe, A.; Moreno, A.J.; Pomposo, J.A.; Radulescu, A.; Colmenero, J. Crowding the Environment of Single Chain Nano-Particles: A Combined Study by SANS and Simulations. Macromolecules 2018, 51, 1573–1585. [Google Scholar] [CrossRef] [Green Version]
- Oyarzún, B.; Mognetti, B.M. Programming configurational changes in systems of functionalised polymers using reversible intramolecular linkages. Mol. Phys. 2018, 116, 2927–2941. [Google Scholar] [CrossRef]
- Cardelli, C.; Bianco, V.; Rovigatti, L.; Nerattini, F.; Tubiana, L.; Dellago, C.; Coluzza, I. The role of directional interactions in the designability of generalized heteropolymers. Sci. Rep. 2017, 7, 4896. [Google Scholar] [CrossRef]
- Pande, V.S.; Grosberg, A.Y.; Tanaka, T. Heteropolymer freezing and design: Towards physical models of protein folding. Rev. Modern Phys. 2000, 72, 259–314. [Google Scholar] [CrossRef]
- Altintas, O.; Lejeune, E.; Gerstel, P.; Barner-Kowollik, C. Bioinspired dual self-folding of single polymer chains via reversible hydrogen bonding. Polym. Chem. 2012, 3, 640–651. [Google Scholar] [CrossRef]
- Hosono, N.; Gillisen, M.A.J.; Li, Y.; Sheiko, S.S.; Palmans, A.R.A.; Meijer, E.W. Orthogonal Self-Assembly in Folding Block Copolymers. J. Am. Chem. Soc. 2013, 135, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Hosono, N.; Stals, P.J.M.; Palmans, A.R.A.; Meijer, E.W. Consequences of Block Sequence on the Orthogonal Folding of Triblock Copolymers. Chem. Asian J. 2014, 9, 1099–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artar, M.; Terashima, T.; Sawamoto, M.; Meijer, E.W.; Palmans, A.R.A. Understanding the Catalytic Activity of Single-Chain Polymeric Nanoparticles in Water. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 12–20. [Google Scholar] [CrossRef]
- Greb, L.; Mutlu, H.; Barner-Kowollik, C.; Lehn, J.-M. Photo- and Metallo-responsive N-Alkyl-Bisimines as Orthogonally Adressable Main-Chain Functional Groups in Metathesis Polymers. J. Am. Chem. Soc. 2016, 138, 1142–1145. [Google Scholar] [CrossRef]
- Matsumoto, K.; Terashima, T.; Sugita, T.; Takenaka, M.; Sawamoto, M. Amphiphilic Random Copolymers with Hydrophobic/Hydrogen-Bonding Urea Pendants: Self-Folding Polymers in Aqueous and Organic Media. Macromolecules 2016, 49, 7917–7927. [Google Scholar] [CrossRef]
- Matsumoto, M.; Terashima, T.; Matsumoto, K.; Takenaka, M.; Sawamoto, M. Compartmentalization Technologies via Self-Assembly and Cross-Linking of Amphiphilic Random Block Copolymers in Water. J. Am. Chem. Soc. 2017, 139, 7164–7167. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Bai, Y.; Li, K.; Garcia, E.S.; Ferguson, A.L.; Zimmerman, S.C. Enzyme-like Click Catalysis by a Copper-Containing Single-Chain Nanoparticle. J. Am. Chem. Soc. 2018, 140, 13695–13702. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Li, K.; Wang, Y.; Gruebele, M.; Ferguson, A.L.; Zimmerman, S.C. Polymeric “Clickase” Accelerates the Copper Click Reaction of Small Molecules, Proteins, and Cells. J. Am. Chem. Soc. 2019, 141, 9693–9700. [Google Scholar] [CrossRef]
- Chen, J.; Li, K.; Shon, J.S.L.; Zimmerman, S.C. Single-Chain Nanoparticle Delivers a Partner Enzyme for Concurrent and Tandem Catalysis in Cells. J. Am. Chem. Soc. 2020, 142, 4565–4569. [Google Scholar] [CrossRef]
- Cao, H.; Cui, Z.; Gao, P.; Ding, Y.; Zhu, X.; Lu, X.; Cai, Y. Metal-Folded Single-Chain Nanoparticle: Nanoclusters and Self-Assembled Reduction-Responsive Sub-5-nm Discrete Subdomains. Macromol. Rapid Commun. 2017, 38, 1700269. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Huang, L.; Ding, Y.; Zhu, X.; Lu, X.; Cai, Y. Compartmentalization and Unidirectional Cross-Domain Molecule Shuttling of Organometallic Single-Chain Nanoparticles. ACS Macro Lett. 2018, 7, 572–575. [Google Scholar] [CrossRef]
- Knöfel, N.D.; Rothfuss, H.; Tzvelkova, P.; Kulendran, B.; Barner-Kowollik, C.; Roesky, P.W. Heterobimetallic Eu(III)/Pt(II) single-chain nanoparticles: A path to enlighten catalytic reactions. Chem. Sci. 2020, 11, 10331–10336. [Google Scholar] [CrossRef]
- Upadhya, R.; Murthy, N.S.; Hoop, C.L.; Kosuri, S.; Nanda, V.; Kohn, J.; Baum, J.; Gormley, A.J. PET-RAFT and SAXS: High Throughput Tools to Study Compactness and Flexibility of Single-Chain Polymer Nanoparticles. Macromolecules 2019, 52, 8295–8304. [Google Scholar] [CrossRef]
- Roy, R.K.; Lutz, J.-F. Compartmentalization of Single Polymer Chains by Stepwise Intramolecular Cross-Linking of Sequence-Controlled Macromolecules. J. Am. Chem. Soc. 2014, 136, 12888–12891. [Google Scholar] [CrossRef]
- Zhang, J.; Gody, G.; Hartlieb, M.; Catrouillet, S.; Moffat, J.; Perrier, S. Synthesis of Sequence-Controlled Multiblock Single Chain Nanoparticles by a Stepwise Folding-Chain Extension-Folding Process. Macromolecules 2016, 49, 8933–8942. [Google Scholar] [CrossRef]
- Heiler, C.; Offenloch, J.T.; Blaco, E.; Barner-Kowollik, C. Photochemically Induced Folding of Single Chain Polymer Nanoparticles in Water. ACS Macro Lett. 2017, 6, 56–61. [Google Scholar] [CrossRef]
- Heiler, C.; Bastian, S.; Lederhose, P.; Blinco, J.P.; Blasco, E.; Barner-Kowollik, C. Folding polymer chains with visible light. Chem. Commun. 2018, 54, 3476–3479. [Google Scholar] [CrossRef]
- Claus, T.K.; Zhang, J.; Martin, L.; Hartlieb, M.; Mutlu, H.; Perrier, S.; Delaittre, G.; Barner-Kowollik, C. Stepwise Light-induced Dual Compaction of Single-Chain Nanoparticles. Macromol. Rapid Commun. 2017, 38, 1700264. [Google Scholar] [CrossRef]
- Ji, X.; Zhang, Y.; Zhao, H. Amphiphilic Janus Twin Single-Chain Nanoparticles. Chem. Eur. J. 2018, 24, 3005–3012. [Google Scholar] [CrossRef]
- Rubio-Cervilla, J.; Frisch, H.; Barner-Kowollik, C.; Pomposo, J.A. Synthesis of Single-Ring Nanoparticles Mimicking Natural Cyclotides by a Stepwise Folding-Activation-Collapse Process. Macromol. Rapid Commun. 2019, 40, 1800491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, H.; Kodura, D.; Bloesser, F.R.; Michalek, L.; Barner-Kowollik, C. Wavelength-Selective Folding of Single Polymer Chains with Different Colors of Visible Light. Macromol. Rapid Commun. 2020, 41, 1900414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Xie, M.; Dou, J.; Li, H.; Huang, X.; Chen, D. Efficient Fabrication of Pure, Single-Chain Janus Particles through Their Exclusive Self-Assembly in Mixtures with Their Analogues. ACS Macro Lett. 2018, 7, 1278–1282. [Google Scholar] [CrossRef]
- Lang, F.; Xiang, D.; Wang, J.; Yang, L.; Qiao, Y.; Yang, Z. Janus Colloidal Dimer by Intramolecular Cross-Linking in Concentrated Solutions. Macromolecules 2020, 53, 2271–2278. [Google Scholar] [CrossRef]
- Keklik, M.; Akar, I.; Temel, B.A.; Balta, D.K.; Temel, G. Single-chain polymer nanoparticles via click crosslinking and effect of photoinduced radical combination on crosslink points. Polym. Int. 2020, 69, 1018–1023. [Google Scholar] [CrossRef]
- Ruiz de Luzuriaga, A.; Perez-Baena, I.; Montes, S.; Loinaz, I.; Odriozola, I.; García, I.; Pomposo, J.A. New Route to Polymeric Nanoparticles by Click Chemistry Using Bifunctional Cross-Linkers. Macromol. Symp. 2010, 296, 303–310. [Google Scholar] [CrossRef]
- Chao, D.; Jia, X.; Tuten, B.; Wang, C.; Berda, E.B. Controlled folding of a novel polyolefin via multiple sequential orthogonal intra-chain interactions. Chem. Commun. 2013, 49, 4178–4180. [Google Scholar] [CrossRef]
- Beck, J.B.; Killops, K.L.; Kang, T.; Sivanandan, K.; Bayles, A.; Mackay, M.E.; Wooley, K.; Hawker, C.J. Facile Preparation of Nanoparticles by Intramolecular Cross-Linking of Isocyanate Functionalized Copolymers. Macromolecules 2009, 42, 5629–5635. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Sanchez, A.; Pomposo, J.A. Efficient Synthesis of Single-Chain Polymer Nanoparticles via Amide Formation. J. Nanomater. 2015, 723492. [Google Scholar]
- Cole, J.P.; Lessard, J.J.; Rodriguez, K.J.; Hanlon, A.M.; Reville, E.K.; Mancinelli, J.P.; Berda, E.B. Single-chain nanoparticles containing sequence-defined segments: Using primary structure control to promote secondary and tertiary structure in synthetic protein mimics. Polym. Chem. 2017, 8, 5829–5835. [Google Scholar] [CrossRef]
- Liu, C.H.; Dugas, L.D.; Bowman, J.I.; Chidanguro, T.; Storey, R.F.; Simon, Y.C. Forcing single-chain nanoparticle collapse through hydrophobic solvent interactions in comb copolymers. Polym. Chem. 2020, 11, 292–297. [Google Scholar] [CrossRef]
- Webb, M.A.; Jackson, N.E.; Gil, P.S.; de Pablo, J.J. Targeted sequence design within the coarse-grained polymer genome. Sci. Adv. 2020, 6, eabc6216. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
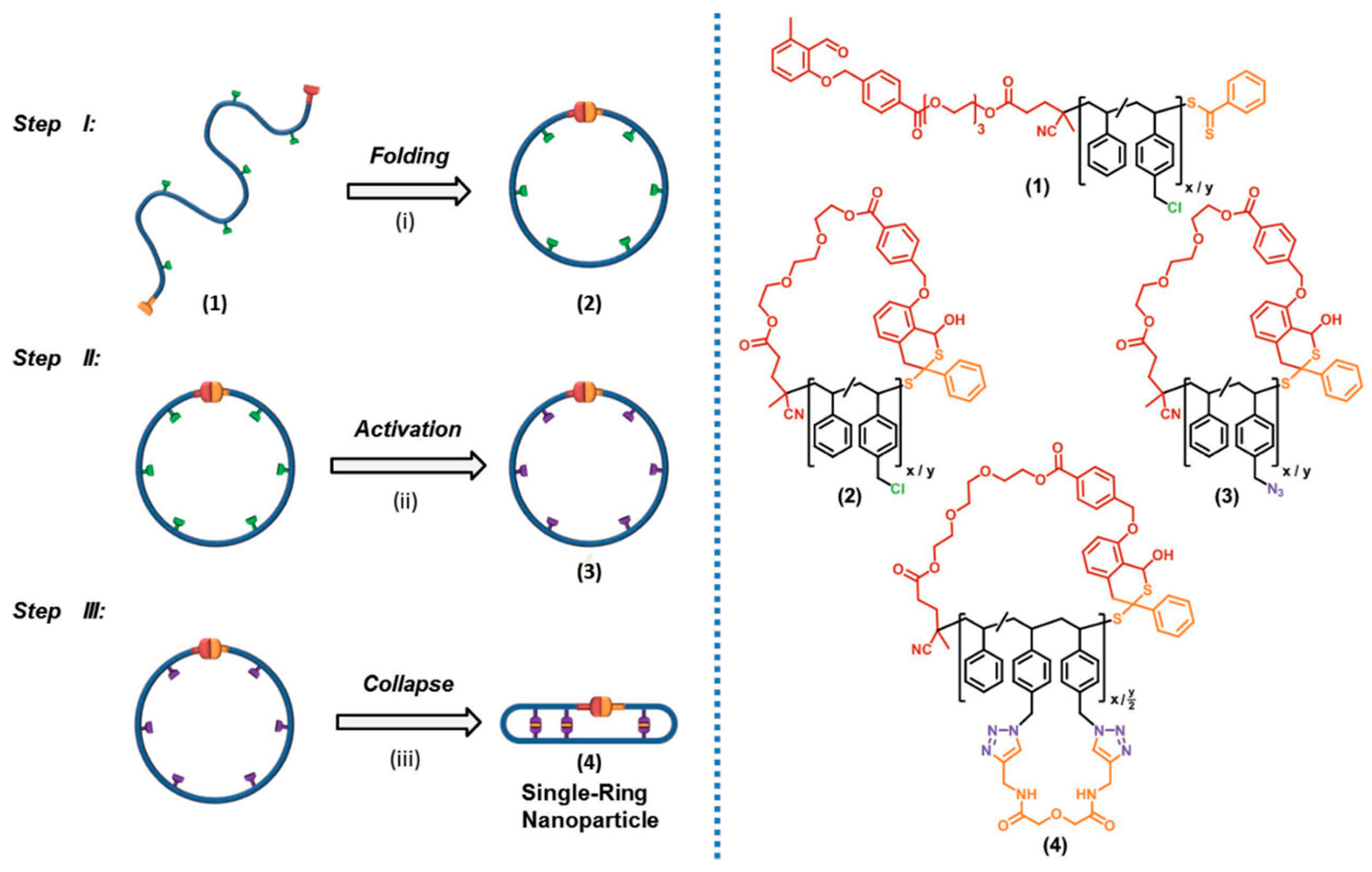{kind=link}
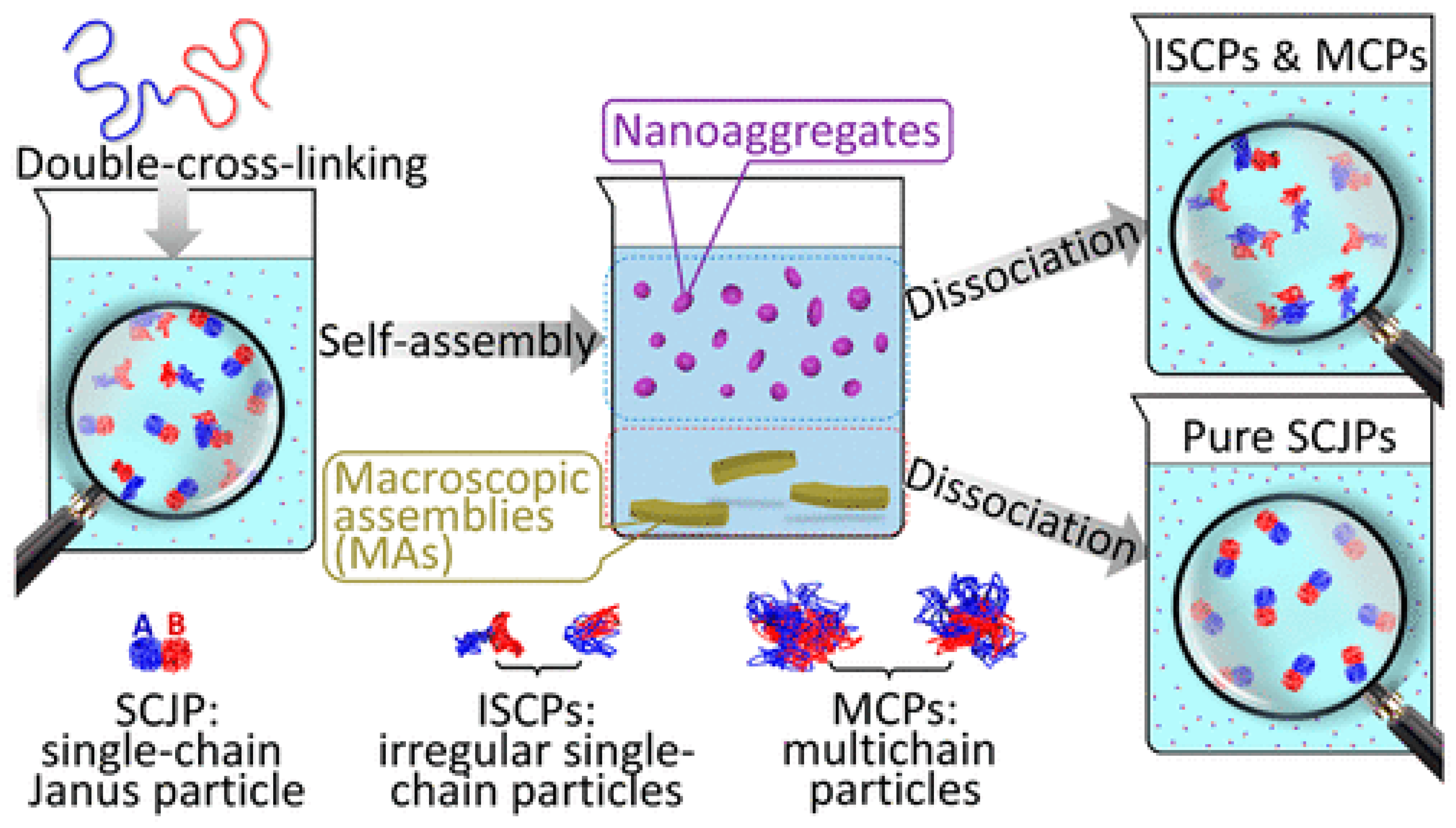{kind=link}
{kind=link}
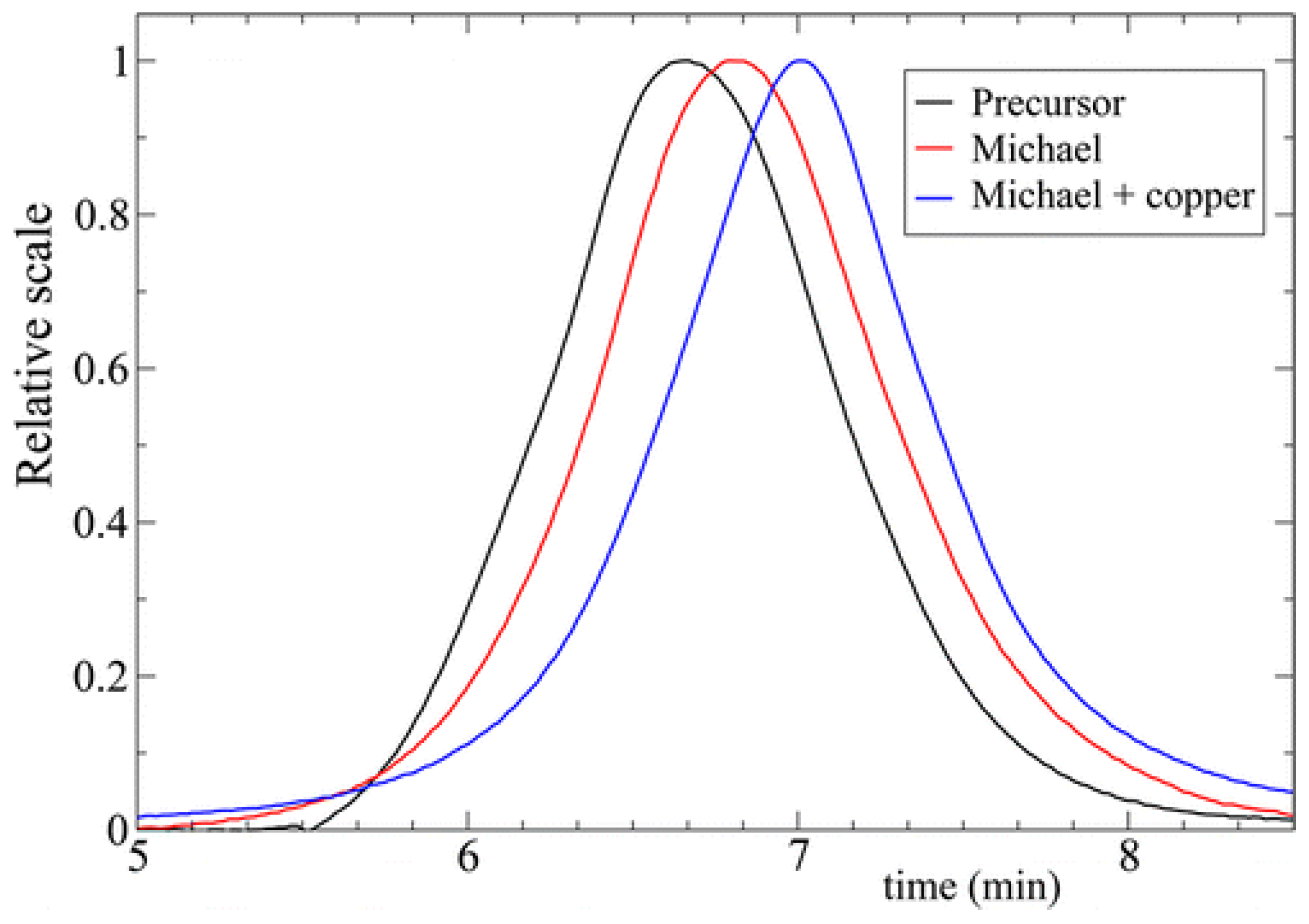{kind=link}
| Type 1 | Folding Interaction 2 | Characterization Technique 3 | References |
|---|---|---|---|
| NCBs | Multiple hydrogen bonding | DLS | [20] |
| NCBs | UPy dimerization | SEC, AFM | [21,22] |
| NCBs | BTA helical stacking | UV, CD | [23] |
| NCBs | CB[8]-viologen complexation | FL | [24] |
| NCBs | Hydrophobic interactions | DLS, FL | [25,26] |
| NCBs | BTA helical stacking & CA−HW dimerization | NMR, CD, DLS, SLS | [27] |
| NCBs | β-CD-FC complexation | DLS, NMR | [28,29] |
| NCBs | B21C7-AS dimerization & CA−HW dimerization | DLS, NMR | [30] |
| NCBs | C4P-TD complexation | FL | [31] |
| NCBs | β-CD-AD complexation | NMR, DLS | [32] |
| DCBs | Hydrazone bonds | SEC | [33] |
| DCBs | Disulfide bonds | SEC | [34,35] |
| DCBs | Enamine bonds | SEC, FTIR | [36] |
| DCBs | Amine quaternization | SEC, DLS | [37] |
| DCBs | Boronate ester formation | SEC, DLS | [38] |
| DCBs | HDA reaction | SEC, DLS, NMR | [39] |
| DCBs | Photoligation of nitroxide radicals | SEC, DLS, NMR, FL | [40] |
| System | υ (Simulations) | υ (SAXS) | R(SEC), nm |
|---|---|---|---|
| Precursor | 0.63 | 0.60 | 25.7 |
| SCNPs (only covalent interactions) | 0.56 | 0.44 | 19.6 |
| SCNPs (covalent & non-covalent interactions) | 0.51 | 0.40 | 10.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blazquez-Martín, A.; Verde-Sesto, E.; Moreno, A.J.; Arbe, A.; Colmenero, J.; Pomposo, J.A. Advances in the Multi-Orthogonal Folding of Single Polymer Chains into Single-Chain Nanoparticles. Polymers 2021, 13, 293. https://doi.org/10.3390/polym13020293
Blazquez-Martín A, Verde-Sesto E, Moreno AJ, Arbe A, Colmenero J, Pomposo JA. Advances in the Multi-Orthogonal Folding of Single Polymer Chains into Single-Chain Nanoparticles. Polymers. 2021; 13(2):293. https://doi.org/10.3390/polym13020293
Chicago/Turabian StyleBlazquez-Martín, Agustín, Ester Verde-Sesto, Angel J. Moreno, Arantxa Arbe, Juan Colmenero, and José A. Pomposo. 2021. "Advances in the Multi-Orthogonal Folding of Single Polymer Chains into Single-Chain Nanoparticles" Polymers 13, no. 2: 293. https://doi.org/10.3390/polym13020293