1. Introduction
Semiflexible polymers are macromolecules with linear chemical architecture in the simplest case, which display considerable bending rigidity along the chain backbone [
1,
2,
3,
4]. In lyotropic solutions of semiflexible polymers, the competition between orientational and translational entropy can induce a transition from an isotropic (i) to a nematic (n) phase. Depending on the chemical nature of the monomeric units, the persistence length
of such macromolecules can vary over several orders of magnitude: For synthetic polymers,
typically lies in the range from
to
[
5], while the distance between repeat units along the backbone,
, is typically on the order of several angstroms. Much larger values for these lengths may occur for biologically relevant polymers, e.g.,
for double stranded DNA [
6] and
for filamentous actin [
7]. Note that
does not only depend on the specific polymer chemistry [
8] but may also depend on external factors, e.g., the nature of the solvent [
6], and the density of monomeric units in a nematically ordered solution or melt [
9].
The ability to control the materials properties of polymeric systems is highly desirable for many practical applications. It is common to do this by mixing two chemically different polymers, e.g., blending poly(phenylene ether) resins with polystyrene results in materials with high heat resistance and strong mechanical stability [
10]. The statistical thermodynamics of such polymer blends is usually studied via the Flory-Huggins (FH) solution theory [
11,
12,
13,
14,
15,
16], which describes the polymer mixture using a lattice model. Here, the entropy of mixing favors homogeneous systems even for very long chains, whereas unmixing is driven by the chemical incompatibility of the polymers, typically quantified through the FH
parameter. One central approximation of the FH solution theory is the mean-field description of the energy of mixing, which completely disregards the (local) chain connectivity. As a consequence, this contribution is identical for regular solutions, polymer solutions, and blends [
15]. Further, chain stiffness does not enter at all in the standard FH solution theory, so that it cannot distinguish between fully flexible and stiff polymers. Therefore, alternative approaches have been developed to describe rod-coil mixtures, where the flexible coil-like polymer is often modeled as an effective soft spherical particle [
17,
18,
19,
20]. Although such coarse-grained descriptions can provide important insights on the (qualitative) phase behavior, they completely lose information on the scale of monomeric units, and are also unsuitable for describing blends of semiflexible polymers, which can neither be described as strictly hard rods nor as soft spheres.
In this work, we consider lyotropic solutions containing two types (A, B) of semiflexible polymers with finite persistence lengths
. This problem has been previously considered by a few analytical approaches, yielding interesting explicit results only for special limiting cases. Semenov and Subbotin [
21] generalized the Onsager-type [
22] treatment of single-component lyotropic dilute polymers solution [
23,
24,
25,
26] to the two-component case. For the limiting case where the contour lengths
and
are in the range
,
, they predicted interesting (qualitative) phase diagrams containing both mixed and coexisting nematic phases [
21]. Experimentally, the coexistence of two nematic phases n
and n
was observed in mixed virus suspensions [
27], which can be modeled theoretically by mixtures of hard rods differing in diameter [
28,
29]. In recent MD simulations, Zhou et al. studied the behavior of mixtures of semiflexible (ring) polymers in spherical [
30] and ellipsoidal [
31] containers, finding a confinement-induced phase separation of the two species. In thermotropic systems, n
–n
coexistence was also predicted for solutions of two kinds of rigid rods with suitable enthalpic interactions [
32], and experimental observations of n
–n
unmixing were also reported for mixtures of side-chain liquid crystalline polymers with small molecule liquid crystals [
33], but all such temperature-driven phase transitions are out of consideration here. Thus, we also do not discuss in detail the theory of Liu and Fredrickson [
34], which was based on a Landau expansion in terms of both the nematic order parameter and deviations of the local volume fraction of monomeric units from its average. Since Landau expansions require that order parameters are small, this approach is not suitable deeply in the nematic phase. In addition, it was assumed that the transitions were driven by standard enthalpic interactions throughout, i.e., i-i unmixing due to a standard FH
parameter, and nematic ordering due to a Maier-Saupe-like term [
35].
In the present work, we focus on systems where transitions are solely driven by entropic interactions, focusing on cases where contour and persistence lengths are of the same order. Preliminary results of our work were already presented as a letter [
36]; in a complementary study [
37], we have described related results for shorter chains, with emphasis on a comparison between polymers with finite stiffness and strictly hard rods, and the classification of various contributions to the Gibbs excess free energy within the Density Functional Theory (DFT) framework. In
Section 2, we briefly characterize the employed models and methods. Following our previous work on one-component solutions of semiflexible polymers [
9,
38,
39,
40], we use both DFT calculations and Molecular Dynamics (MD) simulations for closely related models.
Section 3 gives an overview of the possible phase diagrams, emphasizing their description in different statistical ensembles, as predicted by DFT. In
Section 4, we analyze in more detail selected systems that were studied by both DFT and MD. For one example, we show how a stiffness mismatch leads to an effective
parameter of entropic origin, by applying an approach pioneered by Fredrickson et al. [
41] and Kozuch et al. [
42]. We also discuss the anisotropic character of critical fluctuations for the n-n critical point. Finally,
Section 5 summarizes our results and briefly mentions pertinent experiments [
43,
44].
3. DFT Predictions of Possible Phase Diagrams
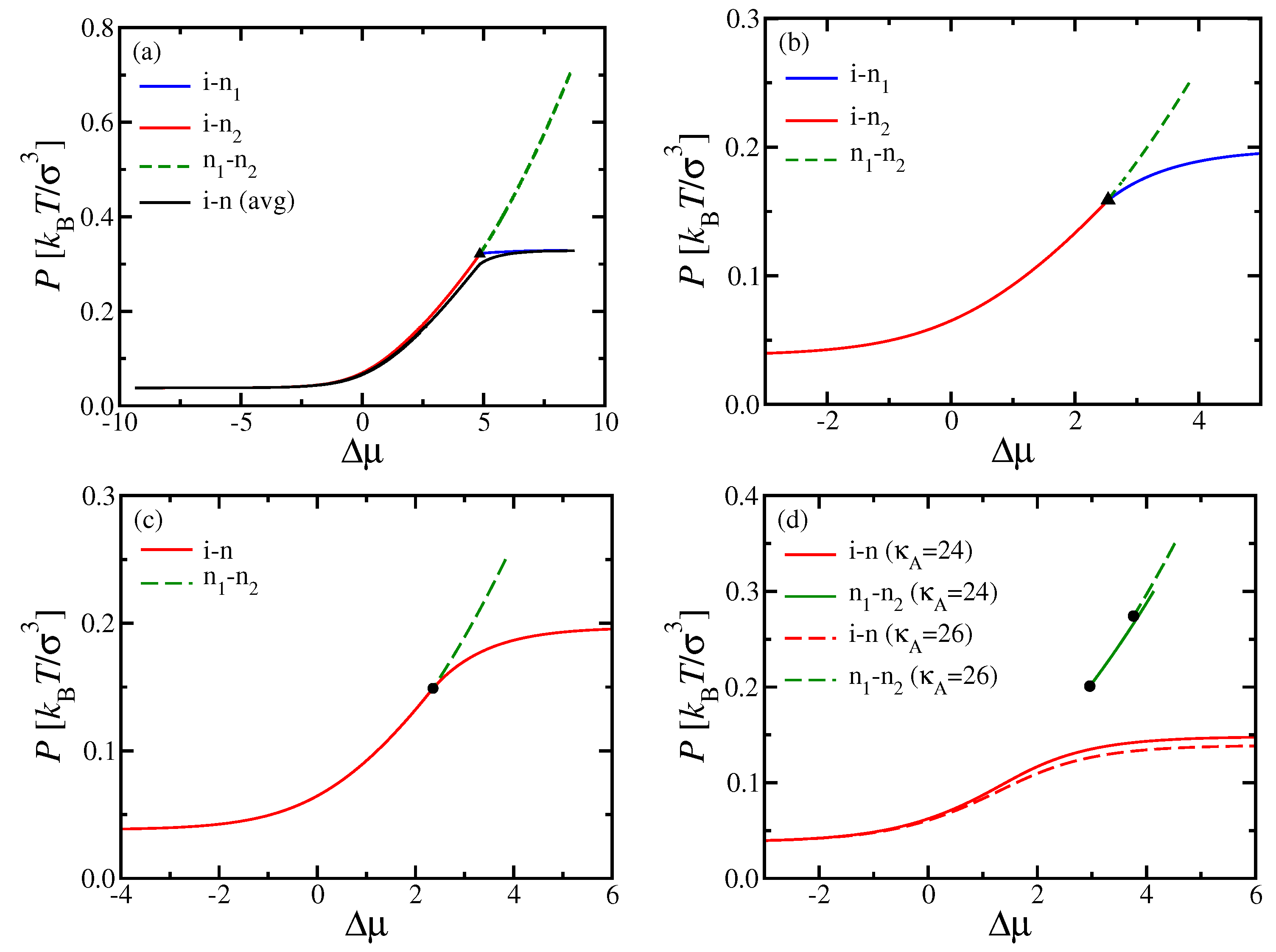
Figure 2 shows phase diagrams in the space of the intensive thermodynamic variables pressure,
P, and chemical potential difference,
, for
. When we fix
and vary
, there is a particular “multicritical” value
(estimated here as
for
and
), where the topology of the phase diagram changes: For
, the (first-order) transition between two distinct nematic phases ends at a triple point, where (first-order) i–n phase separations into A- or B-rich nematic phases set in. However, for
, the first-order n
–n
phase separation ends at a critical point instead.
It is interesting that, for
, the two first-order lines for the i–n transitions meet in the
plane at the triple point under some angle, while, for
, all transition lines seem to have a common tangent at the triple point. Note also that the value
where the phase diagram topology changes is not universal but depends on both
and
N [
36]; qualitatively similar results were found in related work for shorter chains (
) [
37], but there the critical point occurred at distinctly larger pressures, where the unmixing of nematic phases could be potentially preempted by the appearance of smectic and/or crystalline phases. The polymers with
studied here are just large enough so that one can expect n
–n
critical points at monomer densities that are low enough so that simulation studies, and perhaps also experiments, become feasible.
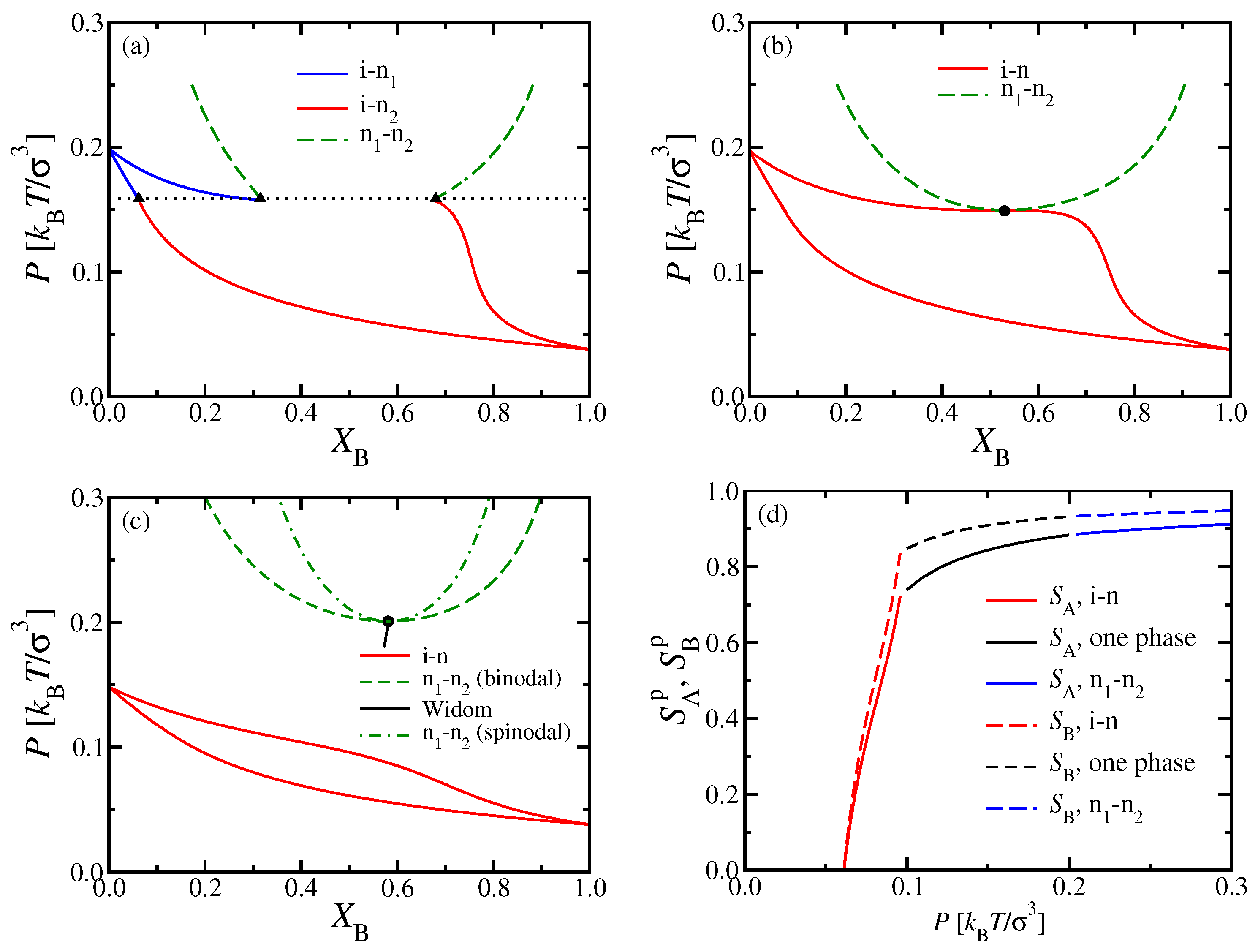
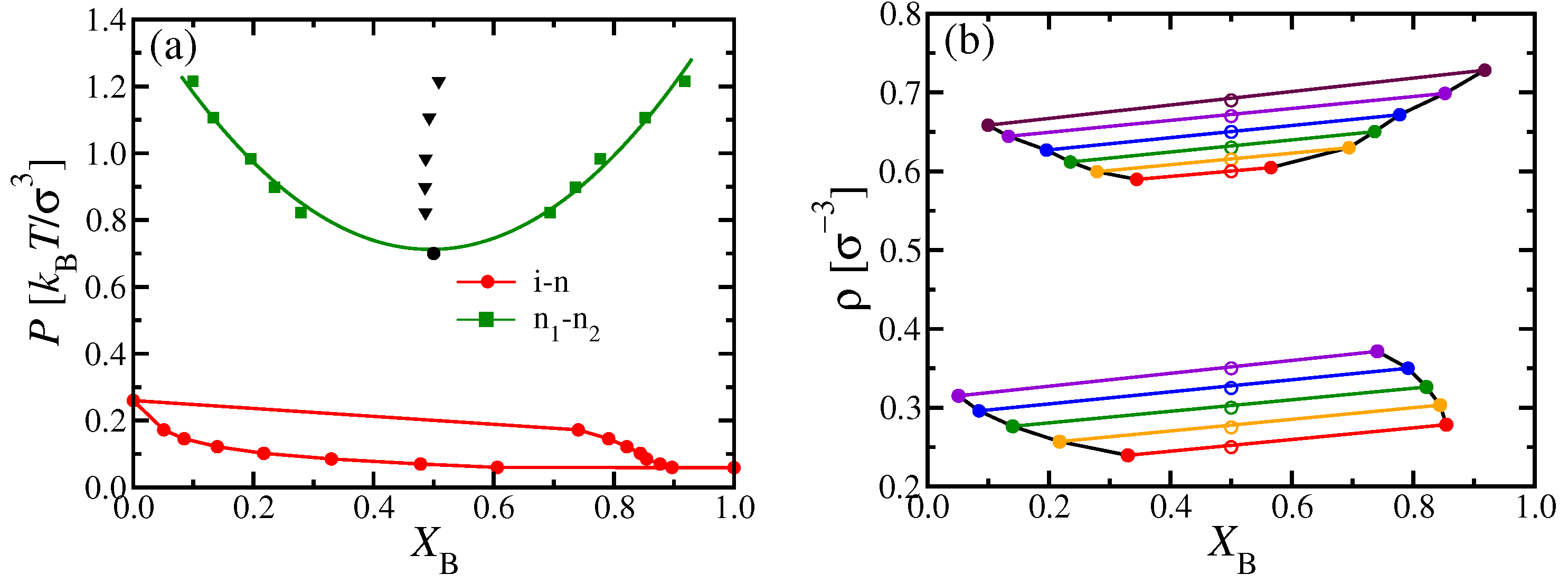
These special features have their counterparts in the phase diagrams, where one chooses
rather than
as a variable (
Figure 3). At the multicritical point
, the phase boundary of the i–n coexistence region has a horizontal tangent on the nematic side and touches the n
–n
two-phase region at its critical point. For
, however, there is a single lens-shaped i–n coexistence region, which extends from the pure A system (
) to the pure B system (
). In this representation, the n
–n
coexistence curve has an approximate parabolic shape near the critical point, which occurs at the minimum of this curve in the
plane. Such a parabolic shape reflects a mean-field critical exponent, as expected for DFT. For
, this coexistence region no longer interferes with i–n coexistence. With increasing
P, the n
–n
coexistence will end when other phases (smectic liquid crystalline or crystalline solid phases) come into play, but such phases can not be captured by the present DFT treatment.
In mean-field theories of critical unmixing of binary mixtures, the response function [
59]
not only diverges at the critical point
, but also along the whole “spinodal curve”
, which touches the coexistence curve at the critical point. In the phenomenological FH theory discussed in
Section 2.3, where
rather than
P was used as a control parameter, the spinodal curve is simply given when we require
in Equation (
13), since
[
59,
60]. Although the concept of a spinodal curve is doubtful outside of mean-field theories [
61], we have included its location in
Figure 3c nevertheless. Generally of interest, however, is the so-called “Widom line”, describing the locus of maxima of
; a short piece of this line is also shown in
Figure 3c.
Figure 3d shows the molecular nematic order parameters
and
of the two types of chains plotted versus the pressure
P, for a mole fraction
.
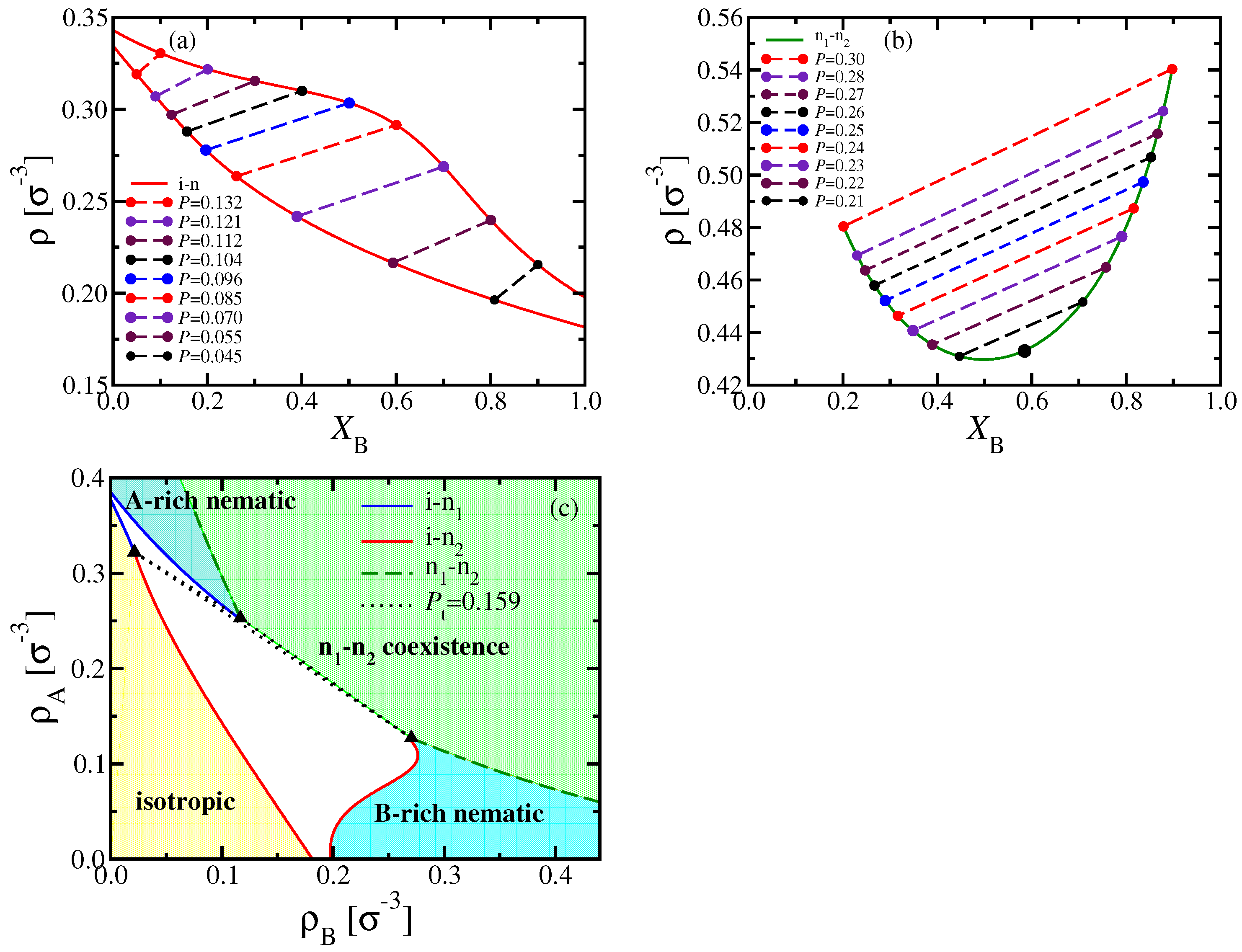
A clear advantage of DFT is that it is straightforward to discuss the phase behavior of the considered systems in various ensembles of statistical mechanics, which is particularly useful for making contact with experiments: The (osmotic) pressure
P of a polymer solution is usually not readily accessible, and one rather uses the polymer concentrations as variables. In our implicit solvent model, these concentrations translate into the monomer number densities
and
, respectively. Alternatively, we may take the total density
and the mole fraction
as variables to draw the phase diagram (
Figure 4a,b).
While coexisting phases in equilibrium must be at the same temperature and pressure, their densities can differ, as clearly shown in
Figure 4a,b. Note also that the critical point no longer coincides with the minimum of the two-phase coexistence curve of n
–n
phase separation in the
phase diagram. While the tie lines, connecting the coexisting phases, are almost parallel to each other for n
–n
coexistence, this is not true for i–n coexistence: In this case, the tie lines must get perpendicular to the
-axis when
and
, but are much flatter in between. To avoid a too confusing picture, we have not shown any tie lines in
Figure 4c, where the phase diagram is shown in the
plane; in that representation, three phases must coexist for any state within the triangle formed by the three dotted lines enclosing the triple region.
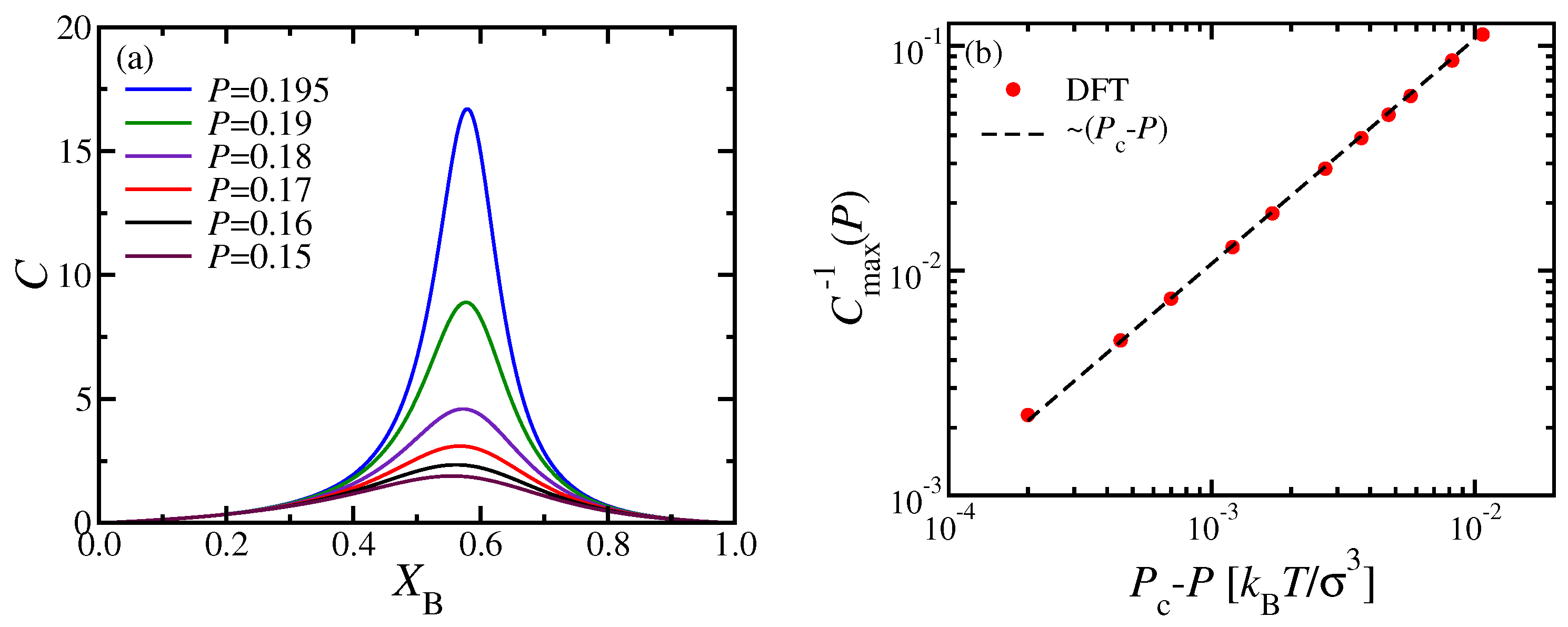
In
Figure 5a, we present the response function
C (Equation (
18)) in the homogeneously mixed nematic phase, plotted versus
for various pressures smaller than the critical pressure
. The coordinates of the maxima of these curves yield the location of the “Widom line” in the
plane, as shown in
Figure 3c. The log-log plot of the inverse height of this maximum,
, reveals the expected critical variation,
(
Figure 5b).
4. Selected MD Results for the Phase Behavior of Mixtures of Semiflexible Polymers
We start this section with a rather simple special case, i.e., two types of chains with the same stiffness,
, but different chain lengths,
and
. We have studied this system in cubic boxes, containing 12544 A and 6272 B chains (
). We systematically varied the pressure
P and measured the average number density of monomeric units.
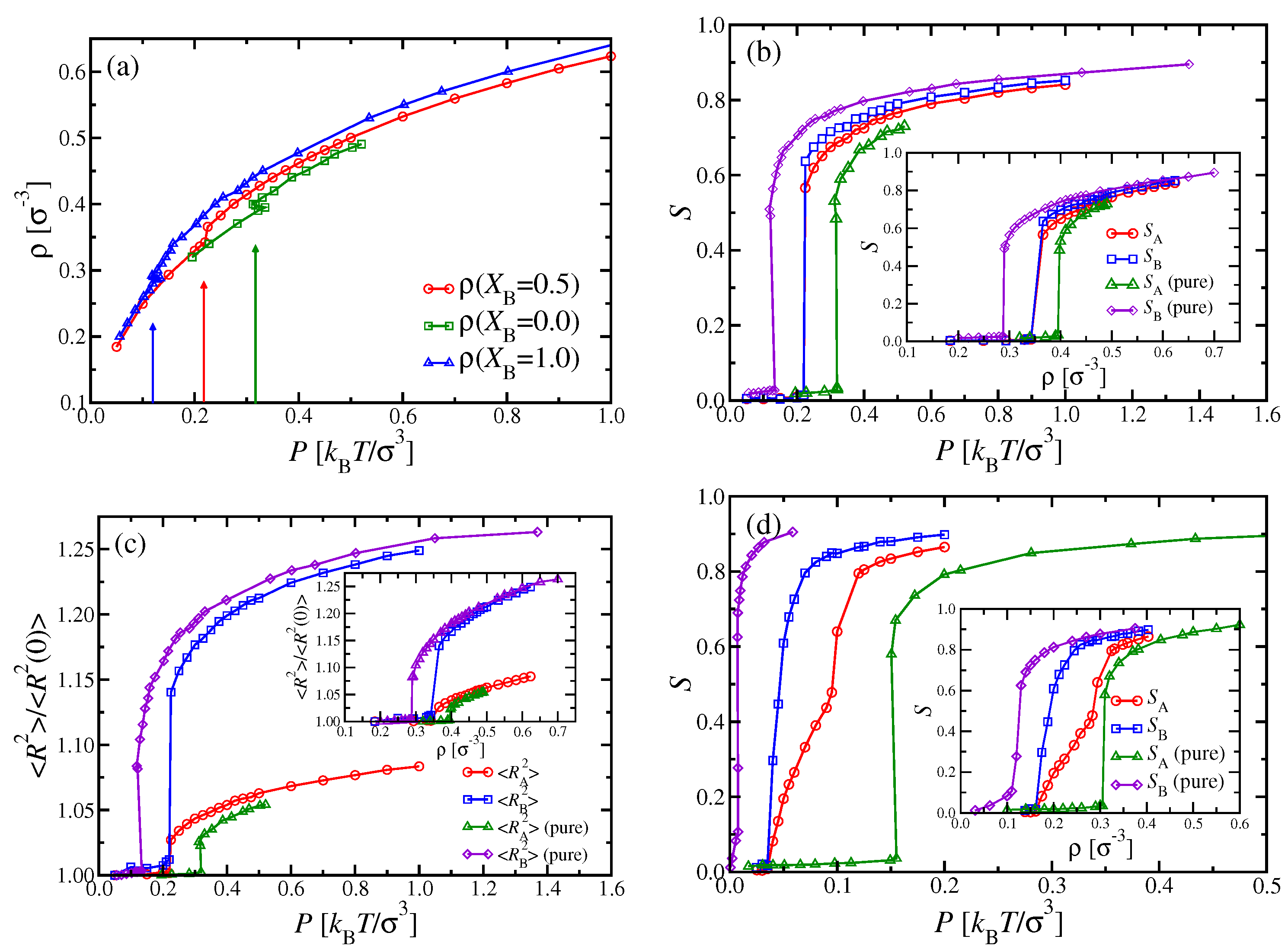
Figure 6a shows the resulting equation of state, compared with the corresponding isotherms of the pure systems (
and
). In the mixed systems, a sudden increase of the density from
to
takes place at
, where the nematic order parameters
and
(
Figure 6b) and end-to-end distances (
Figure 6c) indicate the transition, as well.
While pure systems in the
ensemble should exhibit a sharp first-order i–n transition, where the density
and the nematic order parameter
S have a well-defined jump, this is not the case for mixtures: There, the phase diagram must have in general the shape of a lens, similar to the i–n two-phase coexistence region of
Figure 3c. We would have a unique transition pressure
only for a truly intensive thermodynamic variable, such as
, as in
Figure 2; however, since
is formed from densities
and
of extensive variables, we rather have a two-phase coexistence region again for fixed
, with
. The fact that we seem to observe a unique, well-defined transition pressure in
Figure 6 instead needs to be interpreted by the hypothesis that
and
are so close together that we cannot resolve the difference.
DFT respects these general rules from thermodynamics, but it suffers from other problems: The formulation which we used in this work does not give any information on chain linear dimensions, since it is just based on the distribution
of coarse-grained chains (see
Section 2.2). As a consequence, also the nematic order parameter
computed from DFT (
Figure 3d) refers to the average orientation of the whole chain, and not to the orientation of individual bonds, as defined in Equation (
3) and shown in
Figure 6. It has been demonstrated [
62] that there is a significant difference between the nematic order parameter defined from bond orientations and its counterpart from the orientation of whole chains. Thus, we do not have any DFT counterparts to the simulation data of
Figure 6b,c; with respect to
Figure 6a, even away from the i–n transition, the agreement is only qualitative but not quantitative.
An interesting question concerns the comparison of data for the nematic order parameters and chain radii in the nematic phase for the mixed system with their pure counterparts: We find that , i.e., the admixture of the shorter chains, which exhibit somewhat less nematic order, weakens the order of the longer chains slightly. This is also evident from the fact that throughout the nematic phase. On the other hand, we also have for those pressures where nematic order occurs. Those chains which order better (here, the longer chains) act like a nematic ordering field on the chains which have less tendency to order (in the pure phase). We shall see later that the same phenomenon occurs when we examine mixtures of chains that have identical N but differ in stiffness.
Throughout the isotropic phase, we find here
and
, irrespective of
P and
, and likewise for the mean square end-to-end distances,
and
. Since the persistence length is about twice as large as the contour length for the A chains, only a rather modest increase of
can be observed at the i–n transition (
Figure 6c). In contrast, the longer B chains exhibit a marked jump of their mean square end-to-end distances at the i–n transition. Further note that
for
has already reached
of the contour length, at which this quantity must saturate for large enough pressures.
Now, we turn to some of the cases that were already studied by DFT in
Section 3, where chains have the same chain length
, but differ in stiffness, e.g.,
and
. From DFT, we would expect a transition from the isotropic phase into the i–n two-phase coexistence region with increasing pressure, then a nematic phase to which both types of chains contribute, followed by n
–n
unmixing at still higher pressures (
Figure 3c).
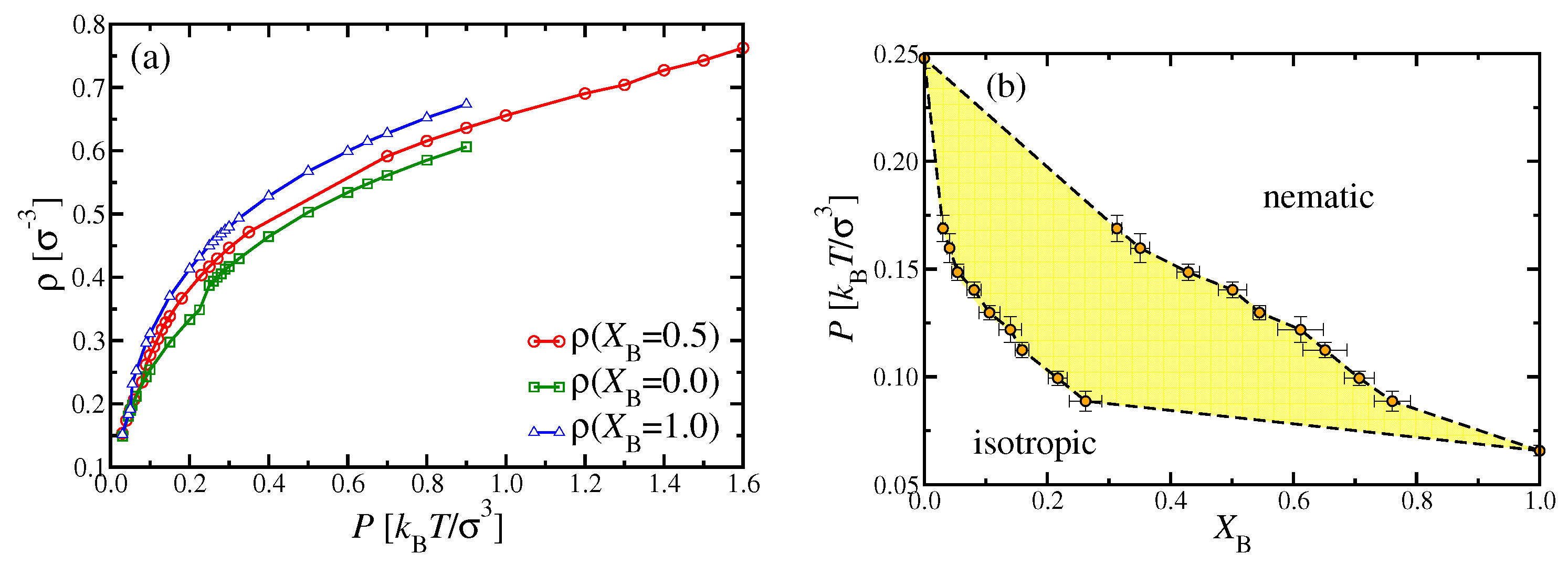
Figure 7a shows the density versus pressure curve of the equimolar mixture (
) from MD simulations, compared to the corresponding pure systems. For the latter, the i–n transitions show up as little kinks at
and
for the A and B chains, respectively, with corresponding monomer number densities being
and
for species A, while
for species B. Unlike DFT, we cannot resolve the density jump at the transition with meaningful accuracy in the MD simulations. Nevertheless, the corresponding DFT results for the transition pressures, i.e.,
and
for the pure A and B systems, respectively, are clearly far outside of the MD error bars. Thus, there is almost a discrepancy by a factor of two in the pressure scale. However, the underestimation by DFT with respect to the densities is much smaller (
Figure 4). When we examine the
versus
P curve for the mixture, the i–n transition cannot be recognized from the data at all (
Figure 7a). This behavior can be expected when the two-phase coexistence region is not very narrow (as it was in
Figure 6), but rather wide, as found by DFT for the present case (
Figure 3c). Then, the density variation in this two-phase region is described by the lever rule,
where
and
describe the curves limiting the two-phase region at the isotropic and nematic side, respectively. At these curves,
has at most a (small) discontinuity of slope, which clearly could not be seen in the simulation.
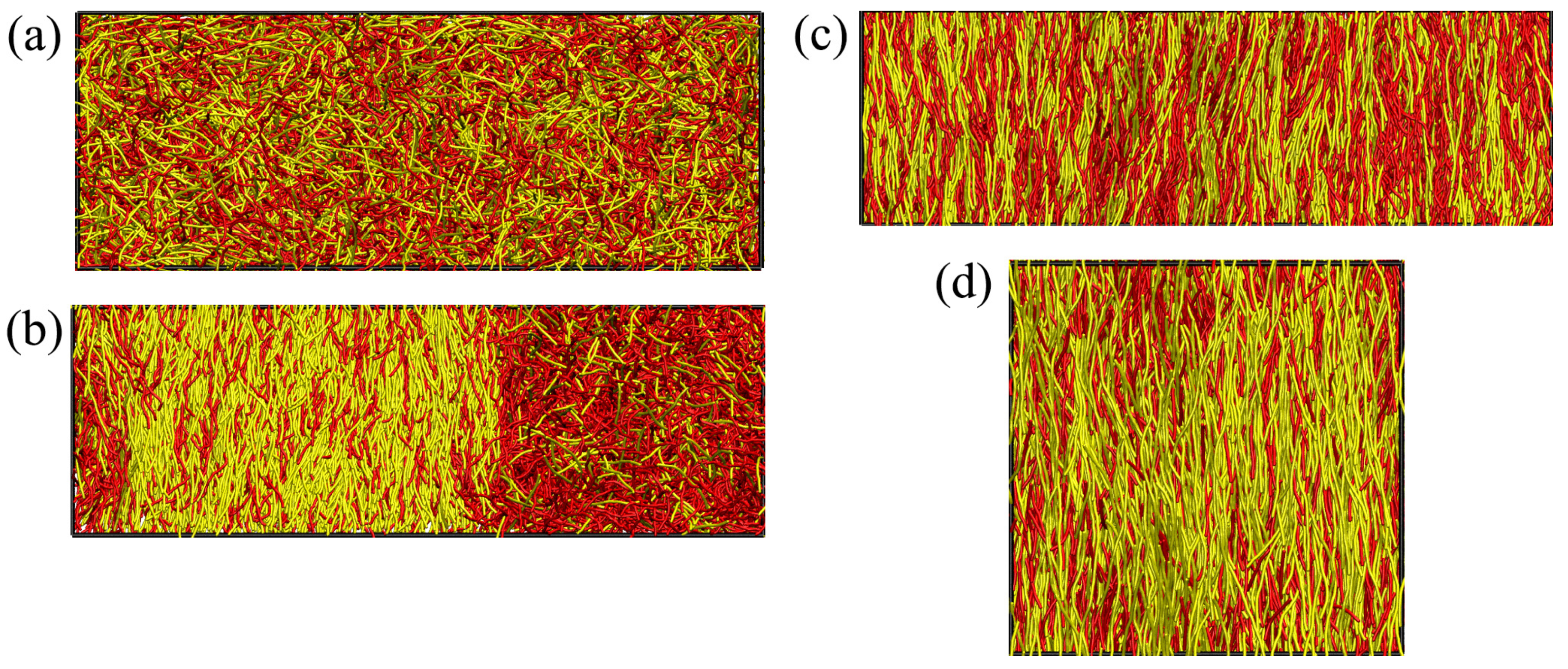
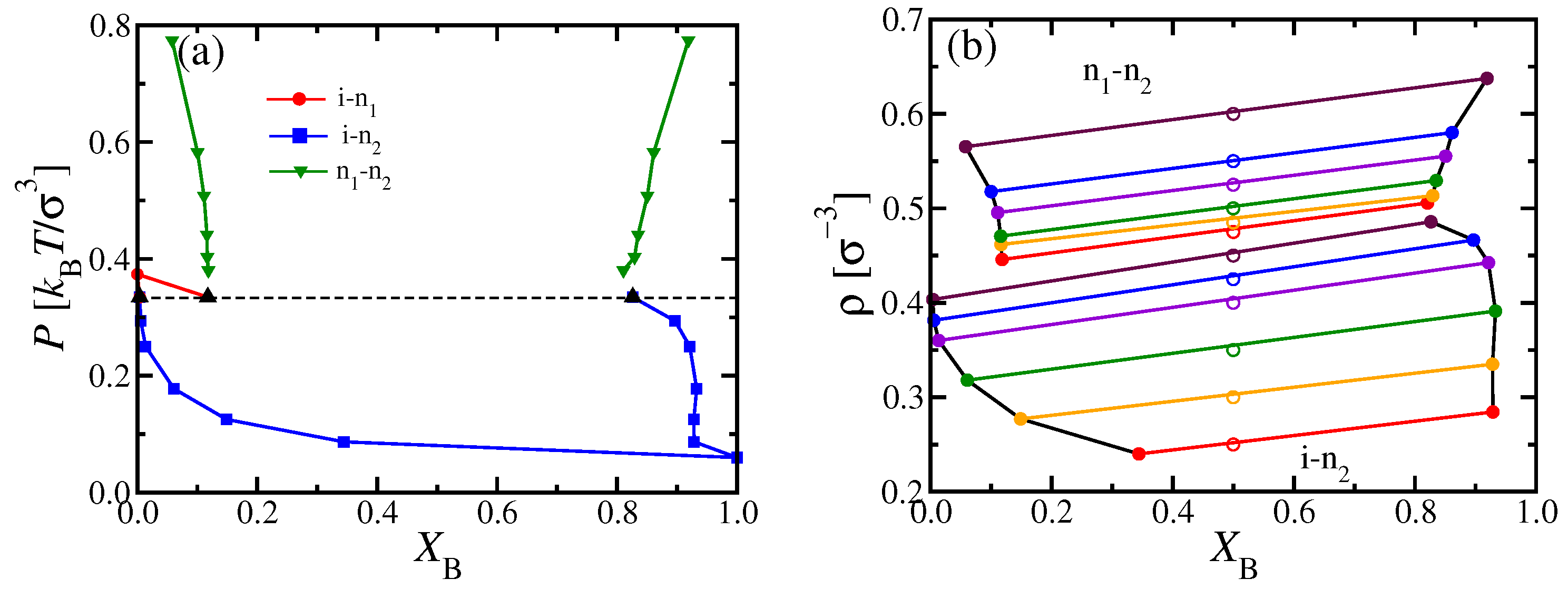
In order to obtain a simulation counterpart (
Figure 7b) to the i–n miscibility gap predicted from DFT in
Figure 3c, we had to carry out MD simulations for various choices of
, and analyze the compositions observed in suitably placed
subsystems with
; “suitably placed” means that simulation snapshots were inspected to check for states where two-phase coexistence occurs with interfaces perpendicular to the
x-axis (see
Figure 1b). The subsystems were then centered far from these interfaces, and compositions (as well as other observables) were recorded separately for the isotropic and nematic subsystems. To avoid systematic errors, we have checked that the diffusion of the domain walls in the
x-direction was small enough during the time interval in which subsystem averages were taken. However, rather large statistical fluctuations of all observables are inevitable in such simulations of two-phase coexistence. Further note that we did not succeed to prepare reasonably stable states of two-phase coexistence for pressures
and
, but rather metastable homogeneous nematic or isotropic states took over in those pressure regions. Hence, the connections of the observed phase boundaries in
Figure 7b to the pure system phase transitions at
and
are tentative interpolations only.
In principle, one could improve the results by running considerably larger systems and/or longer simulation times, which would, however, require a prohibitively large investment of additional computational resources. Thus, while MD simulations yield in principle the exact statistical mechanics of the considered model system, one has to be aware of its practical limitations. Nevertheless, our MD simulations confirm the DFT prediction that a rather wide i–n miscibility gap occurs for intermediate values of
(
Figure 3c), unlike the system shown in
Figure 6.
Figure 7b suggests that, for
, the two-phase region extends from about
to about
, compatible with
Figure 7a.
Since DFT predicted for the case
,
a region of n
–n
unmixing for
(
Figure 3c), or
(
Figure 4b), we extended our MD studies to significantly larger pressures (and corresponding densities) than shown in
Figure 7, in order to search for MD evidence of n
–n
phase separation in this model. To this end, we performed additional simulations in elongated boxes of size
, with hard walls placed at
and
to stabilize nematic order. Starting configurations were generated by orienting all chains along the
z-axis, and placing the A and B chains in the right (
) and left half (
) of the simulation box, respectively. However, we found that the initially separated A- and B-rich phases completely decay by interdiffusion. Even for
, a state deep in the nematic phase for both pure A and pure B systems, the system still develops towards a homogeneous mixture in the region far from the confining walls. At still larger densities, the system is too slowly relaxing to clearly establish equilibrium, and smectic order starts to take over in the pure B phase [
40].
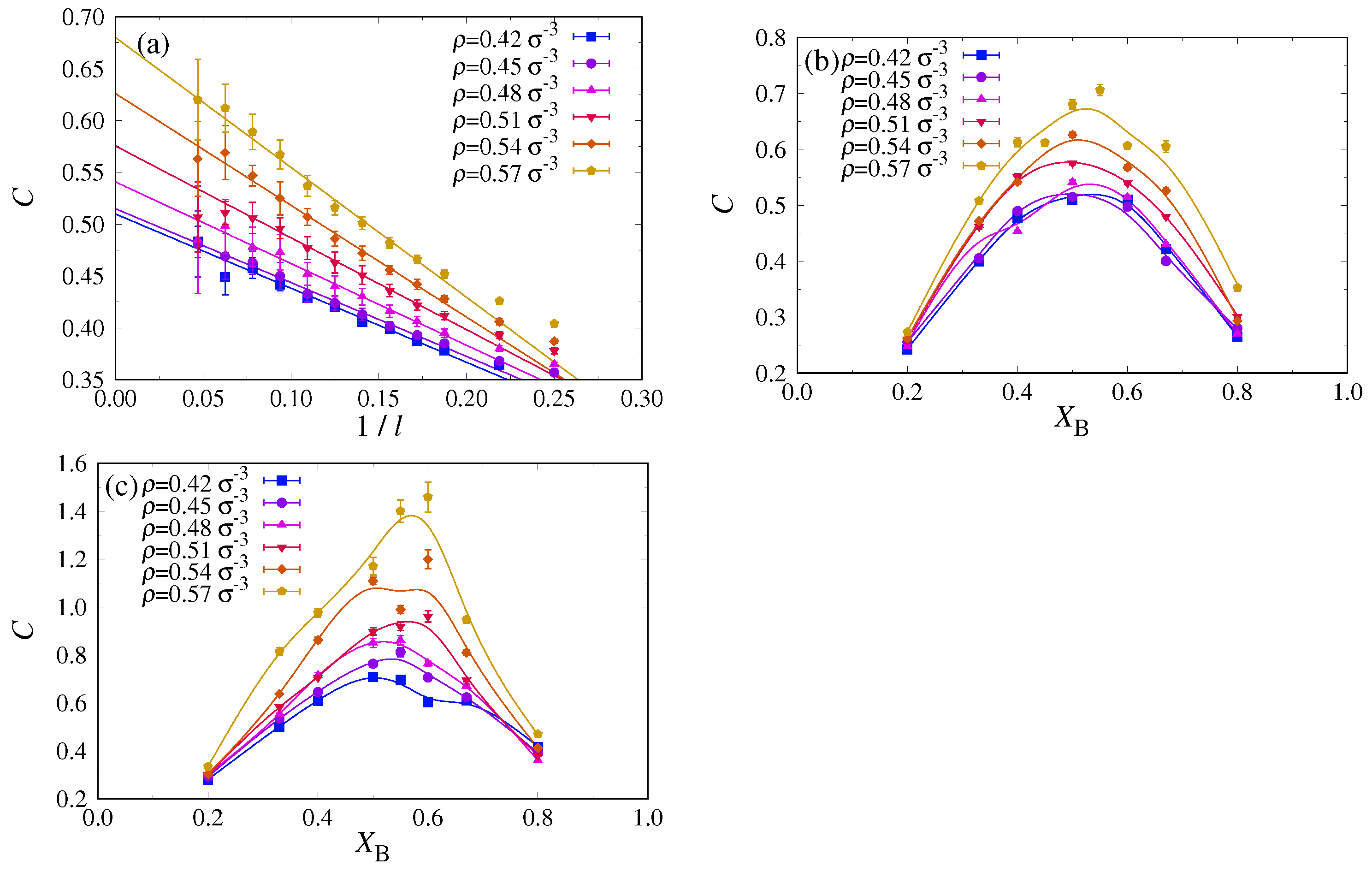
Since the occurrence of n
–n
unmixing can also be seen already in the homogeneous phase by a growth of the response function
as
(Equation (
18)), cf.
Figure 5a, we attempted to estimate this quantity in the density regime where we still can equilibrate well with MD, namely
. To mimic a grandcanonical ensemble in our MD simulations where the total numbers of A and B chains are strictly fixed, we study fluctuations of
, as described by
, in small subsystems of the total system. In any such subsystem, the volume fraction is not conserved: Because the remainder of the system acts like a reservoir on the subsystem, one essentially realizes a grandcanonical ensemble for the subsystem. The feasibility of such an approach has been demonstrated previously for both the Lennard-Jones fluid at constant density [
63] and the Ising/lattice gas model [
64,
65].
For this subsystem analysis, it is most convenient to work in the
ensemble and choose a cubic simulation box with
. For an Ising system, it would be adequate to choose subsystems of the same shape, i.e., a cubic volume
with
. However, in the present case of nematically ordered systems, we must be aware of an extremely strong anisotropy: Since the A and B chains are stretched out over a length of the order of
along the
z-axis, all volume fraction fluctuations in the
z-direction are strongly correlated; on a mean-field level, this effect was already described via the distinction of the correlation lengths
and
(see Equations (
14)–(
17)). To test whether there is a tendency in favor of phase separation in the lateral directions,
x and
y, we found it advantageous to study quasi-two dimensional subsystems perpendicular to the
z-axis. The resulting fluctuations
are plotted in
Figure 8a versus
, since one expects a
-finite size correction due to the subsystem boundary [
63,
64,
65]. The data are compatible with this expectation but also show that there is only a very modest increase of
with increasing density (
Figure 8a). This is also true for other choices of
(
Figure 8b), confirming our conclusion that the blend
,
does not undergo critical n
–n
unmixing at densities where one still has the nematic phase.
Next, we studied systems with slightly more flexible A chains (
,
), which, according to our DFT calculations (cf.
Figure 3a), no longer have a mixed nematic phase extending over the full range of investigated pressures
and densities
. Indeed, now the fluctuation near
increases distinctly stronger with increasing density (
Figure 8c) than for the case
. However, it turns out to be extremely difficult to reach a decent precision of these data; hence, we could still not locate the critical point of n
–n
unmixing of the system with this method. One problem comes from the fact that critical slowing down is known to lead to a systematic bias (underestimation) of
when the runs are not long enough [
66]. This problem is particularly severe for critical binary mixtures, where a finite size scaling of the relaxation time for interdiffusion
is predicted [
67].
However, we found it possible to study n
–n
unmixing for
,
, using the
ensemble and elongated boxes with
,
,
196,608, and a choice of
such that the desired density was realized (e.g.,
for
). The time step here was chosen as
, and runs of
MD steps were carried out for compositions
,
and
. Starting again the simulations with chains oriented along the
z-axis and initially separated in pure A and B domains (according to the chosen
), equilibrium was reached for
for the densities of interest. We then determined the phase diagrams of
Figure 9 and
Figure 10 by analyzing the resulting density profiles
and
. From
Figure 9, it is obvious that the topology of the phase diagram found in MD disagrees with its DFT counterpart for
(
Figure 3a), but rather resembles the DFT phase diagram for
(
Figure 3c). Likewise, the MD phase diagram for
(
Figure 10) is qualitatively similar to the DFT phase diagram found for
(
Figure 3a). Thus, the massive discrepancies between DFT and MD phase diagrams for choices of
and
should not be interpreted as a complete breakdown of the DFT method: Rather, the latter is only somewhat inaccurate with respect to the prediction of the value
, where the phase diagram topology changes. For
, the phase diagram exhibits a triple point, while, for
, two separate coexistence regions occur, i.e., an i–n region at lower pressures and a n
–n
coexistence region (ending in a critical point) at higher pressures. DFT predicts
for
, while MD rather suggests
. Apart from this quantitative mismatch in
, the general sequence of phase diagram changes with increasing
predicted by MD and DFT is the same. Further, these findings are also compatible with the predictions obtained by Semenov and Subbotin [
21] for the limiting case of very large contour lengths. Hence, we conclude that the general features of the phase behavior predicted here are rather robust.
An important aspect of phase coexistence in the studied systems is that only the pressure
P in the coexisting phases is identical, while the density
is not, as evidenced from the nonzero slopes of the tie lines drawn in
Figure 9b and
Figure 10b. This density inhomogeneity could also be seen very well in the density profiles
in the direction perpendicular to the interfaces: Due to the periodic boundary conditions, there must be two interfaces, and the slope of the tie lines tells us that the density of the phase n
is larger than both the density of the phase n
and the isotropic phase. Due to this density variation at the i–n
interfaces, there could occur some excess density (“interfacial adsorption”) associated with these interfaces, which might affect our analysis of the MD results (as a finite size effect of order
). However, a more detailed analysis of our results has shown that this effect (and other finite size effects) is negligible. Rather, we could show that our results on phase coexistence are fully compatible with consequences of the Gibbs phase rule. In fact, when the two A-rich (a) and B-rich (b) phases coexist in our simulation volume, the volume consists of two parts,
, since, per definition, interfaces have zero volume. The particle numbers
and
of the two kinds of monomers can be likewise split into particle numbers in the two phases
with
. The phase boundaries in
Figure 9 and
Figure 10 were based on the four partial densities
,
,
,
, namely
,
, and
, as well as
, with
,
. Note that the volume fraction
must not be confused with the mole fraction
.
Due to Equations (
20) and (21) and the relation for
V, the four partial densities are not all independent of each other. We have found it convenient to formulate this dependence via two equations for the volume fraction of phase b,
, with ratios
and
defined as
with partial densities
and
.
Table A1 and
Table A2 quote the partial densities for
, from which
Figure 9 and
Figure 10 were constructed, as well as the estimates
and
of the corresponding volume fraction of the B-rich phase. Results for
and 2/3 were compatible with these results and are therefore not shown.
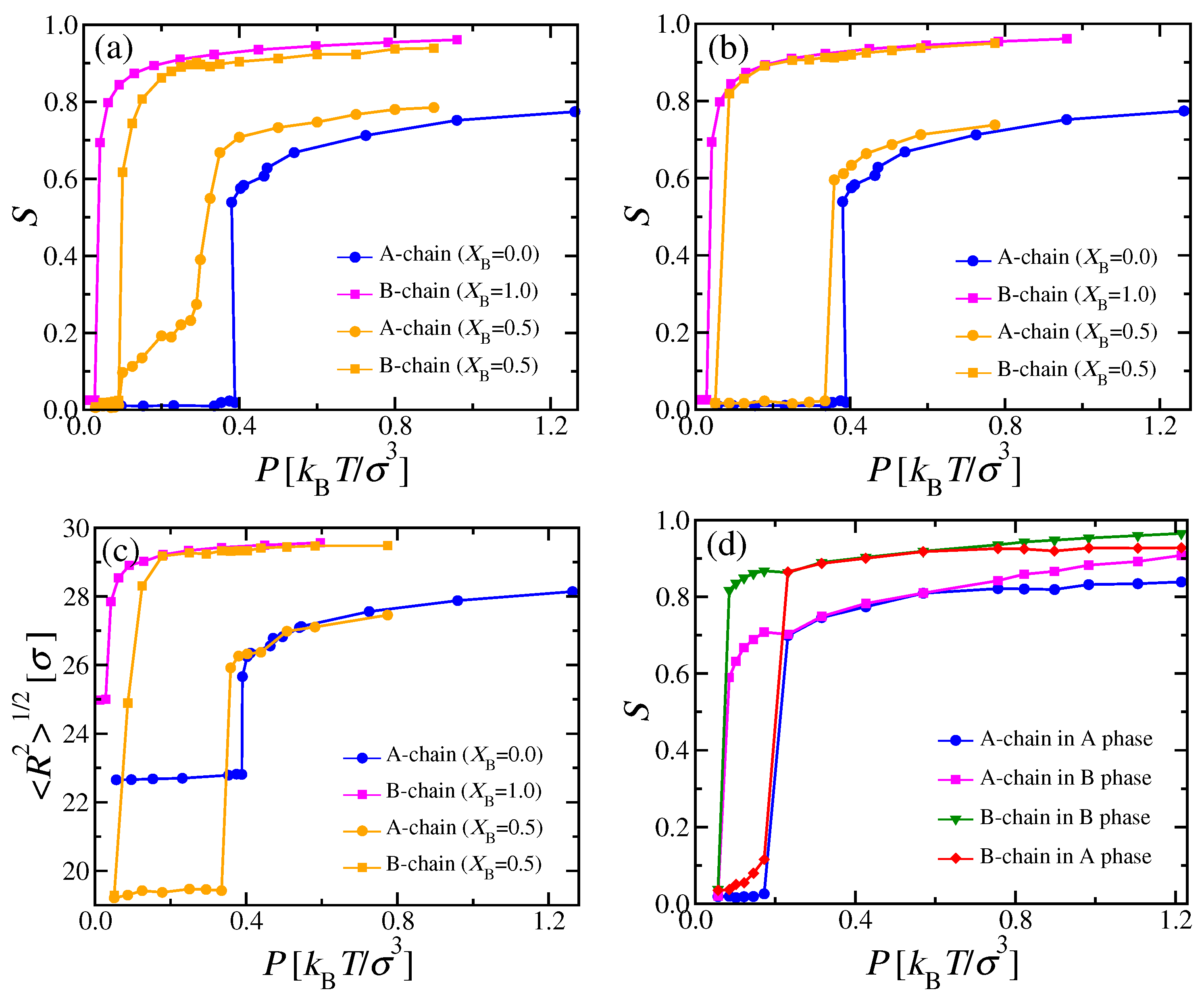
We now discuss the nematic bond order parameters of the two types of chains,
and
, respectively, extracted as usual [
35,
39] as the largest eigenvalue of the tensor
(Equation (
3)). In the pure A and B systems, a discontinuous transition occurs at
and
, respectively, where the nematic order parameter jumps from zero to about
and
[
9,
39], respectively (
Figure 11a). In contrast, the mixed systems exhibit a more gradual behavior, as shown in
Figure 11a. As expected from the phase diagram (
Figure 10a),
starts to rise steeply as soon as the i–n
two-phase coexistence region has entered around
. Unlike the pure B system, the increase of
is continuous, since the mole fraction of the nematic phase grows continuously from zero to one as the two-phase coexistence region is crossed. In addition,
starts to become nonzero together with
, due to A chains that are dissolved in the nematic B-rich phase (at a very small mole fractions, cf.
Figure 10a), and which align due to the nematic ordering field exerted by surrounding majority of B chains. At
, the rise of
is already rather steep: We interpret this phenomenon as a “capillary nematization” effect of the nematic B-rich domains on the remaining A-rich domains, which, in a truly macroscopic system, would still be in the isotropic phase up to a pressure of about
, according to the phase diagram shown in
Figure 10a. This finite size effect will be discussed in more detail below.
In bulk (macroscopic) systems, the gradual increase of the order parameters can be interpreted in terms of the lever rule
Here, the two boundaries of the i–n two-phase coexistence region were denoted as and . If we could cross this region at fixed P by varying , the variation of (or , respectively) with would be simply linear. However, this clearly is not true when we vary P at fixed (different from or 1, of course): Then, and are nontrivial functions, reflecting both the variation of the phase boundaries and , as well as of .
The behavior described in
Figure 11a and Equation (
22) would be observed experimentally by methods that average over all domains in the system, e.g., scattering, optical birefringence, etc. In the simulations, it is possible to resolve the order parameters separately in the two kinds of domains: Then,
measured in the nematic B-rich domain rises much faster with
P (
Figure 11b), similar to the result of the pure B phase. Likewise,
measured in the isotropic A-rich domains stays zero up to
, where also the A-rich phase becomes nematic (
Figure 11b). An analogous interpretation explains the behavior of the end-to-end distance measured separately in the coexisting domains (
Figure 11c). Equation (
23) applies only for pressures
because, for larger pressures, we enter a different two-phase coexistence region between two different nematic phases. The generalization to this case is
In the simulation, the order parameter of the less stiff chains increases indeed steeply around
: According to theory, there should be a clear jump of
at
, since
in the isotropic phase, while
in the phase
. There is also a discontinuous increase from the composition of the isotropic phase
to the composition
of the A-rich nematic phase. The resulting singular behavior of
and
predicted by Equations (
23) and (
24) is somewhat smeared out in the simulations, presumably due to finite size effects. In addition, the polymer end-to-end distances (
Figure 11c) do not show related singularities either. Thus, it clearly would be desirable to study the system using much larger
and much better statistics, which was, however, computationally infeasible for us. One reason for unexpectedly large finite size effects is recognized when we examine the order parameters
,
,
,
,
, and
that belong to the various phases: We find that, in the case where we have i–n
two-phase coexistence in the simulation box, the order parameter
grows gradually with
P and is of order 0.5 already when
is reached. We interpret this finding as a “capillary nematization” effect of the B chains, which have
for
P near
already in the
phase, in the isotropic phase adjacent to the i–n
interfaces. As
, a kind of nematic wetting layer grows in the isotropic phase at the i–n
interface. If the linear dimensions
, this effect will become negligibly small, but it is not for the
values accessible to us. Another interesting feature is that, in the mixed nematic phase, the order parameter of the less stiff phase (
) always exceeds the corresponding value of the pure A phase at the same pressure, and likewise for the stiffer component (
) it is slightly smaller than for its pure counterpart. Thus, the more ordered stiffer chains in the direct neighborhood of less stiff chains enhance the order of the latter; conversely, the less stiff chains somewhat perturb the order of the stiffer ones, but the local nematic order of the mixture is very nonuniform since
is distinctly smaller than
. A similar effect has already been noted for mixtures of chains which have the same stiffness but differ in chain length (where the longer chains are more ordered), as in
Figure 6b,d.
Figure 11d shows a counterpart to
Figure 11b for the case
, where a uniformly mixed nematic phase occurs. While we expect a discontinuous transition from
in the isotropic phase to a nonzero value in the uniformly mixed nematic phase at the transition from the i–n two-phase region, a continuous splitting of both
and
into their different values in the coexisting nematic phase is expected at
. Indeed, the data are compatible with this prediction. We see that
in the A-rich nematic phase grows gradually from zero to distinctly nonzero values, when the transition to the nematic phase is approached: Again, we interpret this precursor effect as a finite size effect, due to “capillary nematization” at the i–n interfaces of the domain of the isotropic phase which has a finite extent in
x-direction (according to
Table A1, the volume fraction taken by the isotropic phase is about 0.39 for
(
)).
Finally, we make contact between the n
–n
critical point found here (
Figure 9 and
Figure 11d) and the phenomenological theory of
Section 2.3: Can one understand the
parameter postulated there from the stiffness asymmetry? To answer this question, we apply the approach of Kozuch et al. [
42], who had considered only isotropic mixtures of rather flexible long polymers with slightly different stiffnesses.
Noting that Equation (
13) also follows from the FH free energy
with volume fractions
[
11,
12,
15], one can see that the excess term
relative to the entropy of mixing terms of the ideal mixture can only result from the difference in bending energies
and
, since both types of chains are identical in all other respects for
. Since the average energy
(with
), one can apply thermodynamic integration methods, based on the appropriate difference in bending energy. Denoting the total free energy of the mixture with
as
, the sought after excess free energy is
Equation (
26) leads to
since the number of bonds in the B chains is only one half of the total number of bonds per chain in the mixed system. Hence,
Here,
is the bending energy per monomer of a B chain,
denotes the thermal average in a 1:1 AB mixture, while
refers to an average in a pure system of B chains. The desired free energy difference then is
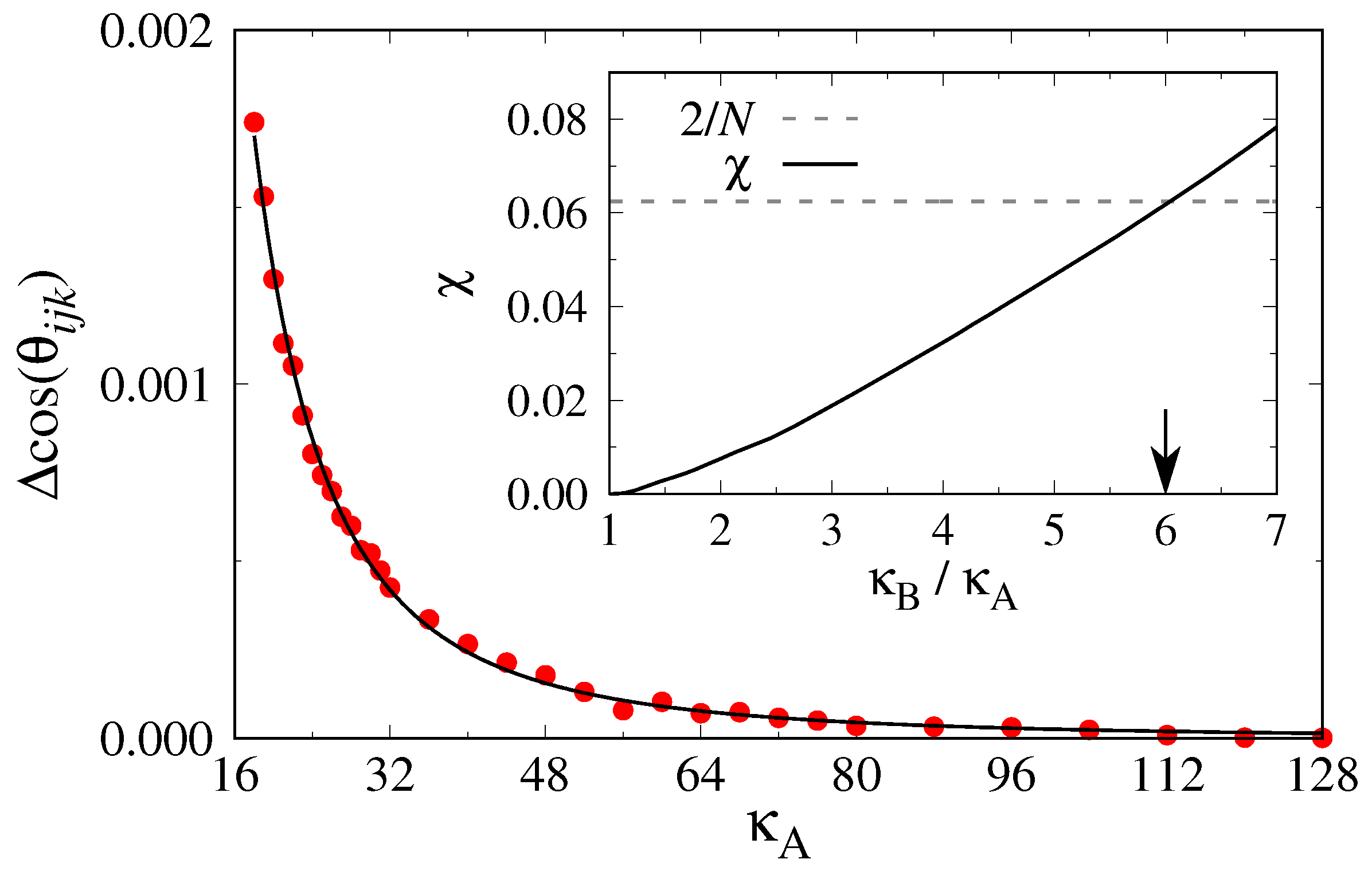
We have performed this thermodynamic integration for a system at
in a cubic box of size
with
monomeric units. We started the numerical integration in a state
, and then lowered the stiffness of the A component to smaller values (
Figure 12). One sees that the integrand is extremely small if both chains have similar stiffness, but it rises steeply for large stiffness mismatches. The resulting effective
parameter is shown in the inset of
Figure 12. Criticality (according to mean-field theory) would be reached for
, i.e.,
. However, while the pressure
for
and
clearly falls inside the i–n
coexistence region, for
, the critical pressure
corresponds to
; hence, this system at
is in the mixed nematic region. Since
can be expected to increase with increasing density, we cannot estimate
for the data shown in
Figure 10, but the order of magnitude of
expected on the basis of
Figure 12 makes sense.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











