Coincident Correlation between Vibrational Dynamics and Primary Relaxation of Polymers with Strong or Weak Johari-Goldstein Relaxation

 , and
, and 
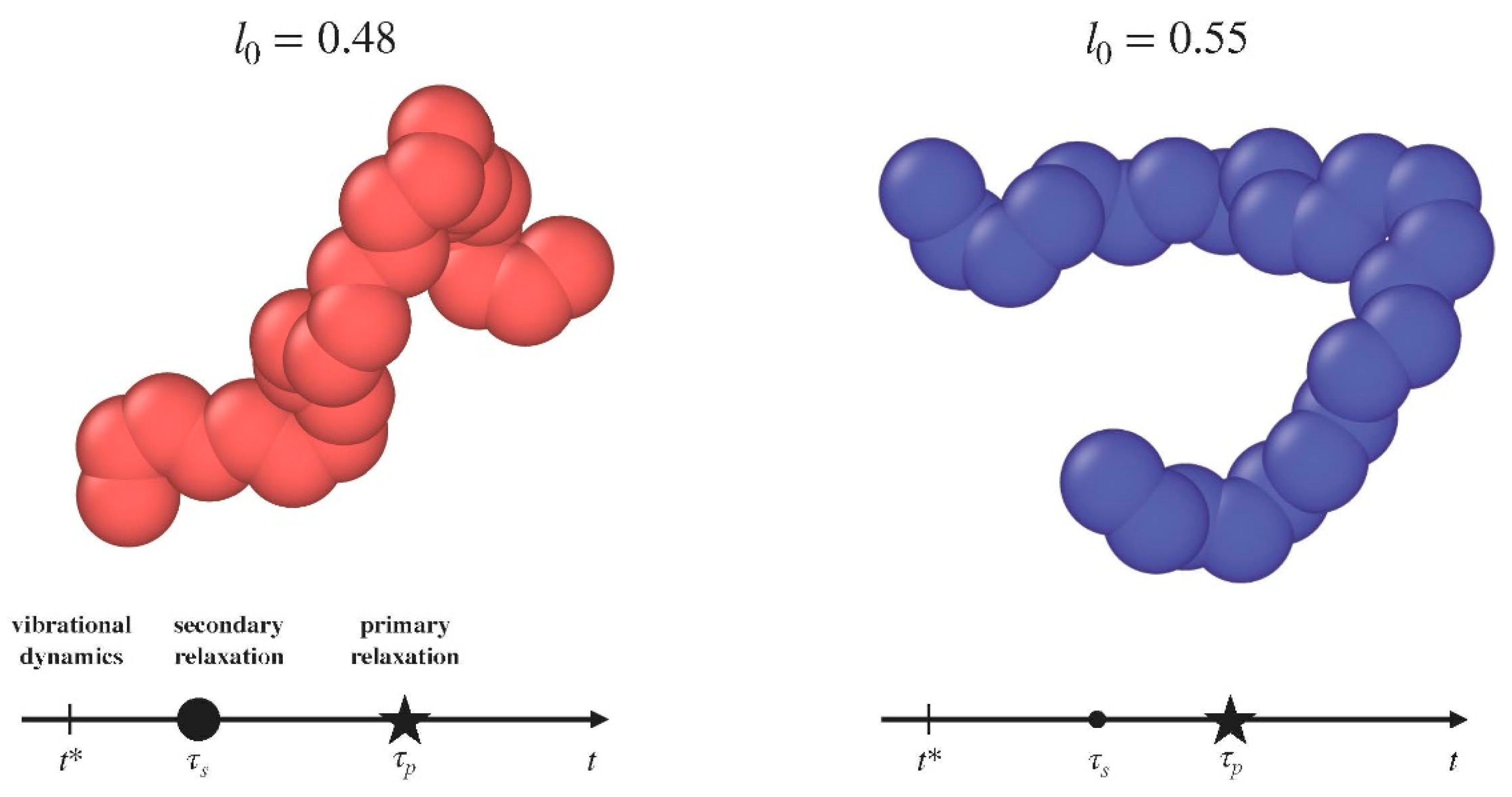{kind=link}
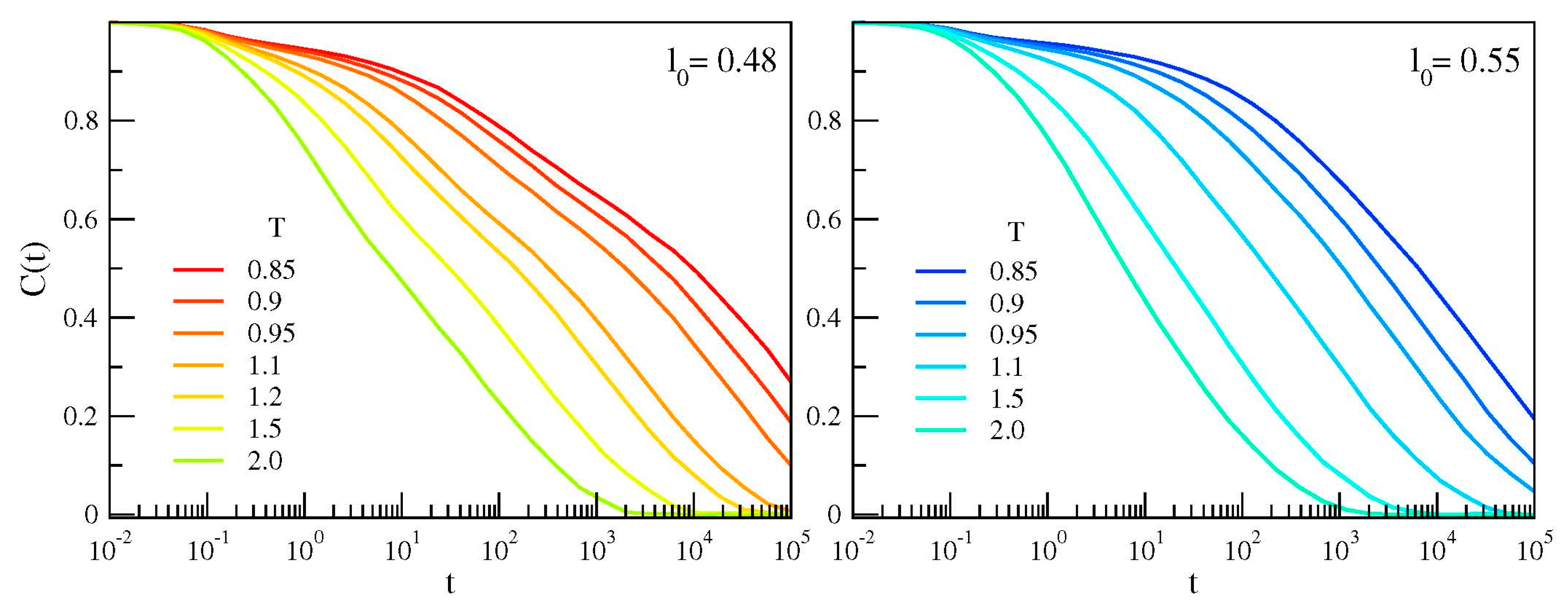{kind=link}
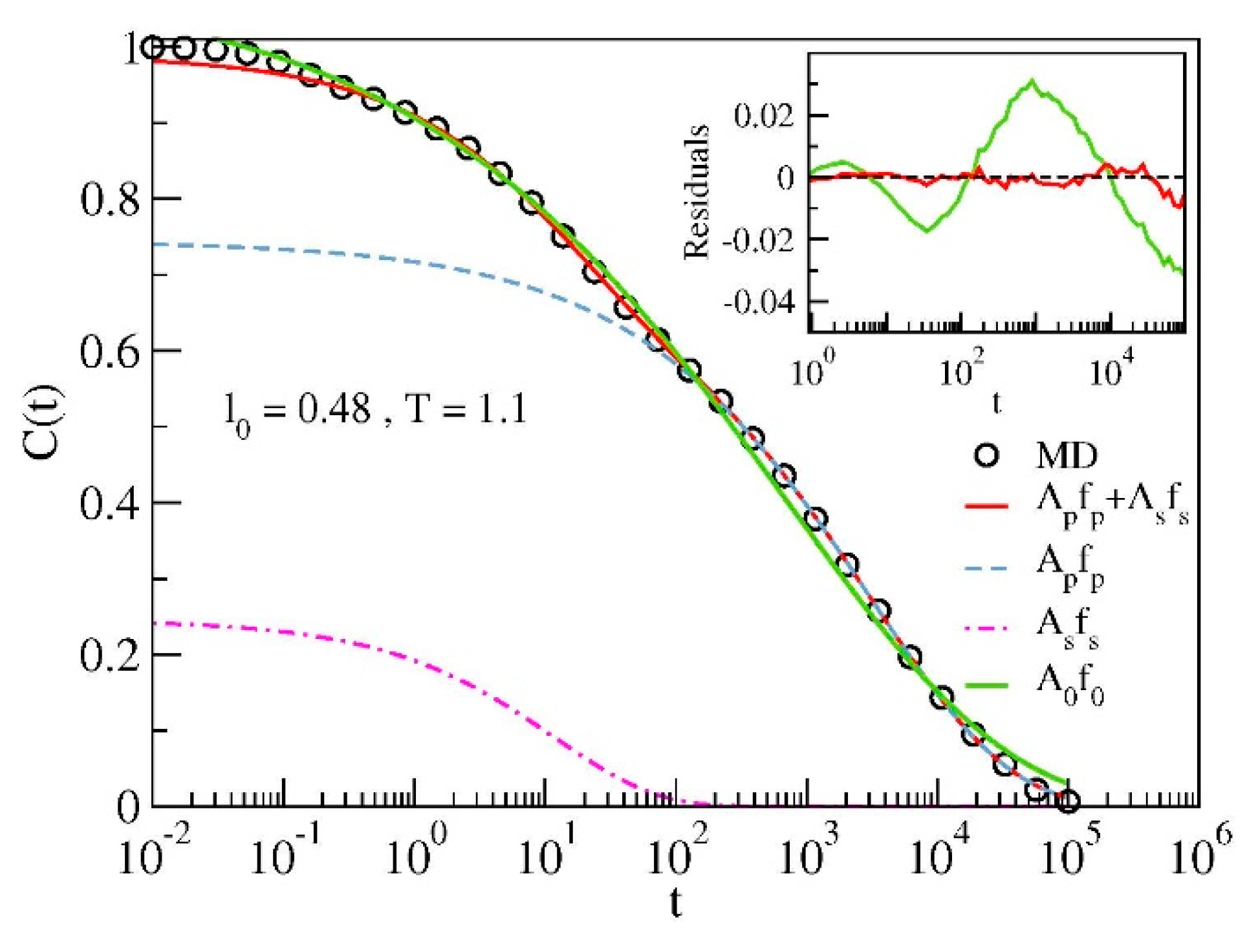{kind=link}
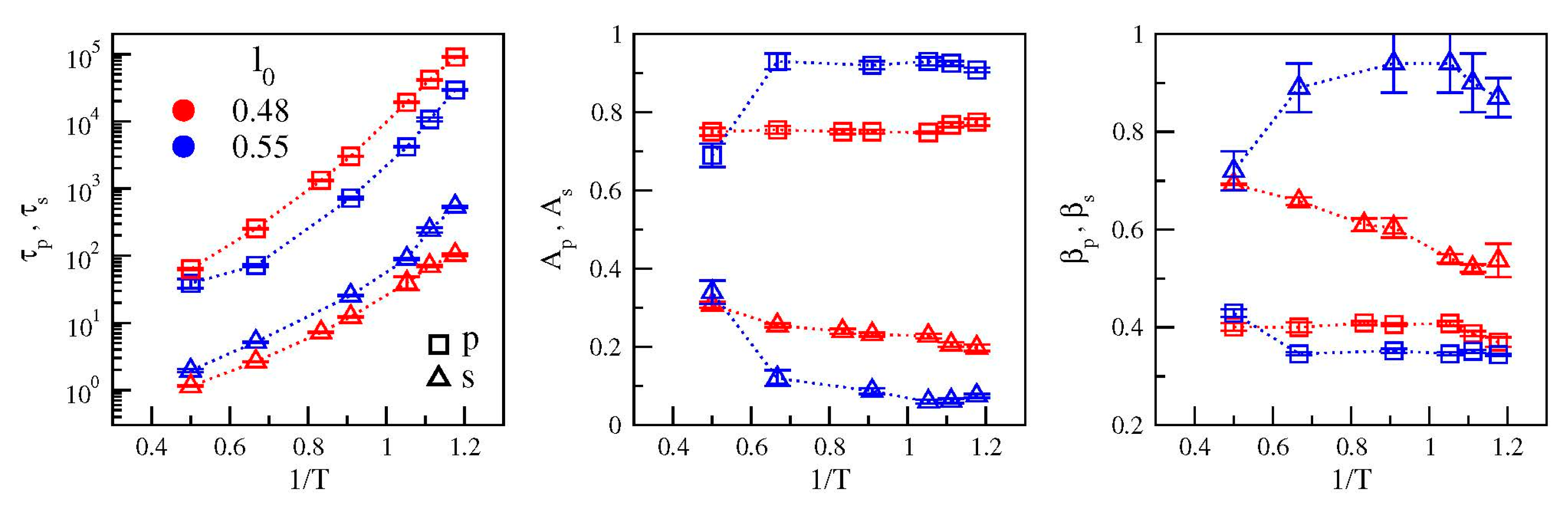{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Numerical Methods
3. Results and Discussion
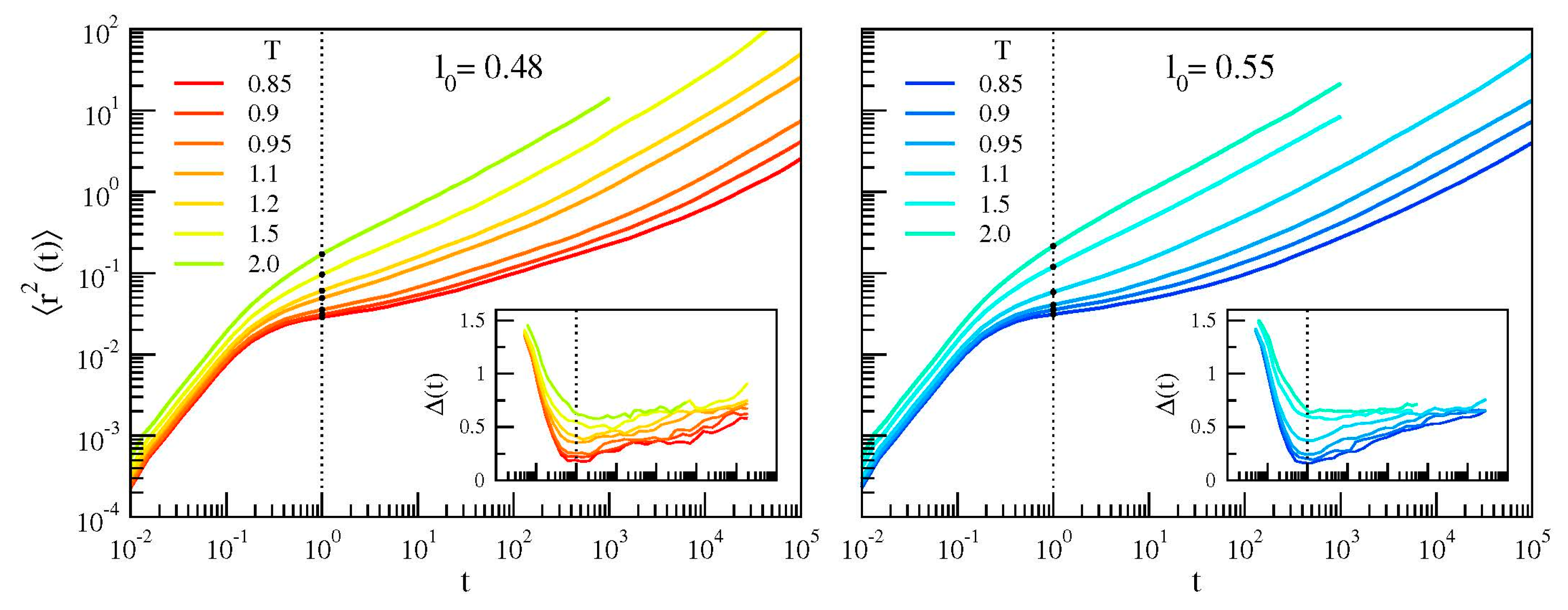
3.1. Bond Reorientation
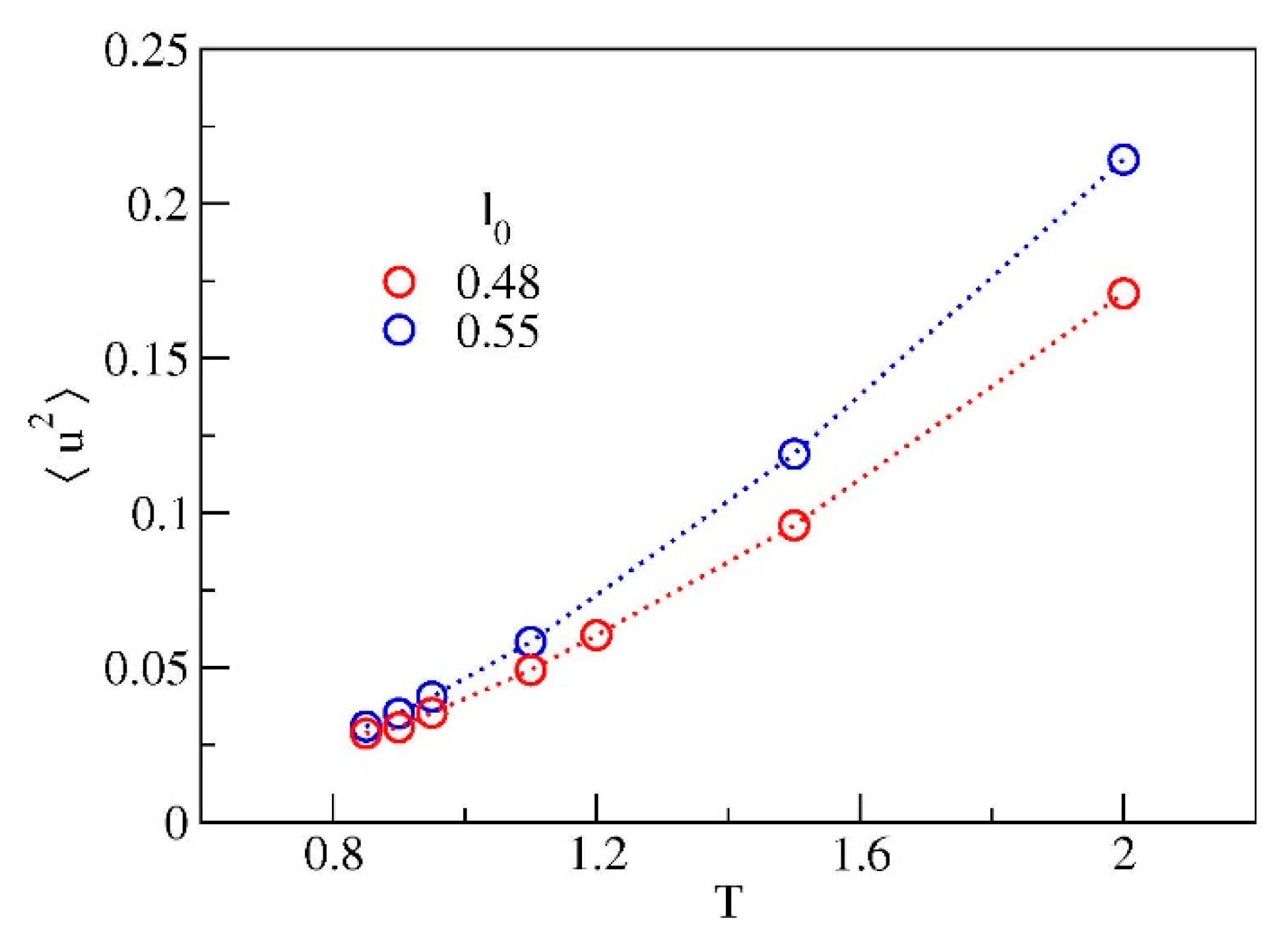
3.2. Monomer Mobility
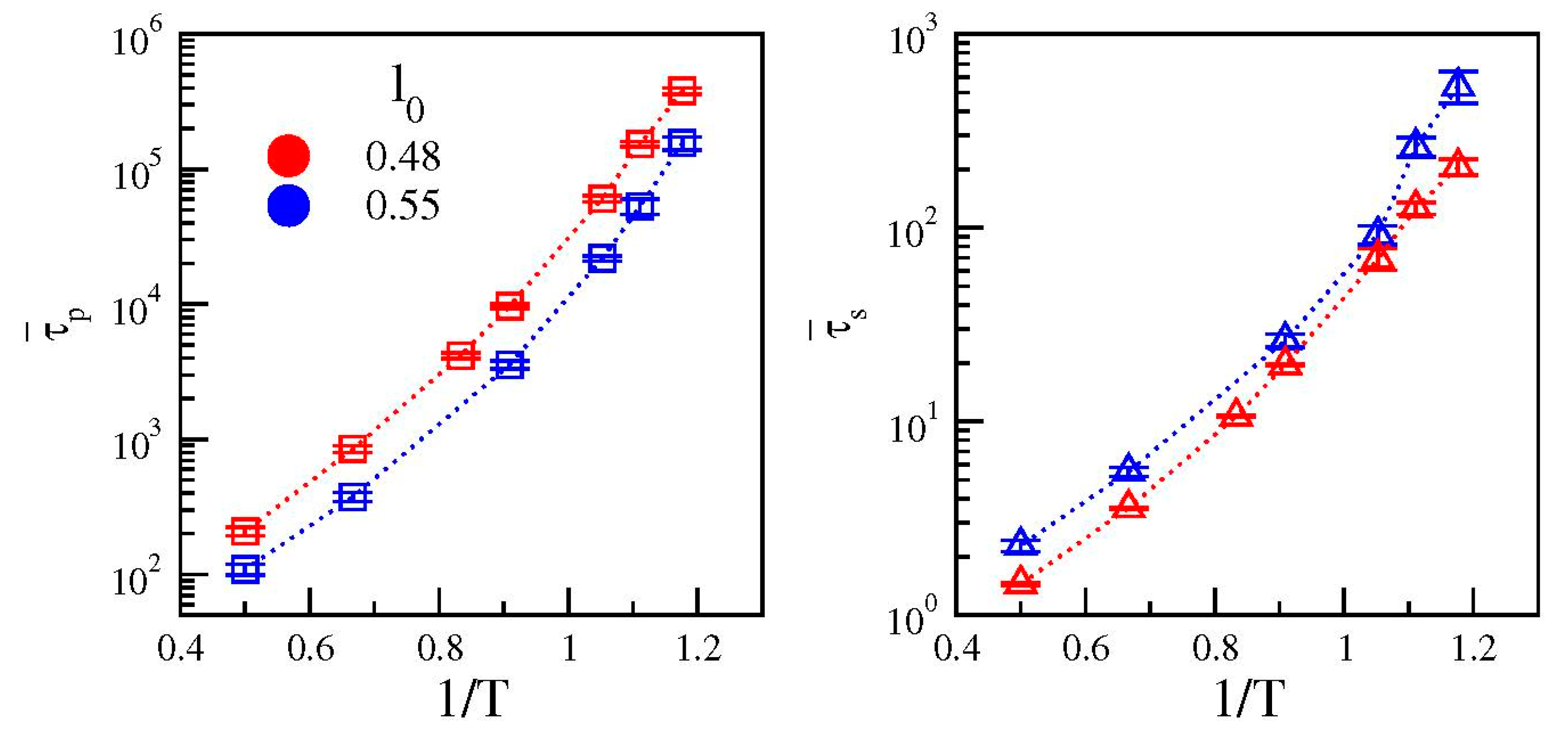
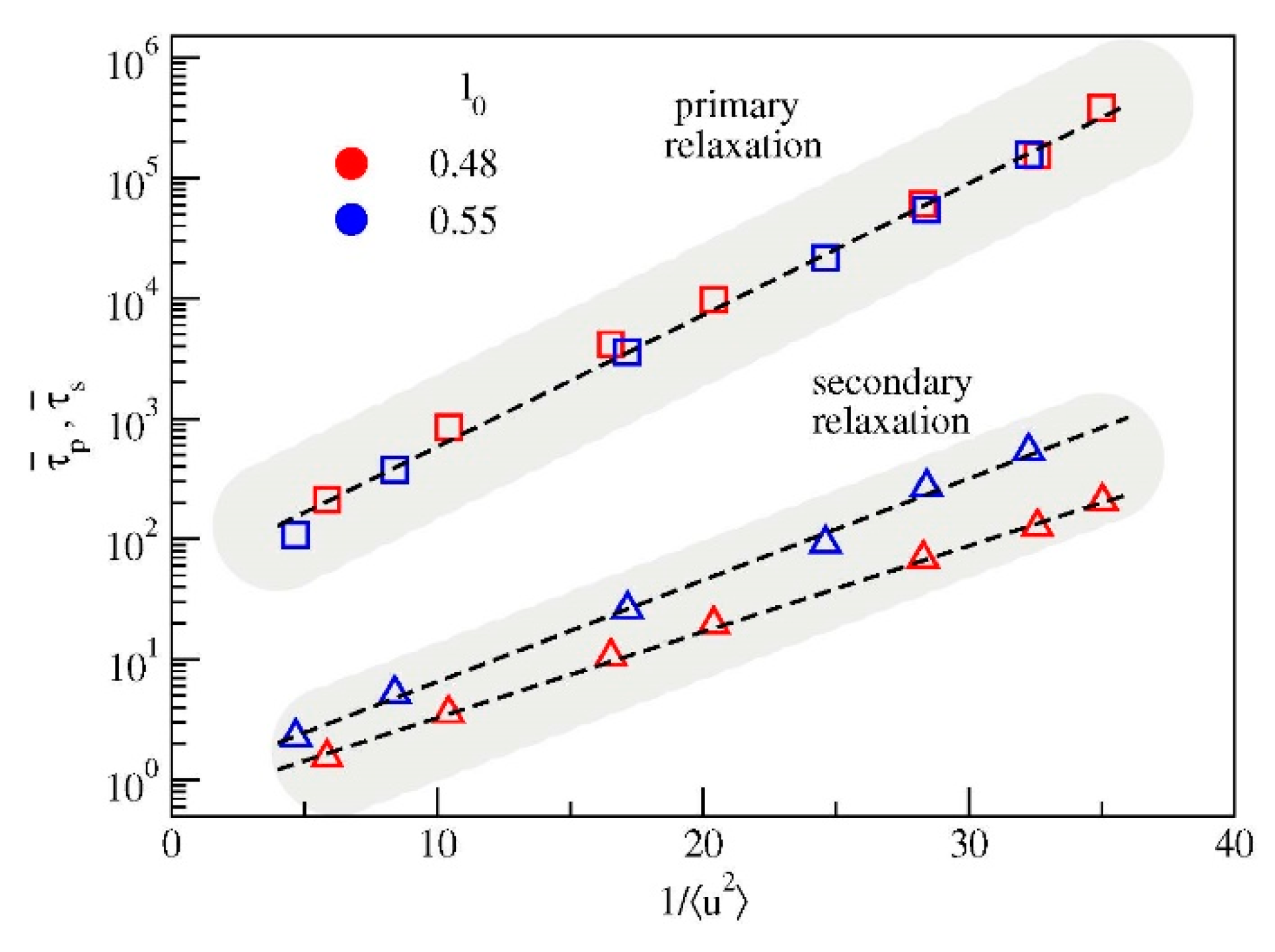
3.3. Cage Dynamics and Correlation with Primary and Secondary Relaxations
3.4. Alternative Probe Functions of Secondary Relaxation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ISF | Intermediate scattering function |
| JG | Johari-Goldstein |
| MD | molecular-dynamics |
| MSD | Mean square displacement |
| DW | Debye-Waller |
References
- Debenedetti, P.G. Metastable Liquids; Princeton University Press: Princeton, NJ, USA, 1997. [Google Scholar]
- McCrum, N.G.; Read, B.E.; Williams, G. Anelastic and Dielectric Effects in Polymeric Solids; Dover Publications: New York, NY, USA, 1991. [Google Scholar]
- Angell, C.A.; Ngai, K.L.; McKenna, G.B.; McMillan, P.; Martin, S.W. Relaxation in glassforming liquids and amorphous solids. J. Appl. Phys. 2000, 88, 3113–3157. [Google Scholar] [CrossRef] [Green Version]
- Ngai, K.L. Relaxation and Diffusion in Complex Systems; Springer: Berlin, Germany, 2011. [Google Scholar]
- Johari, G.P.; Goldstein, M. Viscous Liquids and the Glass Transition. II. Secondary Relaxations in Glasses of Rigid Molecules. J. Chem. Phys. 1970, 53, 2372. [Google Scholar] [CrossRef]
- Ngai, K. Relation between some secondary relaxations and the relaxations in glass-forming materials according to the coupling model. J. Chem. Phys. 1998, 109, 6982–6994. [Google Scholar] [CrossRef]
- Ngai, K.L.; Paluch, M. Classification of secondary relaxation in glass-formers based on dynamic properties. J. Chem. Phys. 2004, 120, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Capaccioli, S.; Paluch, M.; Prevosto, D.; Wang, L.M.; Ngai, K.L. Many-Body Nature of Relaxation Processes in Glass-Forming Systems. J. Phys. Chem. Lett. 2012, 3, 735–743. [Google Scholar] [CrossRef]
- Cicerone, Q.Z.; Tyagi, M. Picosecond Dynamic Heterogeneity, Hopping, and Johari-Goldstein Relaxation in Glass-Forming Liquids. Phys. Rev. Lett. 2014, 113, 117801. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.R.; Samwer, K. Structural rearrangements governing Johari-Goldstein relaxations in metallic glasses. Sci. Adv. 2017, 3, 1701577. [Google Scholar] [CrossRef] [Green Version]
- Larini, L.; Ottochian, A.; De Michele, C.; Leporini, D. Universal scaling between structural relaxation and vibrational dynamics in glass-forming liquids and polymers. Nat. Phys. 2008, 4, 42–45. [Google Scholar] [CrossRef]
- Puosi, F.; Tripodo, A.; Leporini, D. Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids. Int. J. Mol. Sci. 2019, 20, 5708. [Google Scholar] [CrossRef] [Green Version]
- Bedrov, D.; Smith, G.D. Molecular dynamics simulation study of the α and β-relaxation processes in a realistic model polymer. Phys. Rev. E 2005, 71, 050801. [Google Scholar] [CrossRef]
- Bedrov, D.; Smith, G.D. Secondary Johari–Goldstein relaxation in linear polymer melts represented by a simple bead-necklace model. J. Non-Crystal. Solids 2011, 357, 258–263. [Google Scholar] [CrossRef]
- Fragiadakis, D.; Roland, C.M. Participation in the Johari–Goldstein Process: Molecular Liquids versus Polymers. Macromolecules 2017, 50, 4039–4042. [Google Scholar] [CrossRef] [Green Version]
- Fragiadakis, D.; Roland, C.M. Molecular dynamics simulation of the Johari-Goldstein relaxation in a molecular liquid. Phys. Rev. E 2012, 86, 020501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larini, L.; Barbieri, A.; Prevosto, D.; Rolla, P.A.; Leporini, D. Equilibrated polyethylene single-molecule crystals: Molecular-dynamics simulations and analytic model of the global minimum of the free-energy landscape. J. Phys. Condens. Matter 2005, 17, L199–L208. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Lammps. Available online: http://lammps.sandia.gov (accessed on 26 February 2020).
- Prevosto, D.; Capaccioli, S.; Lucchesi, M.; Leporini, D.; Rolla, P. Pressure and temperature dependence of structural relaxation dynamics in polymers: A thermodynamic interpretation. J. Phys. Condens. Matter 2004, 16, 6597–6608. [Google Scholar] [CrossRef] [Green Version]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1988. [Google Scholar]
- Giordano, M.; Leporini, D.; Martinelli, M.; Pardi, L.; Santucci, S.; Umeton, C. Nonlinear electron spin resonance techniques for the study of inhomogeneously broadened spectra. J. Chem. Phys. 1988, 88, 607–616. [Google Scholar] [CrossRef]
- Andreozzi, L.; Giordano, M.; Leporini, D. Efficient characterization of the orientational ordering of ESR-active probes in supermolecular fluids. Appl. Magn. Reson. 1993, 4, 279–295. [Google Scholar] [CrossRef]
- Leporini, D. Relationship between a nonlinear response and relaxation induced by colored noise. Phys. Rev. A 1994, 49, 992–1014. [Google Scholar] [CrossRef]
- Andreozzi, L.; Giordano, M.; Leporini, D. Linear and non-linear electron spin resonance study of the rotational diffusion of a molecular tracer in supercooled o-terphenyl. J. Non-Cryst. Solids 1998, 235, 219–224. [Google Scholar] [CrossRef]
- Andreozzi, L.; Faetti, M.; Giordano, M.; Leporini, D. Scaling between the rotational diffusion of tracers and the relaxation of polymers and glass formers. J.Phys. Condens. Matter 1999, 11, A131–A137. [Google Scholar] [CrossRef]
- Andreozzi, L.; Faetti, M.; Giordano, M.; Leporini, D. Scaling of the Rotational Relaxation of Tracers in o-Terphenyl: A Linear and Nonlinear ESR Study. J. Phys. Chem. B 1999, 103, 4097–4103. [Google Scholar] [CrossRef]
- Barbieri, A.; Gorini, G.; Leporini, D. Role of the density in the crossover region of o-terphenyl and poly(vinyl acetate). Phys. Rev. E 2004, 69, 061509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, A.; Campani, E.; Capaccioli, S.; Leporini, D. Molecular dynamics study of the thermal and the density effects on the local and the large-scale motion of polymer melts: Scaling properties and dielectric relaxation. J. Chem. Phys. 2004, 120, 437–453. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.D.; Bedrov, D. A molecular dynamics simulation study of the α- and β-relaxation processes in 1,4-polybutadiene. J. Non-Crystal. Solids 2006, 352, 4690–4695. [Google Scholar] [CrossRef]
- Fragiadakis, D.; Roland, C.M. Chain Flexibility and the Segmental Dynamics of Polymers. J. Phys. Chem. B 2019, 123, 5930–5934. [Google Scholar] [CrossRef]
- Zorn, R.; Arbe, A.; Colmenero, J.; Frick, B.; Richter, D.; Buchenau, U. Neutron scattering study of the picosecond dynamics of polybutadiene and polyisoprene. Phys. Rev. E 1995, 52, 781–795. [Google Scholar] [CrossRef]
- Gupta, S.; Mamontov, E.; Jalarvo, N.; Stingaciu, L.; Ohl, M. Characteristic length scales of the secondary relaxations in glass-forming glycerol. Eur. Phys. J. E 2016, 39, 40. [Google Scholar] [CrossRef] [Green Version]
- Hilfer, R. Analytical representations for relaxation functions of glasses. J. Non-Crystal. Solids 2002, 305, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Pardo, L.C.; Lunkenheimer, P.; Loidl, A. Dielectric spectroscopy in benzophenone: The beta relaxation and its relation to the mode-coupling Cole-Cole peak. Phys. Rev. E 2007, 76, 030502. [Google Scholar] [CrossRef] [Green Version]
- Angell, C. Relaxation in liquids, polymers and plastic crystals-strong/fragile patterns and problems. J. Non-Crystal. Sol. 1991, 131, 13–31. [Google Scholar] [CrossRef]
- Ottochian, A.; Leporini, D. Scaling between structural relaxation and caged dynamics in Ca0.4K0.6(NO3)1.4 and glycerol: Free volume, time scales and implications for the pressure-energy correlations. Philosophical Magazine 2011, 91, 1786–1795. [Google Scholar] [CrossRef] [Green Version]
- Arbe, A.; Buchenau, U.; Willner, L.; Richter, D.; Farago, B.; Colmenero, J. Study of the Dynamic Structure Factor in the β Relaxation Regime of Polybutadiene. Phys. Rev. Lett. 1996, 76, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids, 3rd ed.; Academic Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Kremer, K.; Grest, G.S.; Carmesin, I. Crossover from Rouse to Reptation Dynamics: A Molecular-Dynamics Simulation. Phys. Rev. Lett. 1988, 61, 566–569. [Google Scholar] [CrossRef]
- Arbe, A.; Richter, D.; Colmenero, J.; Farago, B. Merging of the α and β relaxations in polybutadiene: A neutron spin echo and dielectric study. Phys. Rev. E 1996, 54, 3853–3869. [Google Scholar] [CrossRef] [PubMed]
- Caporaletti, F.; Capaccioli, S.; Valenti, S.; Mikolasek, M.; Chumakov, A.I.; Monaco, G. A microscopic look at the Johari-Goldstein relaxation in a hydrogen-bonded glass-former. Sci. Rep. 2019, 9, 14319. [Google Scholar] [CrossRef] [Green Version]
- Richter, D.; Monkenbusch, M.; Arbe, A.; Colmenero, J.; Farago, B. Dynamic structure factors due to relaxation processes in glass-forming polymers. Phys. B: Condensed Matter 1997, 241, 1005–1012. [Google Scholar] [CrossRef]
- Tölle, A. Neutron scattering studies of the model glass former ortho-terphenyl. Rep. Prog. Phys. 2001, 64, 1473–1532. [Google Scholar] [CrossRef] [Green Version]
- Fragiadakis, D.; Roland, C.M. Dynamic correlations and heterogeneity in the primary and secondary relaxations of a model molecular liquid. Phys. Rev. E 2014, 89, 052304. [Google Scholar] [CrossRef] [Green Version]
- Scopigno, T.; Ruocco, G.; Sette, F.; Monaco, G. Is the Fragility of a Liquid Embedded in the Properties of Its Glass? Science 2003, 302, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Scopigno, D.C.; Ruocco, G. Universal relation between viscous flow and fast dynamics in glass-forming materials. Phys. Rev. B 2010, 81, 100202. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripodo, A.; Puosi, F.; Malvaldi, M.; Capaccioli, S.; Leporini, D. Coincident Correlation between Vibrational Dynamics and Primary Relaxation of Polymers with Strong or Weak Johari-Goldstein Relaxation. Polymers 2020, 12, 761. https://doi.org/10.3390/polym12040761
Tripodo A, Puosi F, Malvaldi M, Capaccioli S, Leporini D. Coincident Correlation between Vibrational Dynamics and Primary Relaxation of Polymers with Strong or Weak Johari-Goldstein Relaxation. Polymers. 2020; 12(4):761. https://doi.org/10.3390/polym12040761
Chicago/Turabian StyleTripodo, Antonio, Francesco Puosi, Marco Malvaldi, Simone Capaccioli, and Dino Leporini. 2020. "Coincident Correlation between Vibrational Dynamics and Primary Relaxation of Polymers with Strong or Weak Johari-Goldstein Relaxation" Polymers 12, no. 4: 761. https://doi.org/10.3390/polym12040761