Polymer Conformations, Entanglements and Dynamics in Ionic Nanocomposites: A Molecular Dynamics Study

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Methodology
3. Results and Discussion
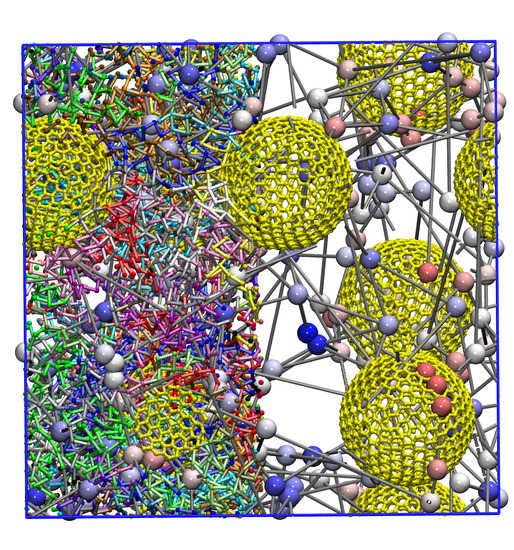
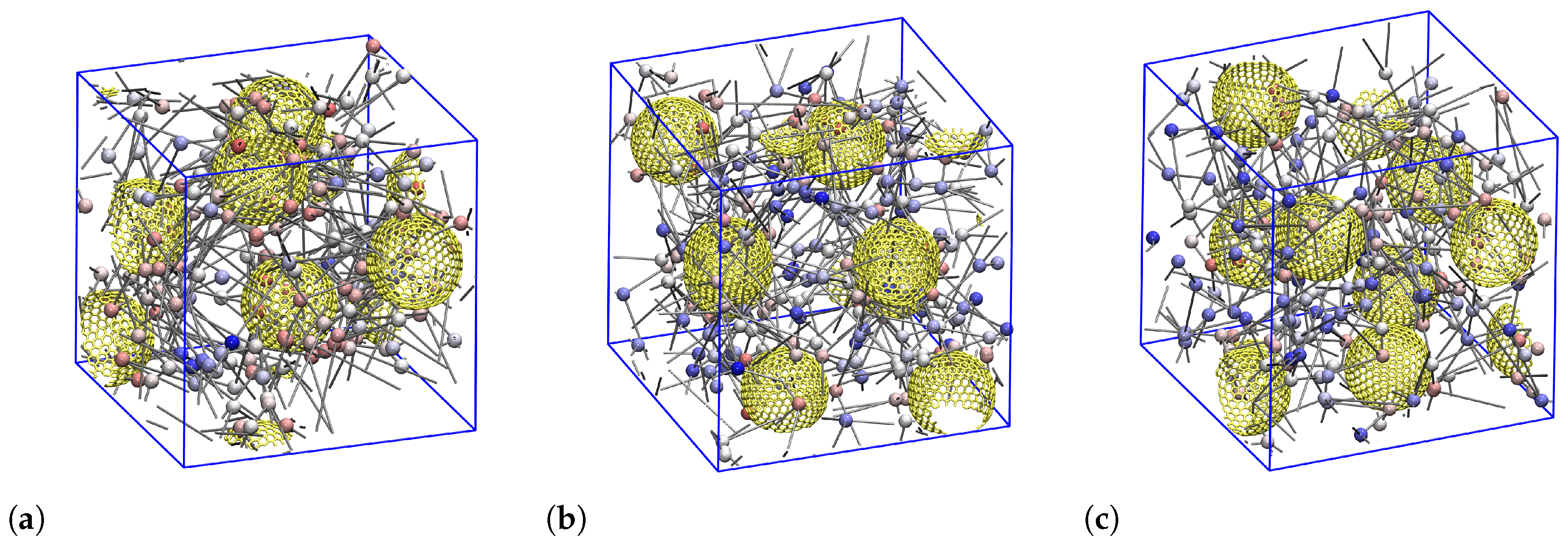
3.1. Nanoparticle and Polymer Structure
3.2. Polymer Dimensions
3.3. Chain Relaxation and Nanoparticle Mobility
3.4. Polymer–Nanoparticle Temporary Crosslinks
3.5. Entanglement Network
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crosby, A.J.; Lee, J.Y. Polymer nanocomposites: The nano effect on mechanical properties. Polym. Rev. 2007, 47, 217–229. [Google Scholar] [CrossRef]
- Hu, H.; Onyebueke, L.; Abatan, A. Characterizing and modeling mechanical properties of nanocomposites. Review and evaluation. J. Miner. Mater. Charact. Eng. 2010, 9, 275–319. [Google Scholar] [CrossRef]
- Suvorova, Y.V.; Alekseeva, S.I.; Fronya, M.A.; Viktorova, I.V. Investigations of physical and mechanical properties of polymeric nanocomposites (Review). Inorg. Mater. 2013, 49, 1357–1368. [Google Scholar] [CrossRef]
- Rong, M.Z.; Zhang, M.Q.; Liu, H.; Zeng, H.; Wetzel, B.; Friedrich, K. Microstructure and tribological behavior of polymeric nanocomposites. Indust. Lubric. Tribol. 2001, 53, 72–77. [Google Scholar] [CrossRef]
- Clancy, T.C.; Frankland, S.J.V.; Hinkley, J.A.; Gates, T.S. Multiscale modeling of thermal conductivity of polymer/carbon nanocomposites. Int. J. Therm. Sci. 2010, 49, 1555–1560. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, V.; Jayaraman, A. Theory and simulation studies of effective interactions, phase behavior and morphology in polymer nanocomposites. Soft Matter 2014, 10, 13–38. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Lin, Q.; Jiang, P.; Li, Y.; Li, J. Ionic liquids incorporating polyamide 6: Miscibility and physical properties. Polymers 2018, 10, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supova, M.; Martynkova, G.S.; Grazina, S.; Barabaszova, K. Effect of nanofillers dispersion in polymer matrices: A review. Sci. Adv. Mater. 2011, 3, 425–431. [Google Scholar] [CrossRef]
- Mackay, M.E.; Tuteja, A.; Duxbury, P.M.; Hawker, C.J.; Van Horn, B.; Guan, Z.; Chen, G.; Krishnan, R.S. General strategies for nanoparticle dispersion. Science 2006, 311, 1740. [Google Scholar] [CrossRef]
- Ferdous, S.F.; Sarker, M.F.; Adnan, A. Role of nanoparticle dispersion and filler-matrix interface on the matrix dominated failure of rigid C60-PE nanocomposites: A molecular dynamics simulation study. Polymer 2013, 54, 2565–2576. [Google Scholar] [CrossRef]
- Cao, X.Z.; Merlitz, H.; Wu, C.X.; Ungar, G.; Sommer, J.U. A theoretical study of dispersion-to-aggregation of nanoparticles in adsorbing polymers using molecular dynamics simulations. Nanoscale 2016, 8, 6964–6968. [Google Scholar] [CrossRef] [Green Version]
- Jouault, N.; Dalmas, F.; Boue, F.; Jestin, J. Multiscale characterization of filler dispersion and origins of mechanical reinforcement in model nanocomposites. Polymer 2012, 53, 761–775. [Google Scholar] [CrossRef]
- Winey, K.I.; Kashiwagi, T.; Mu, M. Improving electrical conductivity and thermal properties of polymers by the addition of carbon nanotubes as fillers. MRS Bull. 2007, 32, 348–353. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Clarke, N. Primitive path network, structure and dynamics of SWCNT/polymer nanocomposites. IOP Conf. Ser. Mat. Sci. Eng. 2012, 40, 012027. [Google Scholar] [CrossRef] [Green Version]
- Samir, E.; Salah, M.; Hajjiah, A.; Shehata, N.; Fathy, M.; Hamed, A. Electrospun PVA polymer embedded with ceria nanoparticles as silicon solar cells rear surface coaters for efficiency improvement. Polymers 2018, 10, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatrantos, A.; Clarke, N.; Kröger, M. Modeling of polymer structure and conformations in polymer nanocomposites from atomistic to mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Jankar, J.; Douglas, J.F.; Starr, F.W.; Kumar, S.K.; Cassagnau, P.; Lesser, A.J.; Sternstein, S.S.; Buehler, M.J. Current issues in research on structure property relationships in polymer nanocomposites. Polymer 2010, 51, 3321–3343. [Google Scholar] [CrossRef]
- Fernandes, N.J.; Akbarzadeh, J.; Peterlik, H.; Giannelis, E.P. Synthesis and properties of highly dispersed ionic silica-poly(ethylene oxide) nanohybrids. ACS Nano 2013, 7, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, N.J.; Koerner, H.; Giannelis, E.P.; Vaia, R.A. Hairy nanoparticle assemblies as one-component functional polymer nanocomposites: Opportunities and challenges. MRS Commun. 2013, 3, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Relinque, J.J.; de Leon, A.S.; Hernandez-Saz, J.; Garcia-Romero, M.G.; Navas-Martos, F.J.; Morales-Cid, G.; Molina, S.I. Development of surface-coated polylactic acid/polyhydroxyalkanoate (PLA/PHA) nanocomposites. Polymers 2019, 11, 400. [Google Scholar] [CrossRef] [Green Version]
- Odent, J.; Raquez, J.M.; Dubois, P.; Giannelis, E.P. Ultra-stretchable ionic nanocomposites: From dynamic bonding to multi-responsive behaviors. J. Mater. Chem. A 2017, 5, 13357–13363. [Google Scholar] [CrossRef]
- Odent, J.; Raquez, J.M.; Samuel, C.; Barrau, S.; Enotiadis, A.; Dubois, P.; Giannelis, E.P. Shape-memory behavior of polylactide/silica ionic hybrids. Macromolecules 2017, 50, 2896–2905. [Google Scholar] [CrossRef]
- Potaufeux, J.E.; Odent, J.; Notta-Cuvier, D.; Delille, R.; Barrau, S.; Giannelis, E.P.; Lauro, F.; Raquez, J.M. Mechanistic insights on ultra-tough polylactide-based ionic nanocomposites. Compos. Sci. Technol. 2020, 191, 108075. [Google Scholar] [CrossRef]
- Donato, K.Z.; Matejka, L.; Mauler, R.S.; Donato, R.K. Recent applications of ionic liquids in the sol-gel process for polymer-silica nanocomposites with ionic interfaces. Colloids Interfaces 2017, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.H.; Wang, D.; Wang, Q.M. Tough and self-healable nanocomposite hydrogels for repeatable water treatment. Polymers 2018, 10, 880. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.T.; Fang, L.; Chen, T.Q.; Sun, M.L.; Lu, C.H.; Xu, Z.Z. Near-infrared light and solar light activated self-healing epoxy coating having enhanced properties using MXene flakes as multifunctional fillers. Polymers 2018, 10, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänninen, A.; Sarlin, E.; Lyyra, I.; Salpavaara, T.; Kellomäki, M.; Tuukkanen, S. Nanocellulose and chitosan based films as low cost, green piezoelectric materials. Carbohyd. Polym. 2018, 202, 418–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz de Luzuriaga, A.; Matxain, J.M.; Ruipérez, F.; Martin, R.; Asua, J.M.; Cabanero, G.; Odriozola, I. Transient mechanochromism in epoxy vitrimer composites containing aromatic disulfide crosslinks. J. Mater. Chem. C 2016, 4, 6220–6223. [Google Scholar] [CrossRef]
- Sessini, V.; Brox, D.; Lopez, A.J.; Urena, A.; Peroni, L. Thermally activated shape memory behavior of copolymers based on ethylene reinforced with silica nanoparticles. Nanocomposites 2018, 4, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Li, Y.; Wang, X.; Li, X.; Sun, J. Thermally and near-infrared light-induced shape memory polymers capable of healing mechanical damage and fatigued shape memory function. ACS Appl. Mater. Interfaces 2019, 11, 9470–9477. [Google Scholar] [CrossRef]
- Wei, T.; Tang, Z.; Yu, Q.; Chen, H. Smart antibacterial surfaces with switchable bacteria-killing and bacteria-releasing capabilities. ACS Appl. Mater. Interfaces 2017, 9, 37511–37523. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, A.; Tan, Y.; Wang, N.; Yu, P. Property-structure relationship of titania ionic liquid nanofluids. Soft Mater. 2013, 11, 315–320. [Google Scholar] [CrossRef]
- Yang, S.; Tan, Y.; Yin, X.; Chen, S.; Chen, D.; Wang, L.; Zhou, Y.; Xiong, C. Preparation and characterization of monodisperse solvent-free silica nanofluids. J. Dispers. Sci. Technol. 2016, 38, 425–431. [Google Scholar] [CrossRef]
- Hong, B.; Chremos, A.; Panagiotopoulos, A.Z. Simulations of the structure and dynamics of nanoparticle-based ionic liquids. Faraday Discuss. 2012, 154, 29–40. [Google Scholar] [CrossRef]
- Hong, B.; Panagiotopoulos, A.Z. Diffusivities, viscosities, and conductivities of solvent-free ionically grafted nanoparticles. Soft Matter 2013, 9, 6091–6102. [Google Scholar] [CrossRef]
- Babayekhorasani, F.; Dunstan, D.E.; Krishnamoorti, R.; Conrad, J.C. Nanoparticle diffusion in crowded and confined media. Soft Matter 2016, 12, 8407–8416. [Google Scholar] [CrossRef]
- Nath, P.; Mangal, R.; Kohle, F.; Choudhury, S.; Narayanan, S.; Wiesner, U.; Archer, L.A. Dynamics of nanoparticles in entangled polymer solutions. Langmuir 2018, 34, 241–249. [Google Scholar] [CrossRef]
- Poling-Skutvik, R.; Krishnamoorti, R.; Conrad, J.C. Size-dependent dynamics of nanoparticles in unentangled polyelectrolyte solutions. ACS Macro Lett. 2015, 4, 1169–1173. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Kröger, M.; Clarke, N. Modeling of entangled polymer diffusion in melts and nanocomposites: A Review. Polymers 2019, 11, 876. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Nguyen, T.D.; Pryamitsyn, V.A.; de la Cruz, M.O. Ionic correlations in random ionomers. ACS Nano 2018, 12, 2311–2318. [Google Scholar] [CrossRef]
- Ting, C.L.; Sorensen-Unruh, K.E.; Stevens, M.J.; Frischknecht, A.L. Nonequilibrium simulations of model ionomers in an oscillating electric field. J. Chem. Phys. 2016, 145, 044902. [Google Scholar] [CrossRef]
- Wu, S.; Xiao, C.; Zhang, Z.; Chen, Q.; Matsumiya, Y.; Watanabe, H. Molecular design of highly stretchable ionomers. Macromolecules 2018, 51, 4735–4746. [Google Scholar] [CrossRef]
- Sampath, J.; Hall, L. Influence of a nanoparticle on the structure and dynamics of model ionomer melts. Soft Matter 2018, 14, 4621–4632. [Google Scholar] [CrossRef] [PubMed]
- Volgin, I.G.; Larin, S.V.; Lyulin, A.V.; Lyulin, S.V. Coarse-grained molecular-dynamics simulations of nanoparticle diffusion in polymer nanocomposites. Polymer 2018, 145, 80–87. [Google Scholar] [CrossRef] [Green Version]
- David, A.; Pasquini, M.; Tartaglino, U.; Raos, G. A coarse-grained force field for silica–polybutadiene interfaces and nanocomposites. Polymers 2020, 12, 1484. [Google Scholar] [CrossRef]
- Brini, E.; Algaer, E.A.; Ganguly, P.; Li, C.; Rodríguez-Ropero, F.; van der Vegt, N. Systematic coarse-graining methods for soft matter simulations—A review. Soft Matter 2013, 9, 2108–2119. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057. [Google Scholar] [CrossRef]
- Kröger, M.; Hess, S. Rheological evidence for a dynamical crossover in polymer melts via nonequilibrium molecular dynamics. Phys. Rev. Lett. 2000, 85, 1128–1131. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press Inc.: New York, NY, USA, 2003. [Google Scholar]
- Miwatani, R.; Takahashi, K.Z.; Arai, N. Performance of coarse graining in estimating polymer properties: Comparison with the atomistic model. Polymers 2020, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Warner, H.R. Kinetic theory and rheology of dilute suspensions of finitely extendible dumbbells. Ind. Eng. Chem. Fund. 1972, 11, 379–387. [Google Scholar] [CrossRef]
- Kröger, M.; Loose, W.; Hess, S. Structural changes and rheology of polymer melts via nonequilibrium molecular dynamics. J. Rheol. 1993, 37, 1057–1079. [Google Scholar] [CrossRef]
- Hagita, K.; Murashima, T.; Iwaoka, N. Thinning approximation for calculating two-dimensional scattering patterns in dissipative particle dynamics simulations under shear flow. Polymers 2018, 10, 1224. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Uehara, E. Statistical and dynamical properties of topological polymers with graphs and ring polymers with knots. Polymers 2017, 9, 252. [Google Scholar] [CrossRef]
- Kröger, M. Simple, admissible, and accurate approximants of the inverse Langevin and Brillouin functions, relevant for strong polymer deformations and flows. J. Non-Newtonian Fluid Mech. 2015, 223, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Moghimikheirabadi, A.; Sagis, L.M.C.; Kröger, M.; Ilg, P. Gas–liquid phase equilibrium of a model Langmuir monolayer captured by a multiscale approach. Phys. Chem. Chem. Phys. 2019, 21, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Hou, J.X.; Svaneborg, C.; Everaers, R.; Grest, G.S. Stress relaxation in entangled polymer melts. Phys. Rev. Lett. 2010, 105, 068301. [Google Scholar] [CrossRef] [Green Version]
- Hoy, R.S.; Foteinopoulou, K.; Kröger, M. Topological analysis of polymeric melts: Chain-length effects and fast-converging estimators for entanglement length. Phys. Rev. E 2009, 80, 031803. [Google Scholar] [CrossRef] [Green Version]
- Norris, A.N.; Sheng, P.; Callegari, A.J. Effective medium theories for two-phase dielectric medium. J. Appl. Phys. 1985, 57, 1990. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, X.; Wu, K.; Wu, S.; Chen, Y.; Meng, Y. Modeling and simulation for effective permittivity of two-phase disordered composites. J. Appl. Phys. 2008, 103, 034111. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Hagita, K.; Morita, H.; Doi, M.; Takano, H. Coarse-grained molecular dynamics simulation of filled polymer nanocomposites under uniaxial elongation. Macromolecules 2016, 49, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Hagita, K.; Morita, H.; Takano, H. Molecular dynamics simulation study of a fracture of filler-filled polymer nanocomposites. Polymer 2016, 99, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Polymer conformations in polymer nanocomposites containing spherical nanoparticles. Soft Matter 2015, 11, 382. [Google Scholar] [CrossRef] [Green Version]
- Moghimikheirabadi, A.; Sagis, L.M.; Ilg, P. Effective interaction potentials for model amphiphilic surfactants adsorbed at fluid–fluid interfaces. Phys. Chem. Chem. Phys. 2018, 20, 16238–16246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Archer, L.A. Poly(ethylene oxide)/silica nanocomposites: Structure and rheology. Langmuir 2002, 18, 10435–10442. [Google Scholar] [CrossRef]
- Koizumi, N.; Hanai, T. Dielectric properties of polyethylene glycols: Dielectric relaxation in solid state. Bull. Inst. Chem. Res. 1964, 42, 115–127. [Google Scholar]
- Porter, C.; Boyd, R. A dielectric study of the effects of melting on molecular relaxation in poly(ethylene oxide) and polyoxymethylene. Macromolecules 1971, 4, 589–594. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte solutions I. Colligative properties. J. Chem. Phys. 1969, 51, 924. [Google Scholar] [CrossRef]
- Crawford, M.K.; Smalley, R.J.; Cohen, G.; Hogan, B.; Wood, B.; Kumar, S.K.; Melnichenko, Y.B.; He, L.; Guise, W.; Hammouda, B. Chain conformation in polymer nanocomposites with uniformly dispersed nanoparticles. Phys. Rev. Lett. 2013, 110, 196001. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, A.; Duxbury, P.M.; Mackay, M.E. Polymer chain swelling induced by dispersed nanoparticles. Phys. Rev. Lett. 2008, 100, 077801. [Google Scholar] [CrossRef] [PubMed]
- Karatrantos, A.; Koutsawa, Y.; Dubois, P.; Clarke, N.; Kröger, M. Miscibility and diffusion in ionic nanocomposites. Polymers 2018, 10, 1010. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Xie, Y.; Kumar, S.K.; Yang, H.; Bansal, A.; Ho, D.L.; Hall, L.; Hooper, J.B.; Schweizer, K.S. Chain conformations and bound-layer correlations in polymer nanocomposites. Phys. Rev. Lett. 2007, 98, 128302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouault, N.; Dalmas, F.; Said, S.; Schweins, R.; Jestin, J.; Boue, F. Direct measurement of polymer chain conformation in well-controlled model nanocomposites by combining SANS and SAXS. Macromolecules 2010, 43, 9881–9891. [Google Scholar] [CrossRef]
- Jouault, N.; Crawford, M.K.; Chi, C.; Smalley, R.J.; Wood, B.; Jestin, J.; Melnichenko, Y.B.; He, L.; Guise, W.E.; Kumar, S.K. Polymer chain behavior in polymer nanocomposites with attractive interactions. ACS Macro Lett. 2016, 5, 523–527. [Google Scholar] [CrossRef]
- Jouault, N.; Kumar, S.K.; Smalley, R.J.; Chi, C.; Moneta, R.; Wood, B.; Salerno, H.; Melnichenko, Y.B.; He, L.; Guise, W.E.; et al. Do very small POSS nanoparticles perturb s–PMMA chain conformations? Macromolecules 2018, 51, 5278–5293. [Google Scholar] [CrossRef]
- Moghimikheirabadi, A.; Ilg, P.; Sagis, L.M.C.; Kröger, M. Surface rheology and structure of model triblock copolymers at a liquid–vapor interface: A molecular dynamics study. Macromolecules 2020, 53, 1245–1257. [Google Scholar] [CrossRef]
- Sagis, L.M.C.; Liu, B.; Li, Y.; Essers, J.; Yang, J.; Moghimikheirabadi, A.; Hinderink, E.; Berton-Carabin, C.; Schroen, K. Dynamic heterogeneity in complex interfaces of soft interface-dominated materials. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Structure, entanglements and dynamics of polymer nanocomposites containing spherical nanoparticles. IOP Conf. Ser. Mat. Sci. Eng. 2014, 64, 012041. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Clarke, N. Polymer and spherical nanoparticle diffusion in nanocomposites. J. Chem. Phys. 2017, 146, 203331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.C.; Gam, S.; Meth, J.S.; Clarke, N.; Winey, K.I.; Composto, R.J. Do attractive polymer-nanoparticle interactions retard polymer diffusion in nanocomposites. Macromolecules 2013, 46, 4502–4509. [Google Scholar] [CrossRef]
- Holt, A.P.; Griffin, P.J.; Bocharova, V.; Agapov, A.L.; Imel, A.E.; Dadmun, M.D.; Sangoro, J.R.; Sokolov, A.P. Dynamics at the polymer/nanoparticle interface in poly(2-vinylpyridine)/silica nanocomposites. Macromolecules 2014, 47, 1837–1843. [Google Scholar] [CrossRef]
- Jouault, N.; Moll, J.F.; Meng, D.; Windsor, K.; Ramcharan, S.; Kearney, C.; Kumar, S.K. Bound polymer layers in nanocomposites. ACS Macro Lett. 2013, 2, 371–374. [Google Scholar] [CrossRef]
- Kröger, M. Shortest multiple disconnected path for the analysis of entanglements in two- and three-dimensional polymeric systems. Comput. Phys. Commun. 2005, 168, 209–232. [Google Scholar] [CrossRef]
- Li, Y.; Kröger, M.; Liu, W.K. Nanoparticle effect on the dynamics of polymer chains and their entanglement network. Phys. Rev. Lett. 2012, 109, 118001. [Google Scholar] [CrossRef] [Green Version]
- Toepperwein, G.N.; Karayiannis, N.C.; Riggleman, R.A.; Kröger, M.; de Pablo, J.J. Influence of nanorod inclusions on structure and primitive path network of polymer nanocomposites at equilibrium and under deformation. Macromolecules 2011, 44, 1034. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Composto, R.J.; Winey, K.I. Entanglements in polymer nanocomposites containing spherical nanoparticles. Soft Matter 2016, 12, 2567. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, M.F.; Phillips, J.C. (Eds.) Phase Transitions and Self-Organization in Electronic and Molecular Networks; Springer: Boston, MA, USA, 2001. [Google Scholar]
- Toepperwein, G.N.; Riggleman, R.A.; de Pablo, J.J. Dynamics and deformation response of rod-containing nanocomposites. Macromolecules 2011, 45, 543–554. [Google Scholar] [CrossRef]
- Schneider, G.J.; Nusser, K.; Willner, L.; Falus, P.; Richter, D. Dynamics of entangled chains in polymer nanocomposites. Macromolecules 2011, 44, 5857–5860. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length | Energy | Mass | Charge | Time | Pressure | Temperature | Spring Coefficient |
|---|---|---|---|---|---|---|---|
| m |
| Charge | Oligomer | Polymer | Polymer | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | Type | n | :: | n | :: | n | :: | ||||||
| 6.2% | ends | 3 | 270 | 180 | ✓ | 4 | 144 | 72 | ✓ | 4 | 72 | 36 | ✓ |
| 6.2% | p3 | 3 | 270 | 1260 | ✓ | 4 | 144 | 1224 | ✓ | 4 | 72 | 1206 | ✓ |
| 11.6% | ends | 6 | 270 | 90 | ✓ | 12 | 216 | 36 | ✓ | 12 | 108 | 18 | ✓ |
| 11.6% | p3 | 6 | 270 | 630 | ✓ | 8 | 144 | 612 | ✓ | 8 | 72 | 603 | ✓ |
| 20.8% | ends | 12 | 270 | 45 | ✓ | 24 | 216 | 18 | × | 24 | 108 | 9 | × |
| 20.8% | p3 | 12 | 270 | 315 | ✓ | 16 | 144 | 306 | ✓ | 16 | 72 | 306 | ✓ |
| 28.3% | ends | 18 | 270 | 30 | ✓ | 36 | 216 | 12 | × | 36 | 108 | 6 | × |
| 28.3% | p3 | 18 | 270 | 210 | ✓ | 24 | 144 | 204 | ✓ | 24 | 72 | 201 | ✓ |
| 37.1% | ends | 27 | 270 | 20 | × | 36 | 144 | 8 | × | 36 | 72 | 4 | × |
| 37.1% | p3 | 27 | 270 | 140 | ✓ | 36 | 144 | 136 | ✓ | 36 | 72 | 134 | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moghimikheirabadi, A.; Mugemana, C.; Kröger, M.; Karatrantos, A.V. Polymer Conformations, Entanglements and Dynamics in Ionic Nanocomposites: A Molecular Dynamics Study. Polymers 2020, 12, 2591. https://doi.org/10.3390/polym12112591
Moghimikheirabadi A, Mugemana C, Kröger M, Karatrantos AV. Polymer Conformations, Entanglements and Dynamics in Ionic Nanocomposites: A Molecular Dynamics Study. Polymers. 2020; 12(11):2591. https://doi.org/10.3390/polym12112591
Chicago/Turabian StyleMoghimikheirabadi, Ahmad, Clément Mugemana, Martin Kröger, and Argyrios V. Karatrantos. 2020. "Polymer Conformations, Entanglements and Dynamics in Ionic Nanocomposites: A Molecular Dynamics Study" Polymers 12, no. 11: 2591. https://doi.org/10.3390/polym12112591