3.1. Chemical-Physical Properties of GO-1 and GO-2
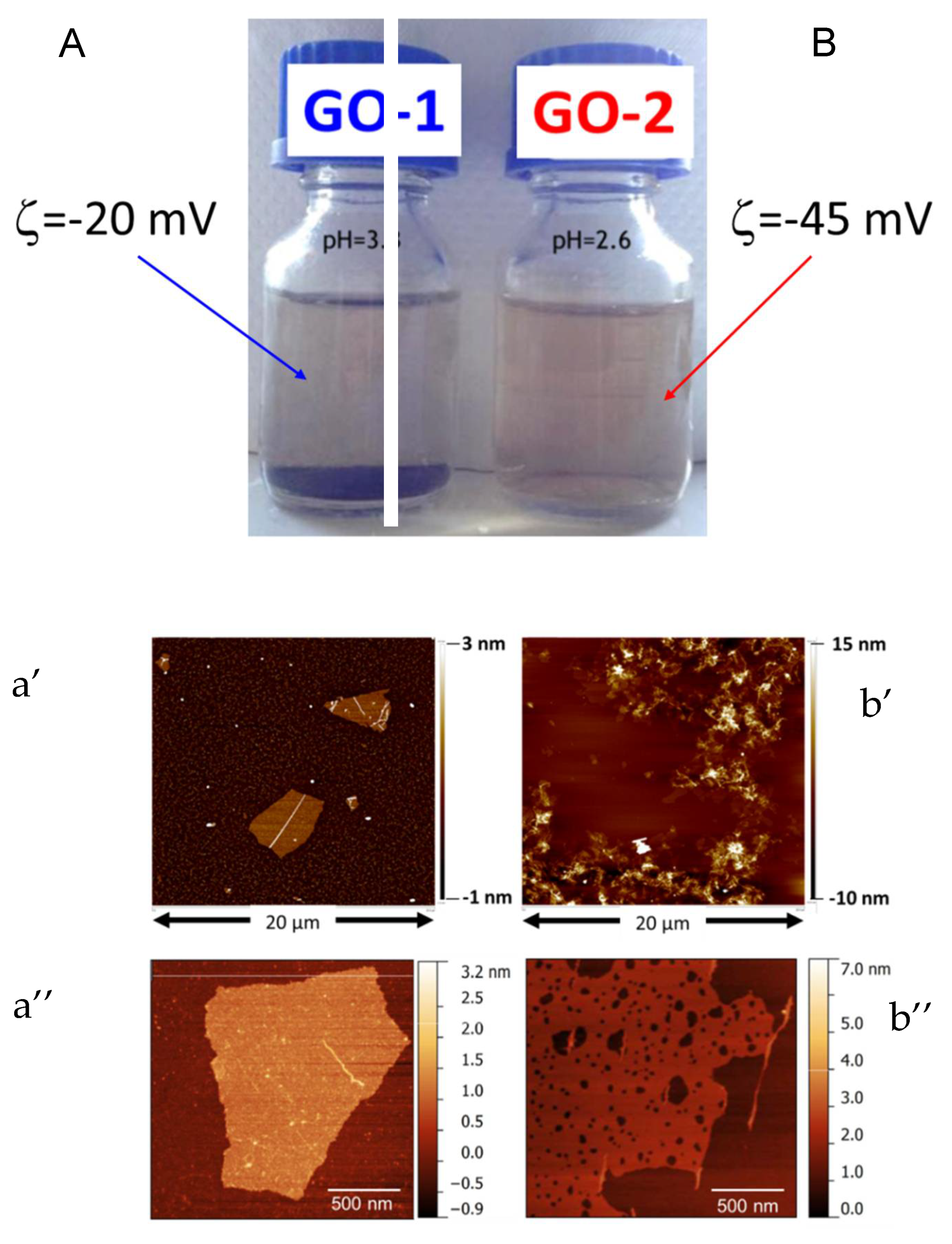
Undoubtedly, the extent of oxidation has strong repercussions on the chemical-physical features of GO. First, a different dispersibility in an aqueous medium was found. GO-1 tends to darken and compact, as a result of progressive dehydration and consequent re-stacking. The highly oxygenated GO-2 retains a light-yellow colour.
In
Figure 1A,B, the dispersions (0.5 mg/l) of GO-1 and GO-2 in water after 10 days are reported, respectively. GO-1 and GO-2 showed dramatic differences from a chemical-physical point of view. The prolonged oxidation provided an impressive, enhanced stability in aqueous suspension over a wide period of time. Moreover, GO-1 (
Figure 1A) required at least 1 h of ultrasonic treatment to achieve good dispersion in water, GO-2 (
Figure 1B) remains in suspension if simply hand-shaken. Furthermore, the two GO samples displayed different acidities, with GO-2 dispersion resulting in a lower pH (2.6) than that of GO-1 (pH = 3.3), being equal to the concentration in water. Zeta potential measurements and AFM observations were performed to provide further insight into the status of lamellae. AFM observation of GO-1 dispersion, provided in
Figure 1a’, revealed that GO-1 lamellae were more regular and smaller sized (
t = 0.7 nm;
D = 5 µm), owing to sonication, whereas GO-2 (
Figure 1b’) displays extremely crumpled and wrinkled lamellae with a larger size distribution, ranging from hundreds of nanometres to several microns. Nevertheless, GO-2 has improved stability in aqueous media and a zeta potential (−45 mV) much more negative than GO-1 (−20 mV). The crumpling could be a direct consequence of the high density of epoxy and lactones moieties in the basal planes, developed within the re-arrangement of the structure during the aging, or due to the presence of oxidation debris. AFM analysis performed at a higher magnification of GO-1 (
Figure 1a’’) and GO-2 (
Figure 1b’’) enabled us to detect the presence of carbonaceous fragments (sub-micrometric sized) in both samples. If, in the case of GO-1, they might have been caused by high sonication times, for the simply hand-shaken GO-2 suspension one can hypothesize that the prolonged oxidation might have chopped up the basal planes of lamellae. As a result, GO-2 possesses an extremely wide lateral size distribution, ranging from 400 nm to 25–30 µm. Herein, we are not interested in explaining whether they are detrimental carbonaceous fragments or oxidative debris.
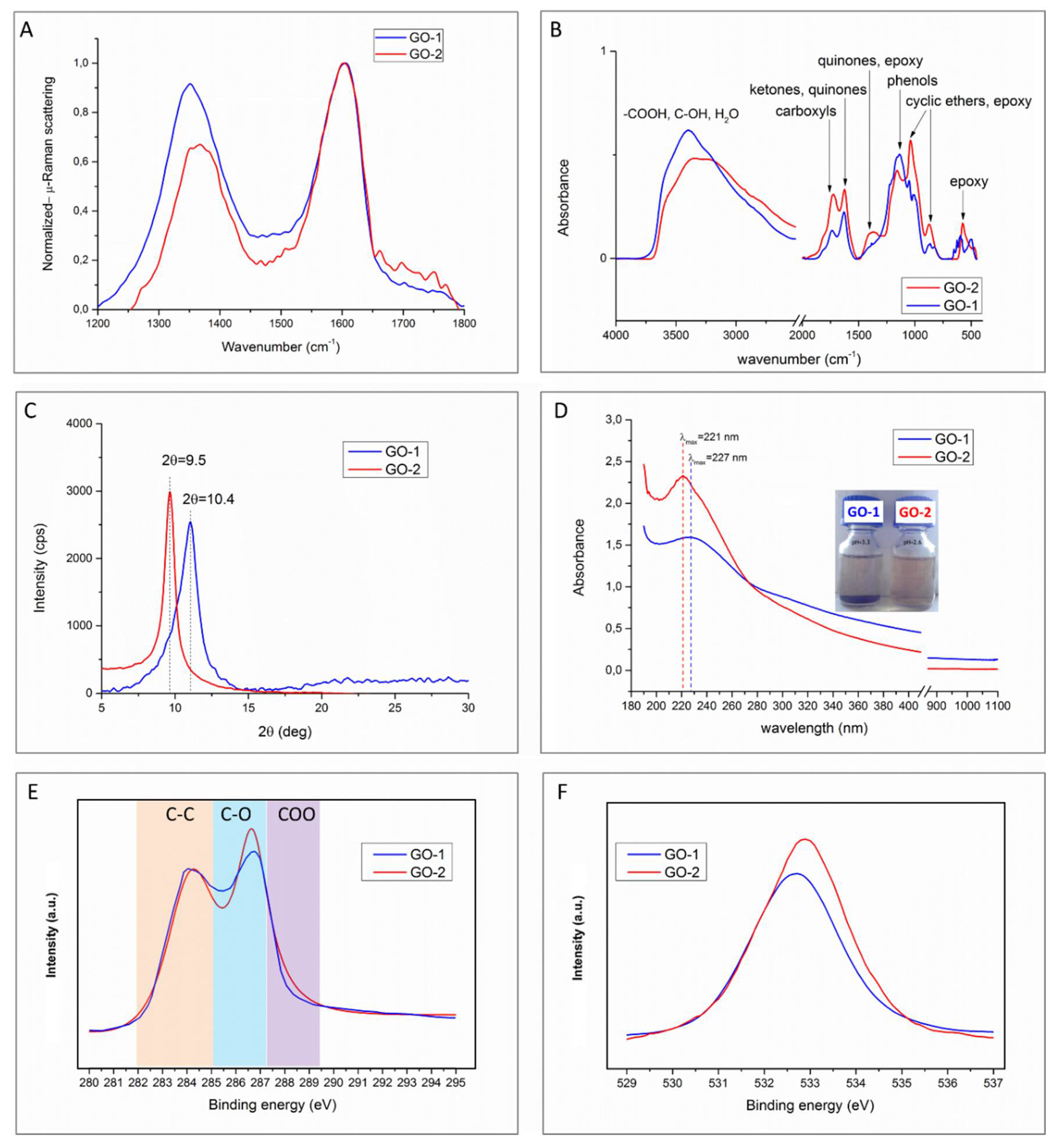
Figure 2A reports μ-Raman plots of GO-1 and GO-2 in the range of 1200–1800 cm
−1, that is, referred to the first order (one phonon) Raman region. The D peak (K point in the Brillouin zone) is located at 1350 cm
−1, whereas the G mode (at point Γ) is usually centred at around 1600 cm
−1. The D-band refers to the sp
3 diamond-like carbons, whereas the G-band is ascribed to the sp
2 hybridized carbons [
21,
22,
23]. The Raman parameters allow the assessment of the oxidation degree and have an intensity ratio between D and G modes (
ID/
IG), which provides information about the size of sp
2 domains and the band located at 1700–1780 cm
−1, which is assigned to non-regular rings (5-7-7-5 Stone Wales defects, 5-8-5 rings resulting from a C di-vacancy, and so on [
9,
24,
25,
26]). By comparing the spectra of GO-1 and GO-2, normalized to G amplitude, strong changes were detected:
ID/
IG ratio for GO-1 is shown to be higher than for GO-2, suggesting that the average size of sp
2 domains dramatically increases in the order GO-1 < GO-2. Moreover, the increased intensity of the bands at around 1700–1780 cm
−1 in GO-2 suggests a higher amount of non-regular rings [
9,
25]. Based on these results, it can be hypothesized that prolonged oxidation in GO-2 involved the amorphous carbon of GO, thus likely resulting in the formation of oxidative debris while preserving its aromaticity [
27,
28,
29,
30].
FTIR analysis of GO-1 and GO-2, reported in
Figure 2B, confirmed that both samples have substantially the same functional groups but in different proportions. The bands related to carboxyl and epoxy moieties, located respectively at 1724 and 1025 cm
−1, were more intense for GO-2. Beyond the little differences observed in the intensity of epoxy and carboxyl signals, they cannot explain the strong differences observed in macroscopic properties, such as stability of suspension and reactivity towards PA6. However, recent studies [
31] put into evidence that the presence of debris interferes with the spectroscopic analysis of GO.
XRD plots are provided in
Figure 2C. GO-1 and GO-2 displayed a broad band centred at 10.4° and 9.5°, respectively, while no diffraction peak at 26° (typical of pristine graphite) was detected. These results put into evidence that both samples underwent a full exfoliation during chemical oxidation and interlamellar spacing was found to be equal to 0.95 nm for GO-1 and 1 nm for GO-2. UV/vis spectra of the samples are provided in
Figure 2D. Both samples display a maximum, which is in the 221–227 nm range, traditionally assigned to the π–π* plasmon peak at the transition of aromatic bonds, as previously reported for GO by several authors [
4,
32,
33]. Furthermore, a shoulder, or at least a broad absorption region, can be detected in the wavelength range of 240–320 nm, likely arising from the overlapping of the modes associated to the absorption of epoxy groups (around 250 nm), to π–π* transition of aromatic C–C bond in partially restored C=C bonds (around 260 nm) [
34], n–π* plasmon peak due to the transition of C=O bonds of the carbonyl groups (around 300 nm) [
4]. This complex, extremely wide distribution is supposed to be due to the contribution of the different functional groups to the microstructural disorder, in agreement with FTIR and Raman spectroscopy results, and it is supposed to affect optical behaviour [
34]. The degree of remaining conjugation can be determined by the λ
max of each UV/vis spectrum. It is observed that higher absorption, related to the π–π* transitions (conjugation), correlates with less energy needed for the electronic transition, which results in higher λ
max. The lower λ
max exhibited by GO-2 suggests that, for an equal amount of each sample, the aromatic carbons of GO-2 are organized in graphenic islands bigger than those of GO-1, thus confirming the
ID/
IG analysis outcome from Raman about the size of the aromatic domains. Furthermore, both GO samples are UV-absorbers, while in the visible range GO-2 display higher transparency than GO-1.
XPS results are provided in
Figure 2E,F. C1s spectra of both samples (
Figure 2E) display a peak centred at 284.5 eV that represents the graphitic carbon skeleton, a band centred at 287 eV, assigned to C–OH, –C–O–C– and a shoulder located at 289.4 eV attributable to –COO– bonds. The spectra, normalized to C–C signal, put into evidence that GO-2 possesses a higher oxygen content. A further confirmation of the higher oxygen content of GO-2 is provided by analysis of the O1s signal (
Figure 2F), which was found to be stronger for the most oxidized GO sample. By comparing these findings with those carried out in FTIR, μ-Raman and UV-vis experiments, we can hypothesize that aging under acidic conditions leads to a considerably lower content of sp
2 domains but larger in size [
35].
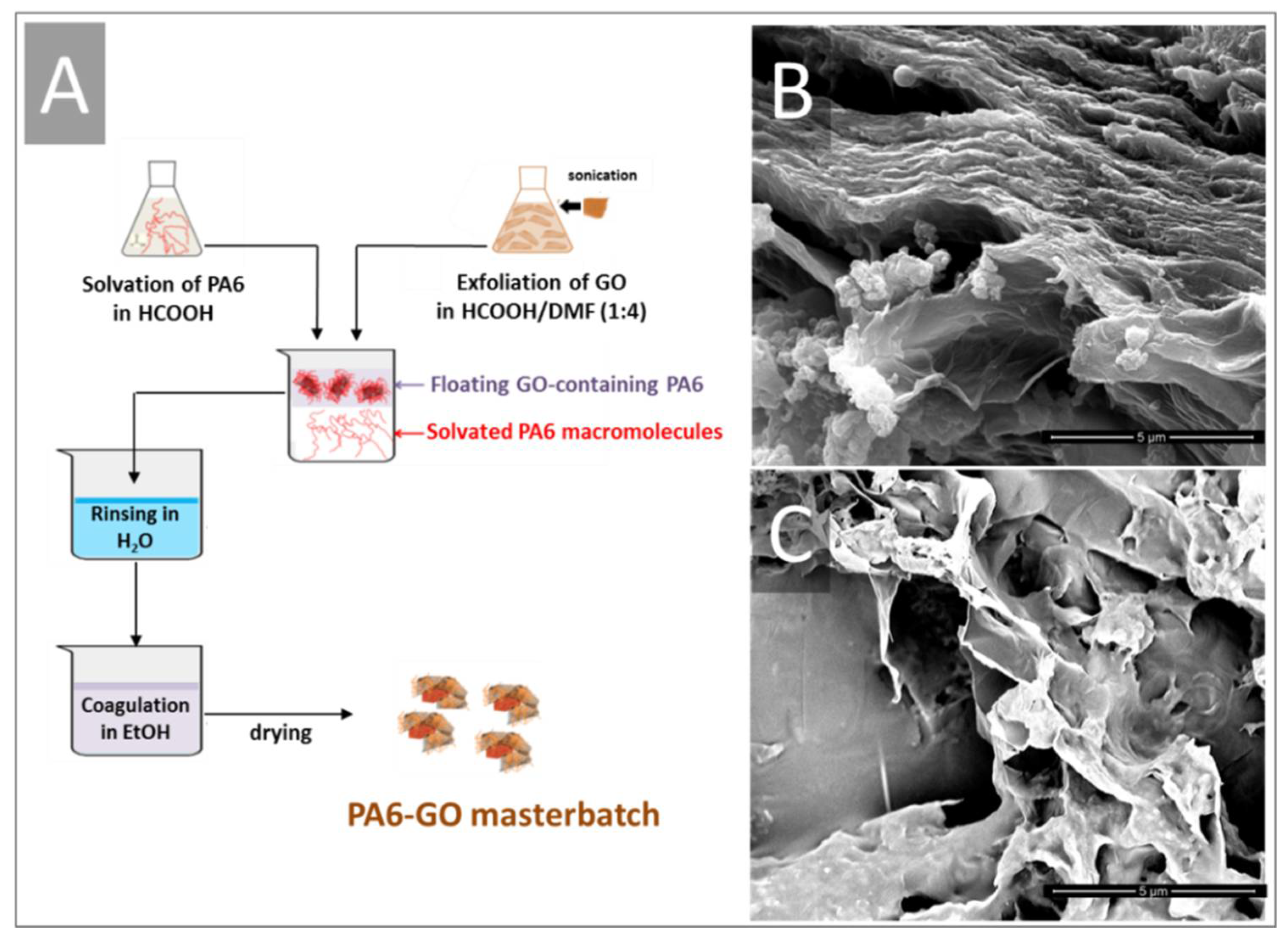
3.2. Chemical-Physical Properties of PA6-GO Nanohybrids
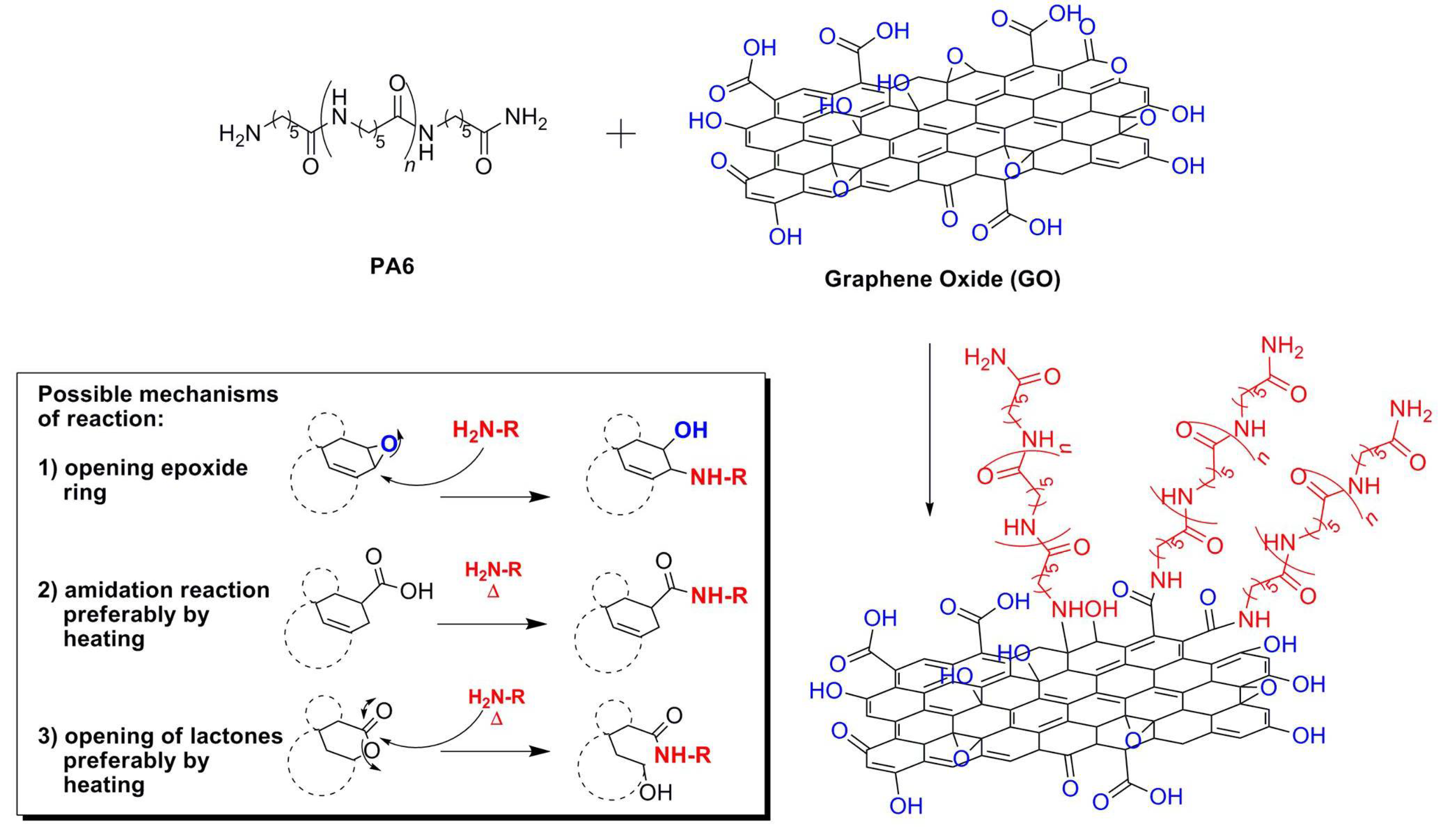
The great differences observed in the two batches of GO obviously affected both the structure and the properties of the PA6-based composites. During the wet phase inversion step, GO-2 showed higher affinity towards PA6, as it is able to drag a higher amount of PA6 into the aqueous phase during the coagulation. When GO-1(0.5 g) is used, the amount of dragged PA6 (1.24 g) is less than that (1.69 g) dragged by GO-2 (see again experimental part). At 60 °C, that is, during wet phase inversion, only epoxy moieties could react by opening the –C–NH– bond of the polymer or by directly reacting with –NH
2 groups. Although it is quite difficult to measure the exact content of epoxy groups prone to react with PA6 moieties, we can suggest that the debris likely play a crucial role. They are supposed to act either as surfactants, thus stabilizing the macromolecules, and/or as nucleophilic agents, thus reacting with the amino and amide groups of the polymer, according to the schematics depicted in
Scheme 2.
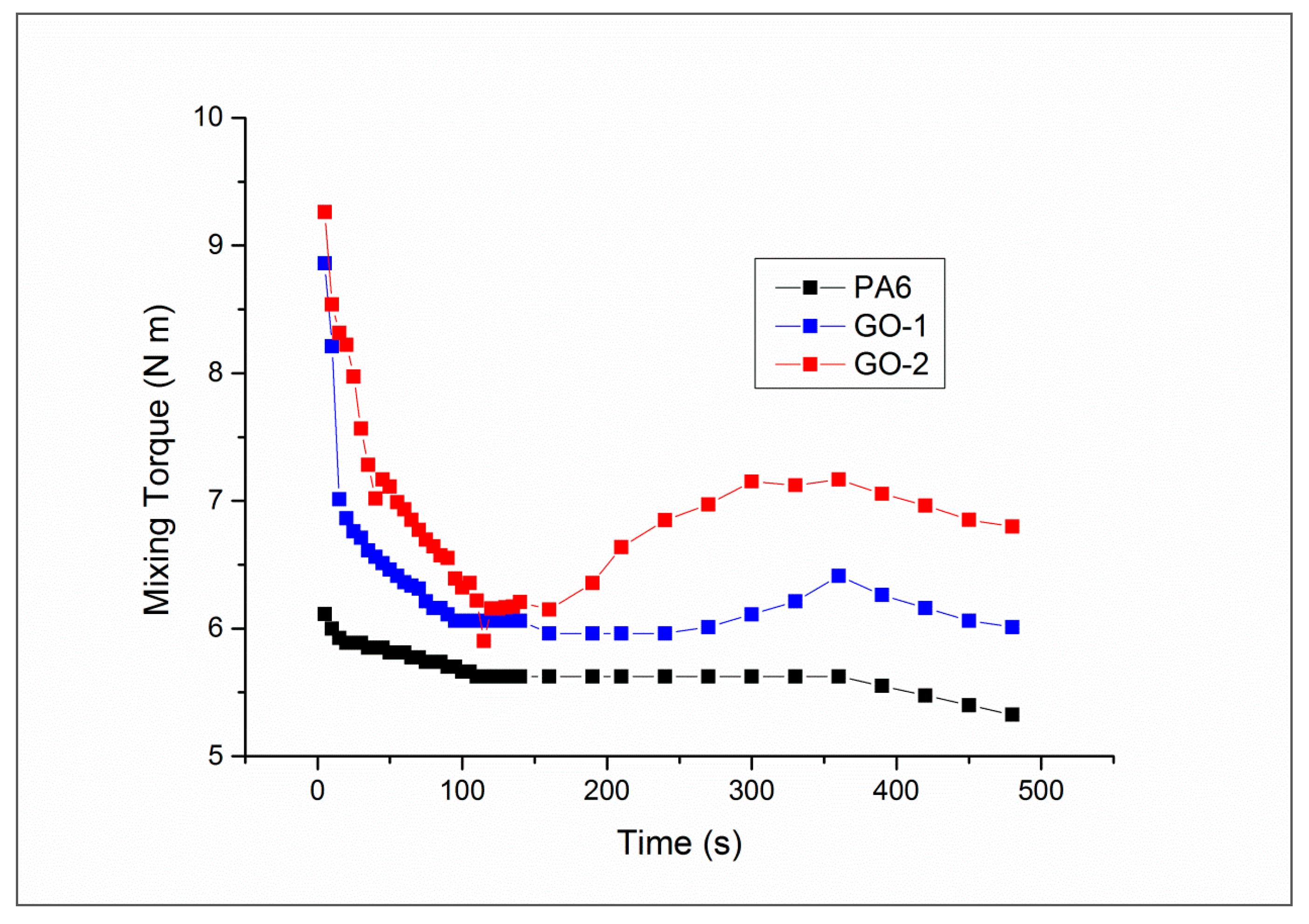
Figure 3 presents the torque as a function of the mixing time of neat PA6 and its composites containing GO-1 and GO-2. As regards PA6, after an initial transitory in which the polymer decreases its viscosity, likely due to thermo-mechanical degradation, the torque achieves a plateau at about
t = 100 s. When GO-1 is added, the torque, the initial value of which is higher than that of pure PA6 due to the presence of the rigid filler, decreases upon increasing mixing time, as observed in most polymer-filler systems investigated [
36,
37,
38], until achieving a constant value of approximately 6 N·m at
t = 150 s. At
t = 240 s, torque starts increasing, achieving a maximum (6.3 N·m) at
t = 360 s. When GO-2 is used, this behaviour appears more pronounced. In fact, after the typical transitory, at
t = 150 s the torque starts increasing in a more remarkable way upon reaching a maximum (7 N·m) at
t = 300 s. The final values of the torque were found to be equal to 5.4, 6.2 and 6.8 N·m for neat matrix, PA6/GO-1 and PA6/GO-2, respectively. The different behavior observed in these materials can be explained by considering the presence of phenomena with opposite repercussions on this property. In fact, thermomechanical degradative phenomena result in chain scissions pathways, which lead to a torque reduction. By contrast, eventual chemical reactions between filler and polymer, as well as filler exfoliation (although to a lesser extent), can lead to an increase of melt viscosity [
13,
14]. In fact, a maximum in mixing torque is typically observed in the case of reactive processing [
13,
14]. In the PA6/GO system, chemical reactions may involve –NH
2 and –COOH end groups of polyamide and epoxy, lactones and carboxyl moieties abundantly present in GO, as schematized in
Scheme 2. Beyond this, we cannot exclude the possibility that debris attach onto PA6 macromolecules, thus acting as bridge-molecules [
31]. By virtue of its higher oxygen content, GO-2 has proven to be more reactive during the melt mixing. In fact, both onset and maximum in the torque plot occur earlier and to a greater extent if compared to the system containing GO-1.
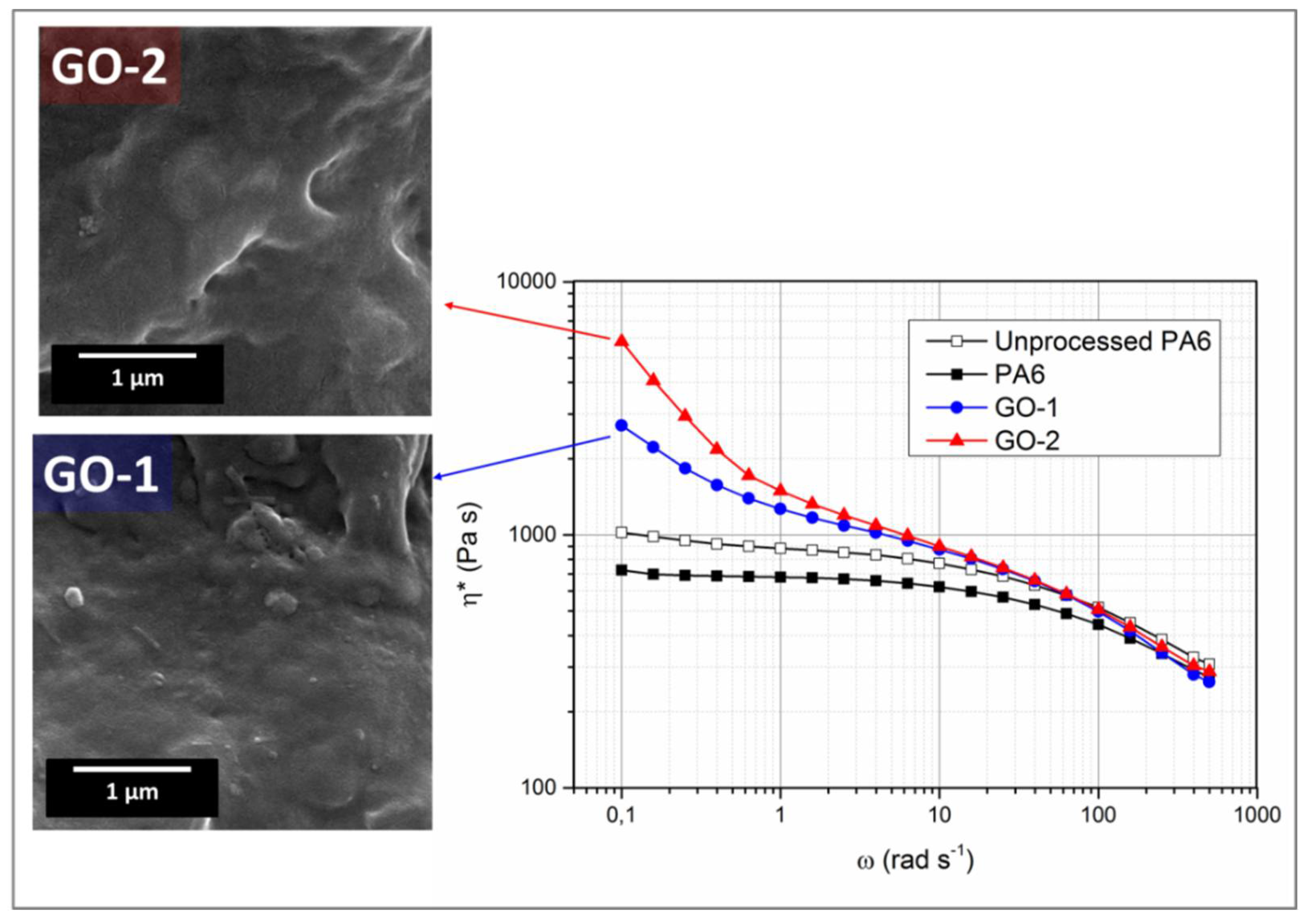
This assumption was corroborated by rheological analysis. The flow curves of nanocomposites prepared with GO-1 and GO-2 are provided in
Figure 4, together with those of unprocessed and melt processed PA6. Both PA6 samples display Newtonian behaviour in the frequency range of 0.1–10 with a slight shear thinning at higher frequencies. However, melt-compounded PA6 shows a viscosity lower than unprocessed polymer, likely suggesting the occurrence of thermomechanical oxidative phenomena due to the long residence time in the mixer. By contrast, the two nanocomposite samples exhibited non-Newtonian behaviour within the whole frequency range investigated and viscosity values higher than those of neat polymer. Notably, the reactive melt mixing in the case of GO-2 results in more pronounced non-Newtonian behaviour and, if the yield stresses in the low frequency region in a nanocomposite system are usually governed by the nanofiller structured in a network [
7,
36], at higher frequencies (usually dominated by the polymer behaviour) the non-Newtonian behaviour persists and the complex viscosity of nanocomposites containing GO-2 was found to be higher than that of PA6-GO-1 system, also confirming the values of final torque discussed before. SEM micrographs of cryofractured samples of PA6/GO-1 and PA6/GO-2, provided in
Figure 4, points out that a high level of matrix-filler adhesion was achieved in both cases, with lamellae totally embedded and surrounded by the polymer matrix. However, the differences observed in terms of rheological behaviour, among nanocomposites containing the same filler amount, suggests that interphase, in terms of interfacial adhesion, filler dispersion and chemical reactions is affected by the type of GO.
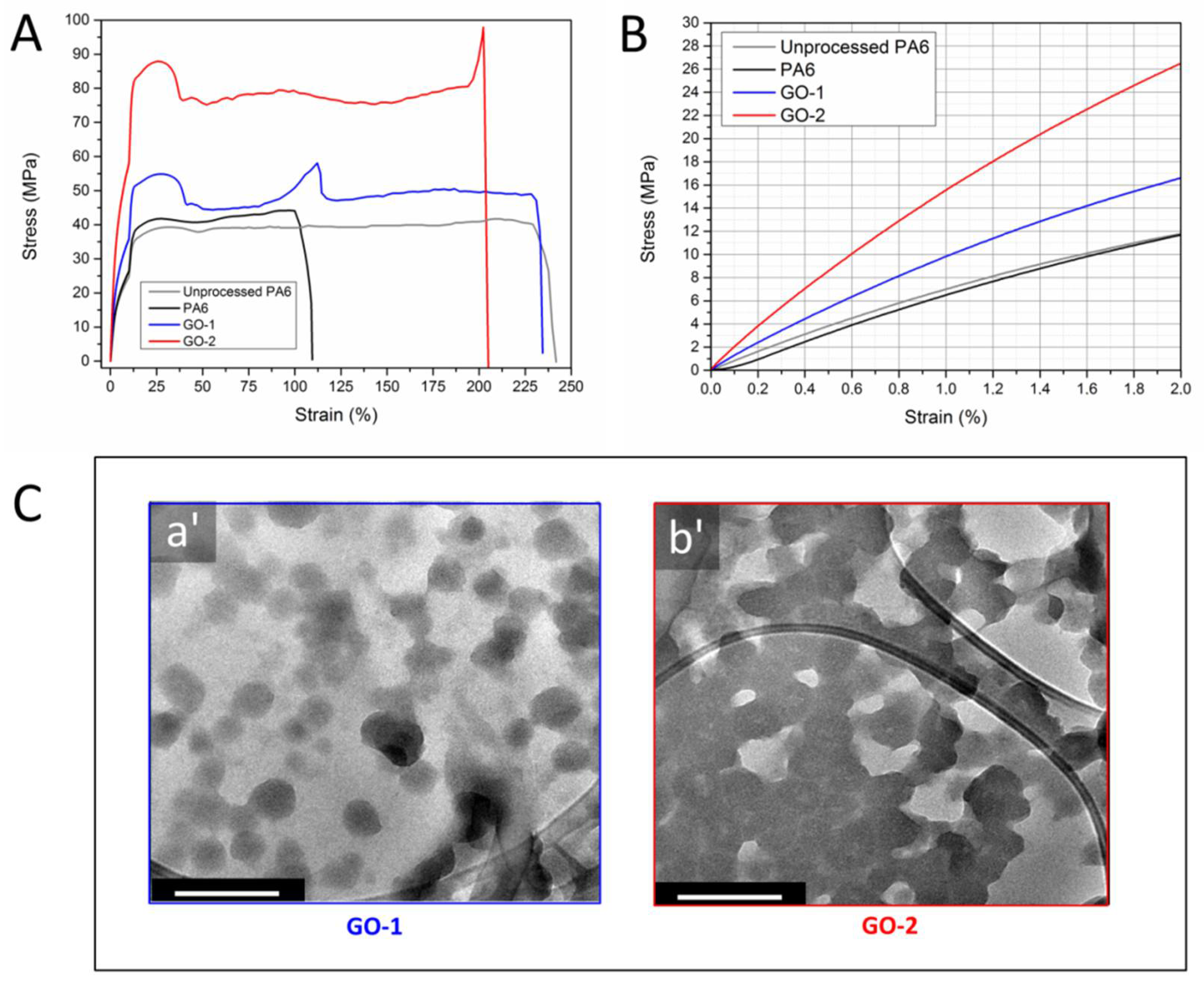
Figure 5A shows the representative stress-strain curves of nanocomposites containing GO-1 and GO-2 together with those of unprocessed and melt compounded PA6 for sake of comparison.
Figure 5B provides a close-up of the low-strains region of each stress-strain curve. The main tensile properties, that is, elastic modulus (
E), ultimate tensile stress (
TS) and elongation at break (
EB) of the samples investigated are reported in
Table 1.
The analysis of stress-strain curves put into evidence that all the materials display ductile behaviour, characterized by a necking region after yield strain; thereafter, a plateau is observed for neat PA6 samples, whereas strain-hardening phenomena were detected for nanocomposites before failure. As regards neat PA6, melt processing was found to have negligible influence on
E (from 638 to 668 MPa) and
TS (from 40 to 42 MPa), while affecting
EB, which proved to vary from 237% to 109%. This feature could likely be ascribed to thermo-mechanical degradation occurred during melt-compounding, due to the long residence time inside the batch mixer (8 min). Indeed, this result strongly agrees with the results of mixing torque and melt rheological analysis previously discussed. Adding GO-1 to PA6 determined an increase of stiffness and
TS (~+40% relative increments), while preserving the stretchability of unprocessed PA6. The incorporation of GO-2 led to larger increments in terms of elastic modulus (+110%) and
TS (+130%), whereas deformability slightly decreased (~−15%) with respect to unprocessed PA6, while being almost two-fold with respect to melt-processed PA6. First, the extremely high stretchability of nanocomposite films could be reasonably ascribed to the stabilising activity of carbonaceous fillers that prevent the thermo-oxidation of polymer during melt processing [
39]. Moreover, the wrinkling of GO lamellae was found to be responsible for catalytic and antioxidant activity of these nanoparticles, since it enables them to absorb a wide variety of degradation products [
40]. As regards the changes in terms of stiffness and tensile strength, it should be highlighted that elastic modulus is directly related to the volume of interphase, while
TS is governed by interfacial strength [
41,
42,
43]. Although both types of GO resulted in stiffer materials, the differences observed among nanocomposites possessing the same filler content suggest that the chemical-physical characteristics of the nanoparticles dramatically affect the interphase region, by influencing crucial features such as the extent of the interphase, the interfacial adhesion and the chain mobility of the polymer [
7,
44].
Panel C provides TEM micrographs of films containing GO-1 (C-a’) and GO-2 (C-b’). It can be seen that, in the case of GO-1 (lower aspect ratio, lower oxidation degree with respect to GO-2), a uniform dispersion was achieved with particle size ranging in the order of 200−300 nm. When GO-2 is used as a filler, it was observed the formation of a network, thus resulting in a huge increase of interphase volume. This feature can be explained by considering the much higher aspect ratio of GO-2 and the eventual reactions occurred between highly oxygenated lamellae and PA6 macromolecules, according to
Scheme 2. A closer investigation of interphase was performed by coupling filler extraction with morpho-spectroscopic analysis, according to our previous work [
6,
45]. The nanocomposite samples were dissolved in HCOOH at
T = 80 °C and repeatedly centrifuged aiming to extract free PA6 chains, whereas the insoluble fraction of each sample was collected and analysed.
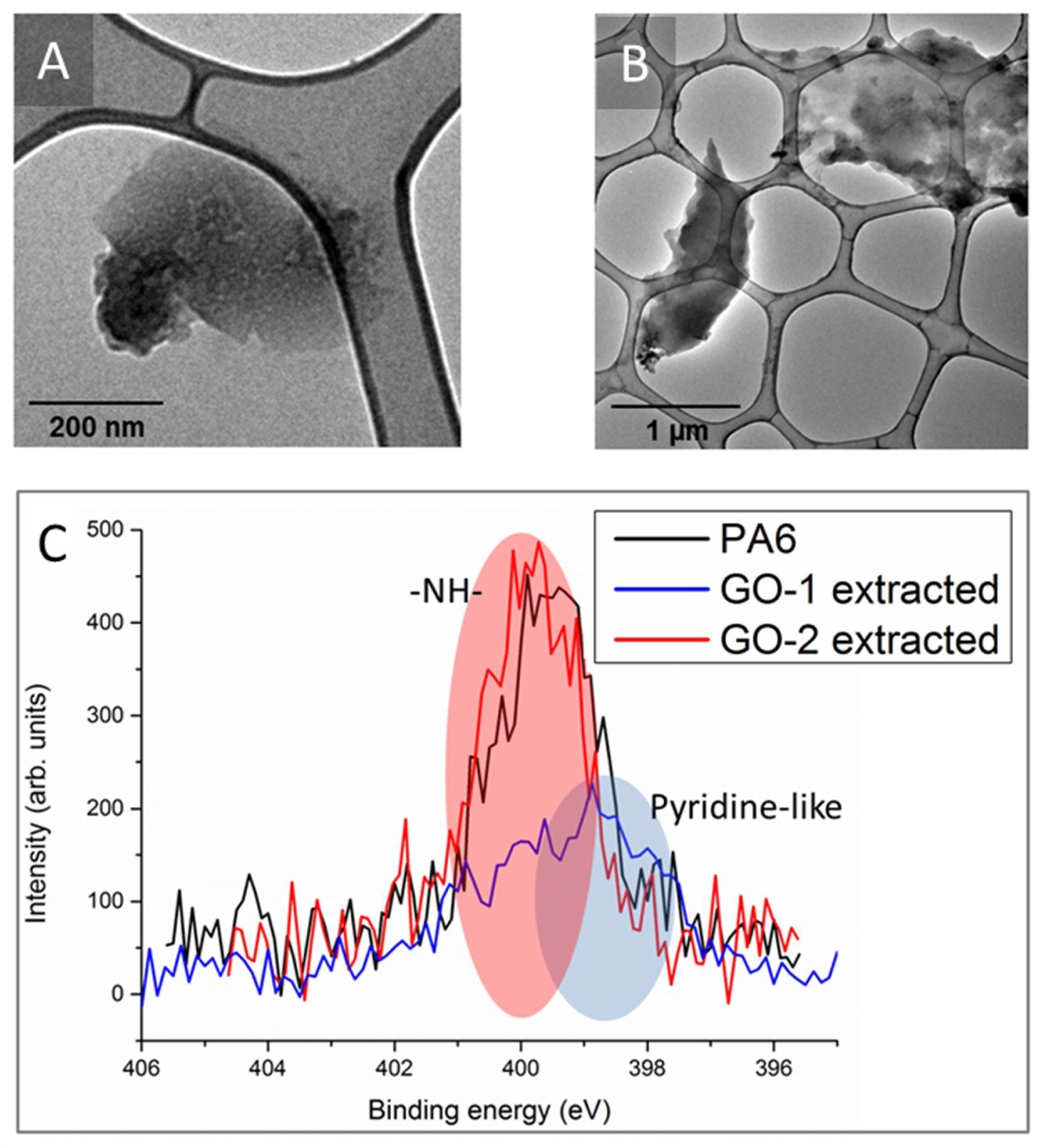
Figure 6 reports TEM micrographs of GO-1 (panel A) and GO-2 (panel B) extracted from their respective nanocomposites, together with N1s XPS spectra (panel C). Both samples proved to contain PA6, presumably reacted with graphene oxide, and this feature was easily monitored by N1s signal which, of course, are not present in graphene oxide. In particular, GO-1 lamellae extracted show polymer islands arranged into a pyridine-like structure [
18], whereas GO-2 lamellae extracted display a uniform polymer-coating. In the latter case, the presence of thick polymeric layers covering GO lamellae is uniform, as already envisaged by the TEM analysis of nanocomposites (see again
Figure 5C). Of course, the formation of a chemical interphase may have played a key-role in the extremely high
TS increments observed in nanocomposite films containing GO-2, thus explaining the presence of strain hardening before failure and even the reduced ductility.
3.3. Photo-Oxidation Behaviour
According to Cerruti et al. [
46], it is possible to get information from FTIR, monitoring the build-up of carbonyl absorption within the range of 1700–1780 cm
−1 due to primary and secondary photo-oxidation products. For this purpose, a detailed analysis of this spectral region was carried out and
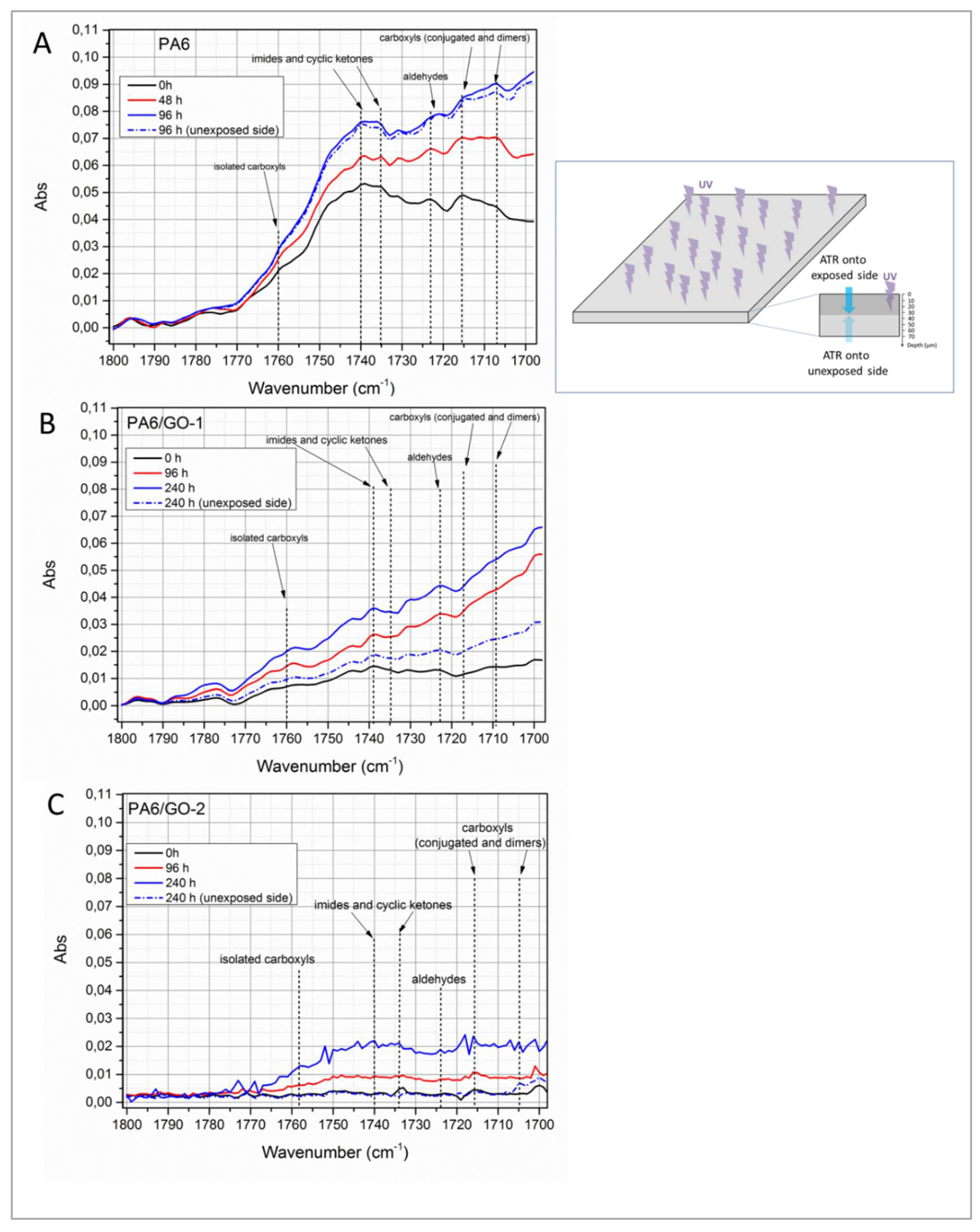
Figure 7A−C presents carbonyl domains at different irradiation times of the spectra recorded for PA6, PA6/GO-1 and PA6/GO-2, respectively.
Unirradiated PA6 (
Figure 7A) presents a variegated spectrum, where it is possible to recognize a bunch of overlapped absorption bands. The presence of carboxylic acids, in dimeric, conjugated or isolated form, is confirmed by the bands centred at 1708 cm
−1, 1715 cm
−1 and 1760 cm
−1, respectively [
46]. Moreover, the modes centred at 1740 cm
−1, 1735 cm
−1 and 1723 cm
−1 can be respectively ascribed to imides, cyclic ketones and aldehydes [
46]. Upon increasing UV-exposure time, the same characteristic peaks can be detected in the spectra, but their intensities increase in a different manner. In fact, the kinetics of the formation and accumulation of photo-oxidation products are different each other, since—for instance—carboxylic acids (present in dimeric, conjugated or isolated form) are stable products, whose concentration grows owing to the oxidation of intermediate moieties, such as aldehydes and imines, and to the hydrolysis of amide groups [
47]. Aldehydes can be already present in unexposed PA6 as a consequence of thermal oxidation during processing, as previously discussed, and can be then converted into carboxylic acids and α,β-unsaturated carbonyls [
47]. On the other hand, imides and cyclic ketones are intermediates, less stable compounds, hence their presence could suggest that the reaction of photo-oxidation is ongoing [
46,
47]. Based on these considerations, one could measure the intensity ratio between the bands respectively centred at 1735 cm
−1 (imides and ketones) and 1725 cm
−1 (aldehydes). As clearly visible in
Figure 7A, the intensity at 1735 cm
−1 (
I1735) is higher than that at 1725 cm
−1 (
I1725) in unirradiated PA6, whereas at 48 h and especially at 96 h of UV-exposure,
I1725 becomes higher than
I1735. Interestingly, PA6 film irradiated for 96 h shows practically the same spectrum on both sides, suggesting that photo-oxidation propagated in the bulk.
As regards nanocomposites containing GO-1 (
Figure 7B), all the carbonyl species are scarcely present in unirradiated samples, likely due to the well-known antioxidant activity of nanocarbons [
48,
49,
50,
51], which could have protected the polymer from thermo-oxidative reactions during mixing, in fully agreement with the results of rheological and mechanical tests previously discussed. The build-up of carbonyl domain upon UV-exposure time in this case follows a different pathway, with the signals attributable to conjugated and dimeric carboxylic acids being almost indistinguishable each other but however more intense than those ascribed to imides and ketones. Notably, in unirradiated samples
I1735 is close to
I1725 value, whereas this latter becomes higher after 96 h and 240 h, thus suggesting that reaction is ongoing. Interestingly, after 240 h of exposure to UV radiation, the opposite side of the film displays a spectrum quite similar to that of unirradiated sample.
The nanocomposites containing GO-2 (
Figure 7C) display a carbonyl domain deeply different from PA6 and PA6/GO-1. In fact, the chemical moieties typically detected in the other samples are scarcely present in this latter sample. The increment of intensities in the carbonyl region tends to zero even in the spectrum of films irradiated for 96 h and slightly increased after 240 h exposure, with values that—however—fall in the range of experimental noise. More interestingly, the opposite side of such films was found to be unaffected by UV exposure.
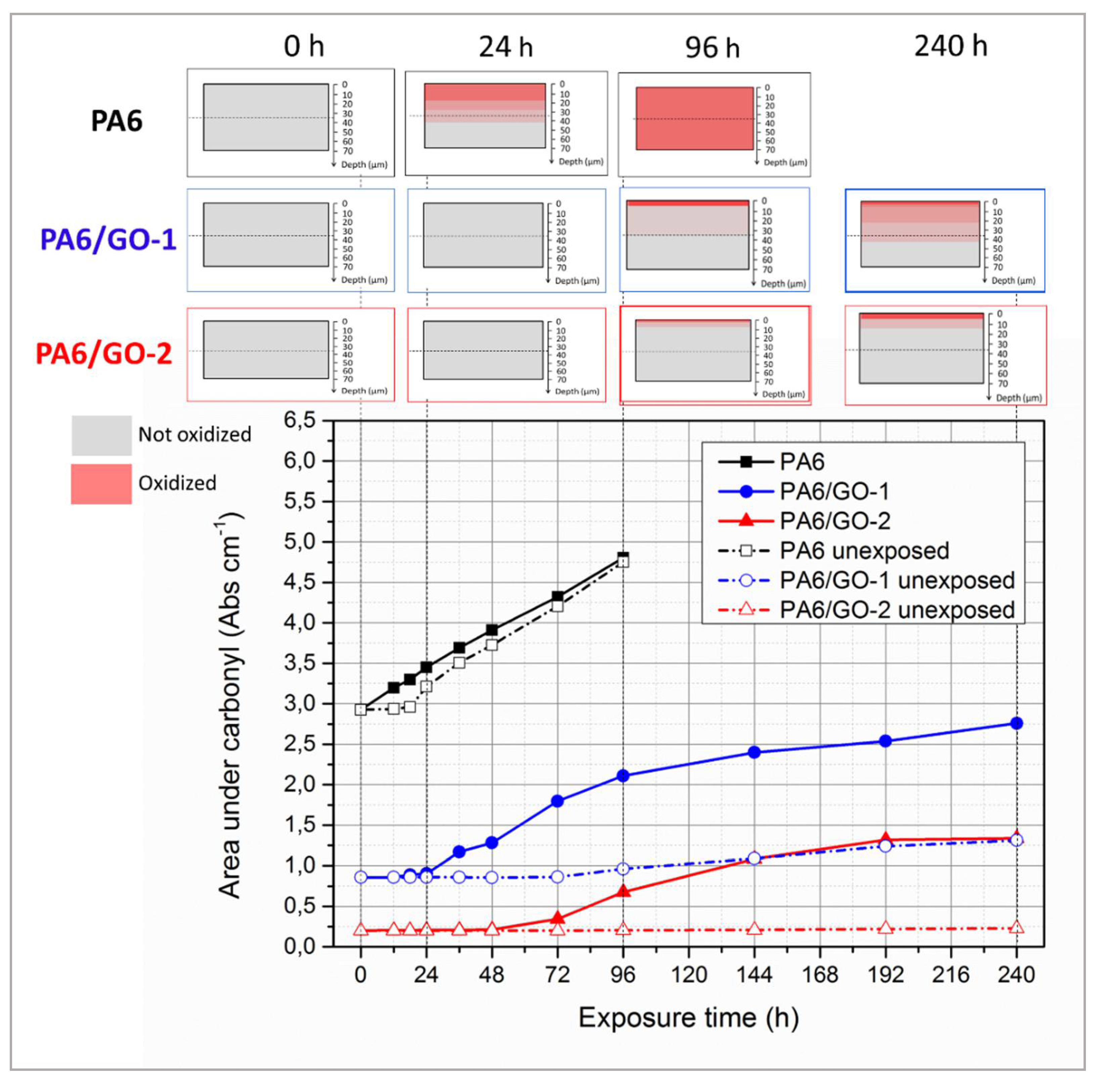
The quantitative analysis of carbonyl build-up is provided in
Figure 8, which reports the integrated areas under carbonyl, measured both onto exposed and unexposed side, plotted as a function of exposure time for all the samples, together with a pictorial representation of the propagation of photochemical reactions throughout the samples at certain time intervals. The first consideration concerns the initial values of this property, since unirradiated PA6 has an area of 3 Abs·cm
−1, whereas those of PA6/GO-1 and PA6/GO-2 are respectively equal to 0.86 and 0.24 Abs·cm
−1. This feature clearly shows once again that GO-1 and GO-2 prevented PA6 oxidation during melt processing. Furthermore, in PA6 this property is found to linearly increase upon irradiation time, and no induction time is detected in the exposed side of the films. In the unexposed side, a small induction time is observed, since area under carbonyl remains constant up to
t = 24 h, thereafter the onset of carbonyl area occurs rapidly upon reaching the same value as the exposed side at
t = 96 h. The onset of this property in the unexposed side suggests that photochemical reactions propagated in the bulk, whereas when the carbonyl area displays the same values in the exposed and unexposed face, it means that the reaction involves the entire sample (see pictorial representation of the propagation of photochemical reactions in
Figure 8).
The system containing GO-1 displays a sigmoid curve in the exposed side, characterized by an induction time of 24 h, followed by a fast grow-up until reaching 2.2 Abs·cm
−1 at
t = 96 h. Thereafter, modest increments were observed, with this property being equal to 2.5 Abs·cm
−1 after 240 h. The area measured in the unexposed side, instead, has proven to remain constant up to 72 h, thereafter it slight increased from 0.86 to 1.3 Abs·cm
−1 after 240 h. This feature suggests that GO-1 nanofillers act as UV absorbers, resulting in UV shielding that, while being unable to arrest the propagation of photochemical reactions onto exposed surface, is capable of preserving the bulk. The area under carbonyls as a function of irradiation time in the exposed side of PA6/GO-2 films displays the same shape as that of PA6/GO-1 but in this latter case the induction time increased to 48 h, the grow-up proved to be slower and the final value was equal to 1 Abs·cm
−1, i.e., practically the same as that of unexposed side of PA6/GO-1. More interestingly, in the unexposed side, no changes were detected within the whole time interval investigated. This behaviour is symptomatic of a multifunctional antioxidant activity. In fact, beyond the UV-shielding effect typical of carbonaceous nanofillers, GO-2 somehow hindered the propagation of photochemical reactions even throughout the exposed surface of films. A possible explanation of this multi-level protection may involve several factors: (i) GO-2 was found to absorb more than GO-1 in UV range (see again
Figure 2C), thus resulting in a UV-shielding activity, (ii) GO-2 presents a larger amount of cavities, holes (
Figure 1) and Stone-Wales defects (detected by Raman spectroscopy,
Figure 2A) that are known to act as radical scavengers [
49,
50], (iii) also the extended chemical interphase may have prevented the propagation of degradation pathways, thus acting as a further barrier.
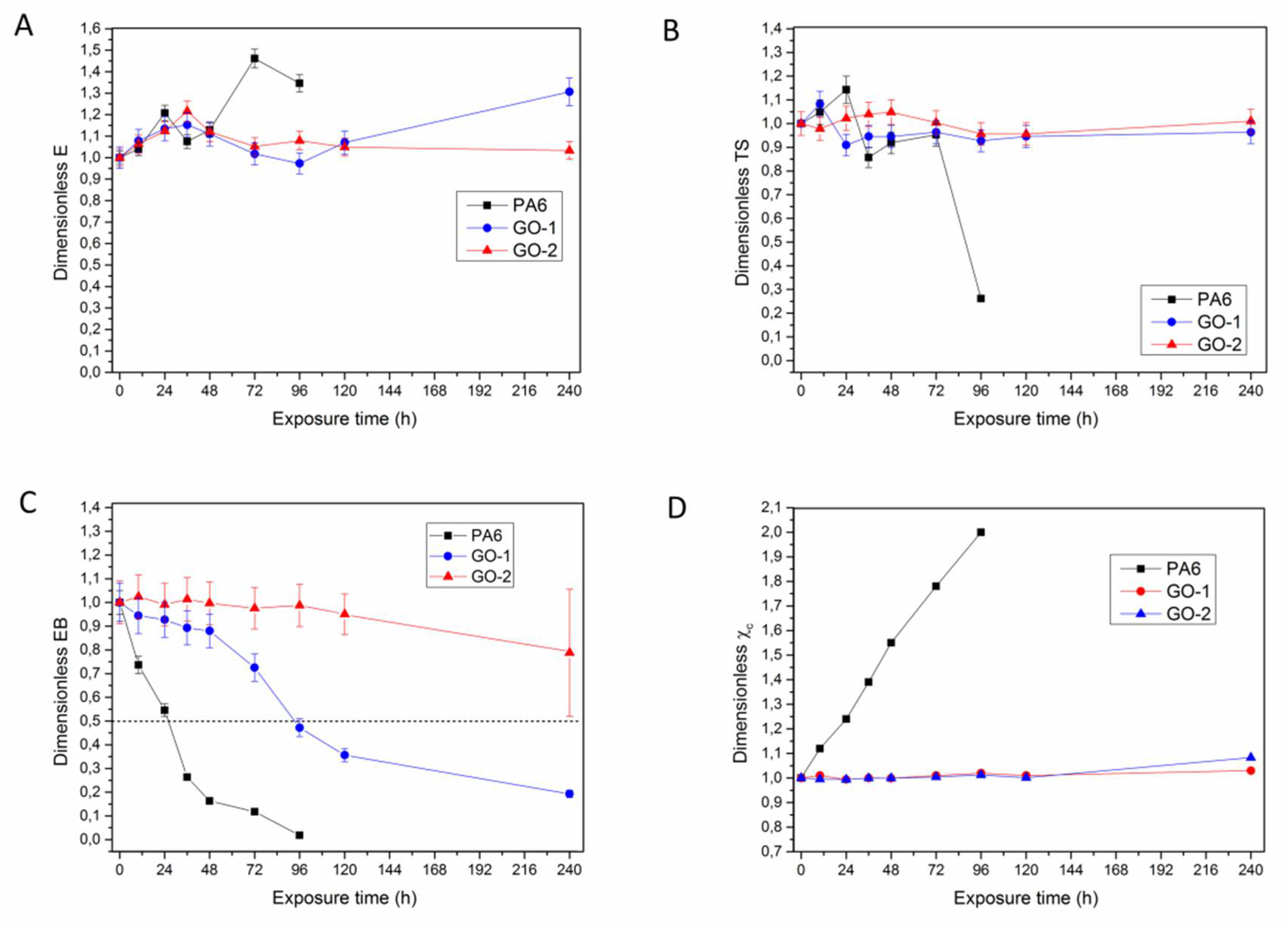
The evolution of mechanical properties, as well as eventual changes in crystallinity, during UV-exposure were monitored.
Figure 9A–D respectively reports the dimensionless values of
E,
TS,
EB and χ plotted as a function of the irradiation time. For all the samples, the dimensionless values of each property were calculated as the ratio between the value of the property at a given UV-exposure time interval and the value of that property at
t = 0 h.
Elastic modulus (
Figure 9A) was found to increase upon UV exposure time in the case of PA6, whereas no appreciable changes were detected for nanocomposites, regardless of the type of GO used. The increase of elastic modulus during photo-oxidation for a semi-crystalline polymer is attributable to the increase of crystallinity, due to the molecular weight reduction. The behaviour of dimensionless values of the crystallinity degree, χ, reported in
Figure 9D, confirms this hypothesis, since this property proved to increase only in the case of PA6, while being substantially unaltered for nanocomposites containing either GO-1 or GO-2.
The same consideration can be made for dimensionless
TS,
Figure 9B. in fact,
TS was found to increase for PA6 at low irradiation times, due to the higher crystallinity degree, thereafter this property was found to decay due to the increasing brittleness of the samples, which resulted in a premature failure. Nanocomposites display negligible changes during UV-exposure (±10–12% with respect to the initial value).
The analysis of dimensionless
EB, instead, is a main concern, since this parameter is correlated with the oxidation level of a material. In fact, as the reaction propagates in the bulk,
EB decays in such a rapid way [
52].
As regards PA6 films,
EB was found to decrease rapidly and monotonically with irradiation time. After 96 h UV-exposure, the excessive brittleness of the specimens made it impossible to continue the experiments. For nanocomposites containing GO-1, a slow decrease was detected until 96 h, thereafter dimensionless
EB reduced to 0.2. However, the samples irradiated at 240 h retained a ductile behaviour. The nanocomposites containing GO-2 showed impressive stretchability retention, since dimensionless
EB was found to remain close to 1 within the time investigated, with a slight reduction (0.8) observed only after 240 h of UV-exposure. Even in this case, a ductile mode in the failure was observed in all the irradiated samples, although the data scattering at this exposure level was found to increase. Furthermore, it was determined the half life time (HLT), i.e., the time necessary to achieve the halving of initial
EB value [
48]. This is a technological parameter to quantitatively assess the effect of the type of filler on the photo-oxidation kinetics. HLT proved to be equal to 26 h for PA6, while being enhanced to 96 h in the case of PA6/GO-1 nanocomposite. Notably, for the materials containing GO-2 no HLT was observed within the time interval investigated, even if some specimens experienced HLF at 240 h (see error bars in
Figure 9D). The data point out that there is a correlation between HLT and ATR analysis of unexposed side of the films. In fact, it can be observed that HLT occurs when carbonyl band area of the unexposed side of the films begins to grow, with this latter feature indicating that photochemical reactions propagated into the bulk.
Although the energy of solar radiation would not be sufficient to cause the direct homolytic scission of C–N bonds, the light absorption can promote homolytic scission of the carbon-hydrogen bonds, owing to the presence of impurities such as catalyst residues, defects in the specimens, and carbonyl or peroxide species formed during melt processing [
47,
53]. As reported in scientific literature [
48,
54], carbonaceous fillers may act as UV absorbers, as chain breaking donor/acceptor of free radical species, as quencher of excited states such as active carbonyl species, and as hydroperoxides decomposer. The oxidation rate in PA6 is mainly determined by radical-forming reactions during UV-exposure, owing to the direct photolysis of the amide bonds [
53]. These hypotheses clearly explain why GO-based nanocomposites showed an improved photo-stability compared to neat PA6. Indeed, due to the presence of aromatic species, defects and cavities within its honeycomb, GO-2 could act as radical scavenger and the formation of an extended chemical interphase in the materials loaded with GO-2 resulted in a sort of impermeability towards the propagation of oxidation reactions, as confirmed by ATR performed on the opposite sides of the samples. The concomitance of all these factors could explain the excellently enhanced photo-stability.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}