Self-Healable Supramolecular Vanadium Pentoxide Reinforced Polydimethylsiloxane-Graft-Polyurethane Composites


Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials and Chemicals
2.2. Synthesis
2.2.1. Vanadium Pentoxide Nanofiber Synthesis
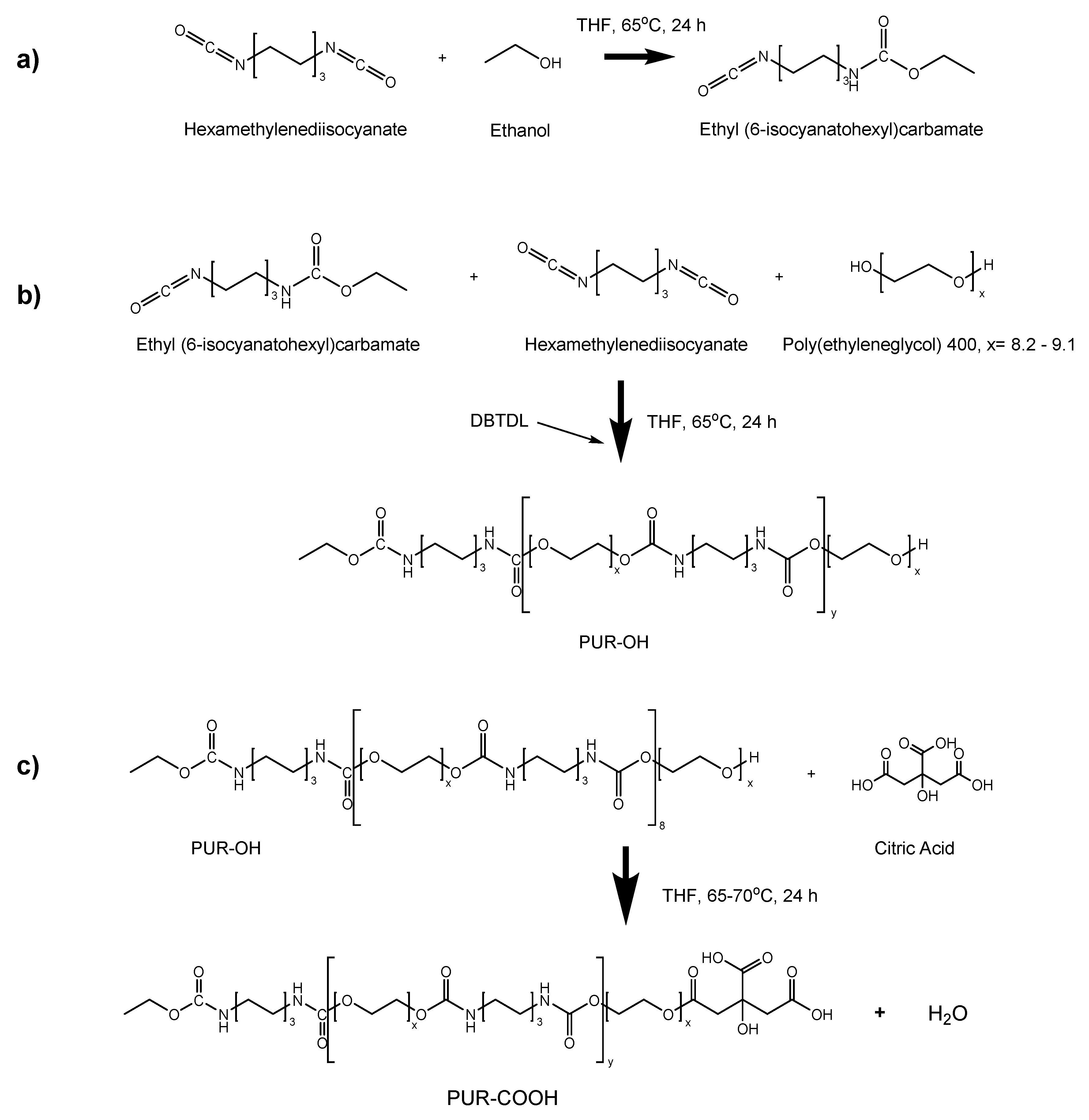
2.2.2. Synthesis of Polyurethane Having One –COOH Functional End Group
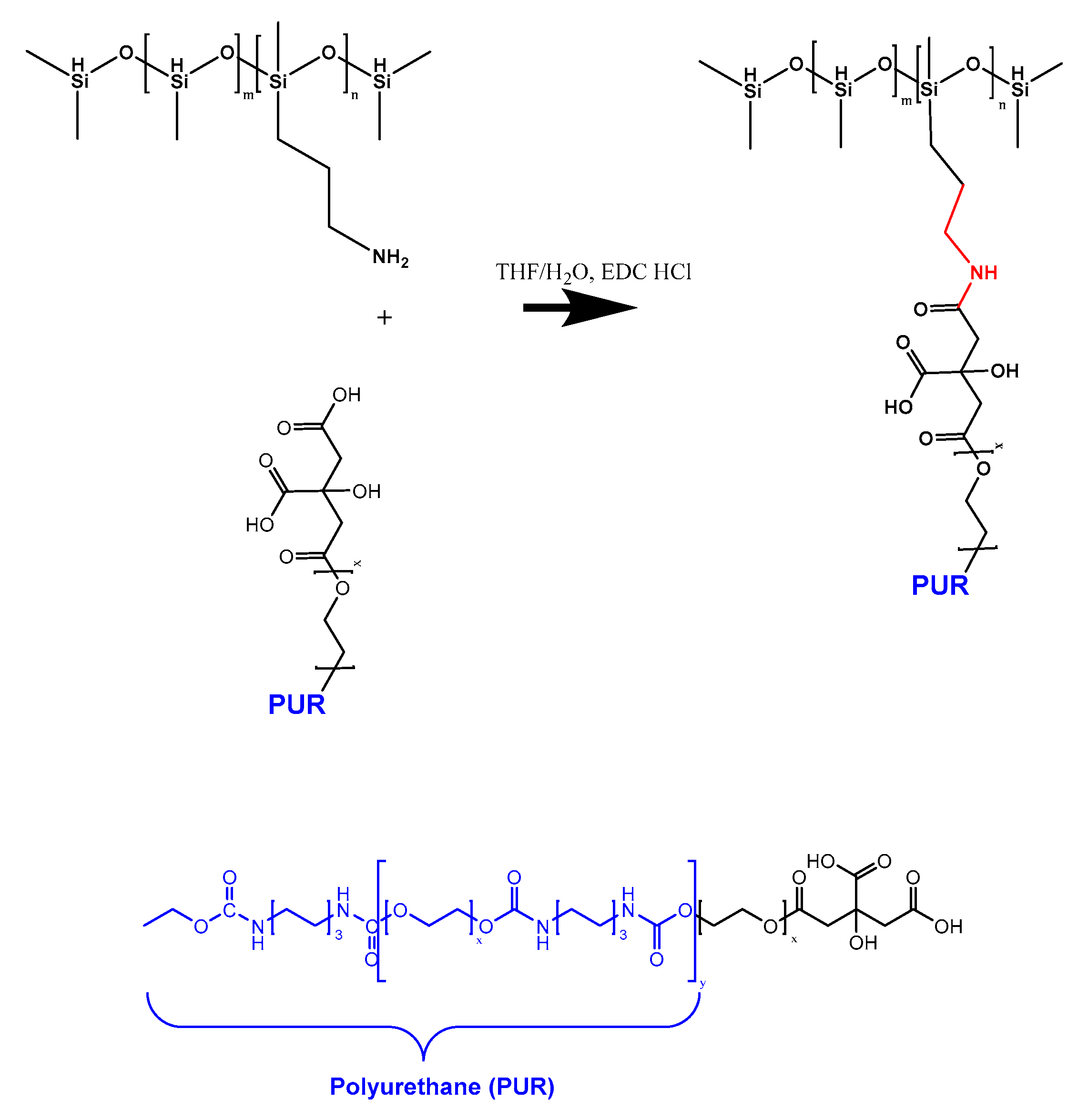
2.2.3. Grafting of PUR-COOH onto AP-PDMS
2.2.4. Preparation of PDMS-g-PUR /V2O5 Composites
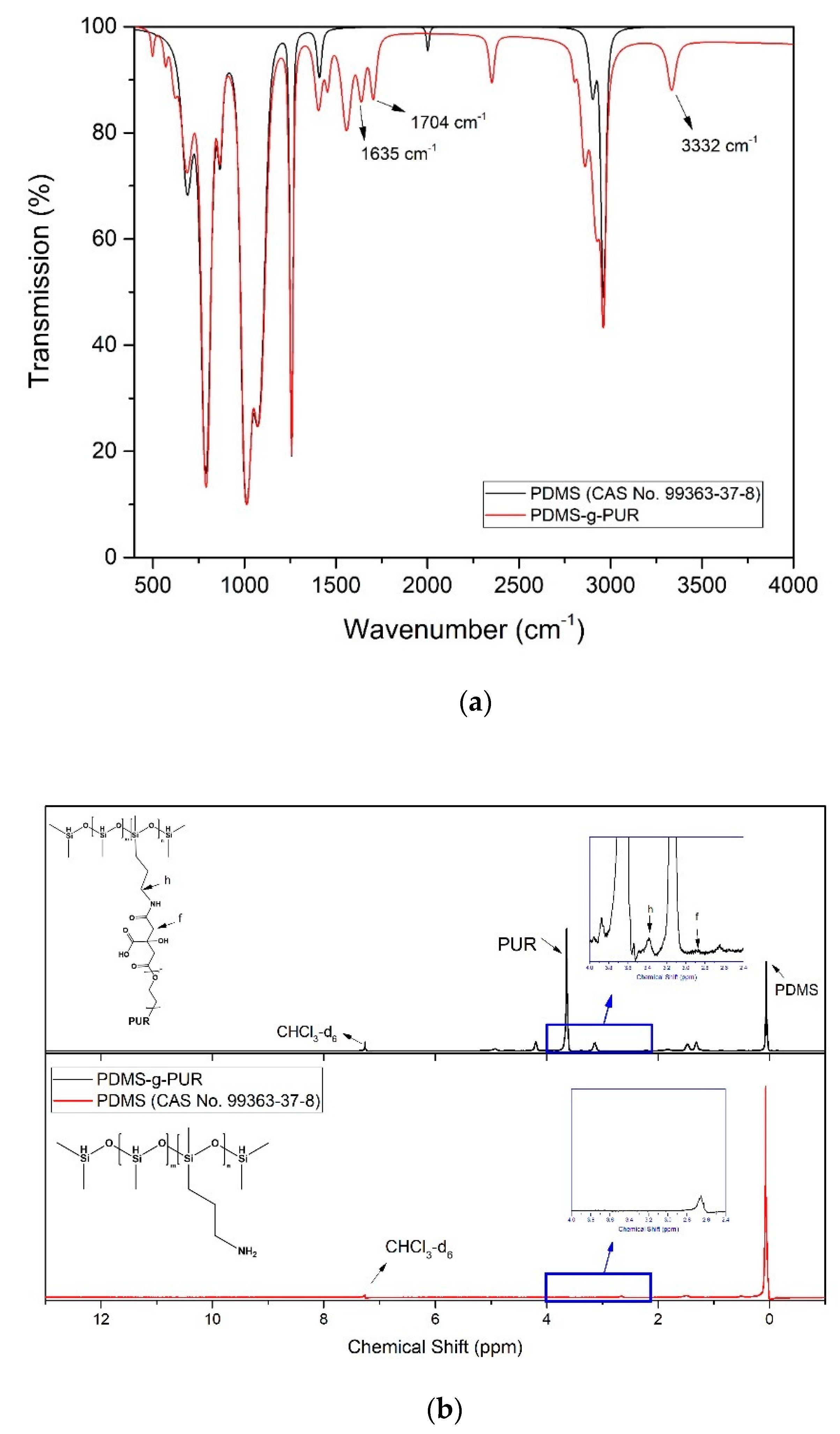
2.3. Characterization
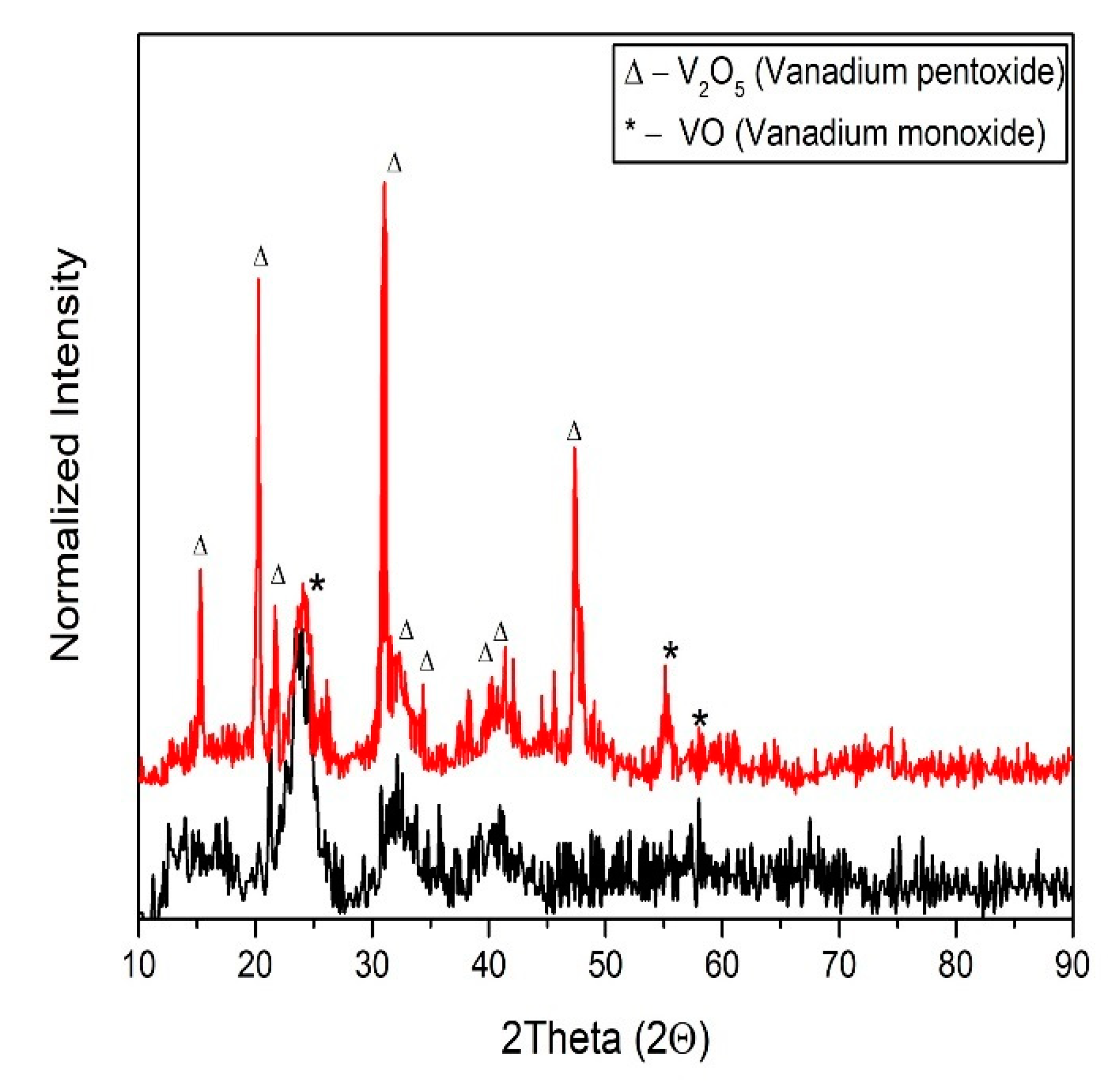
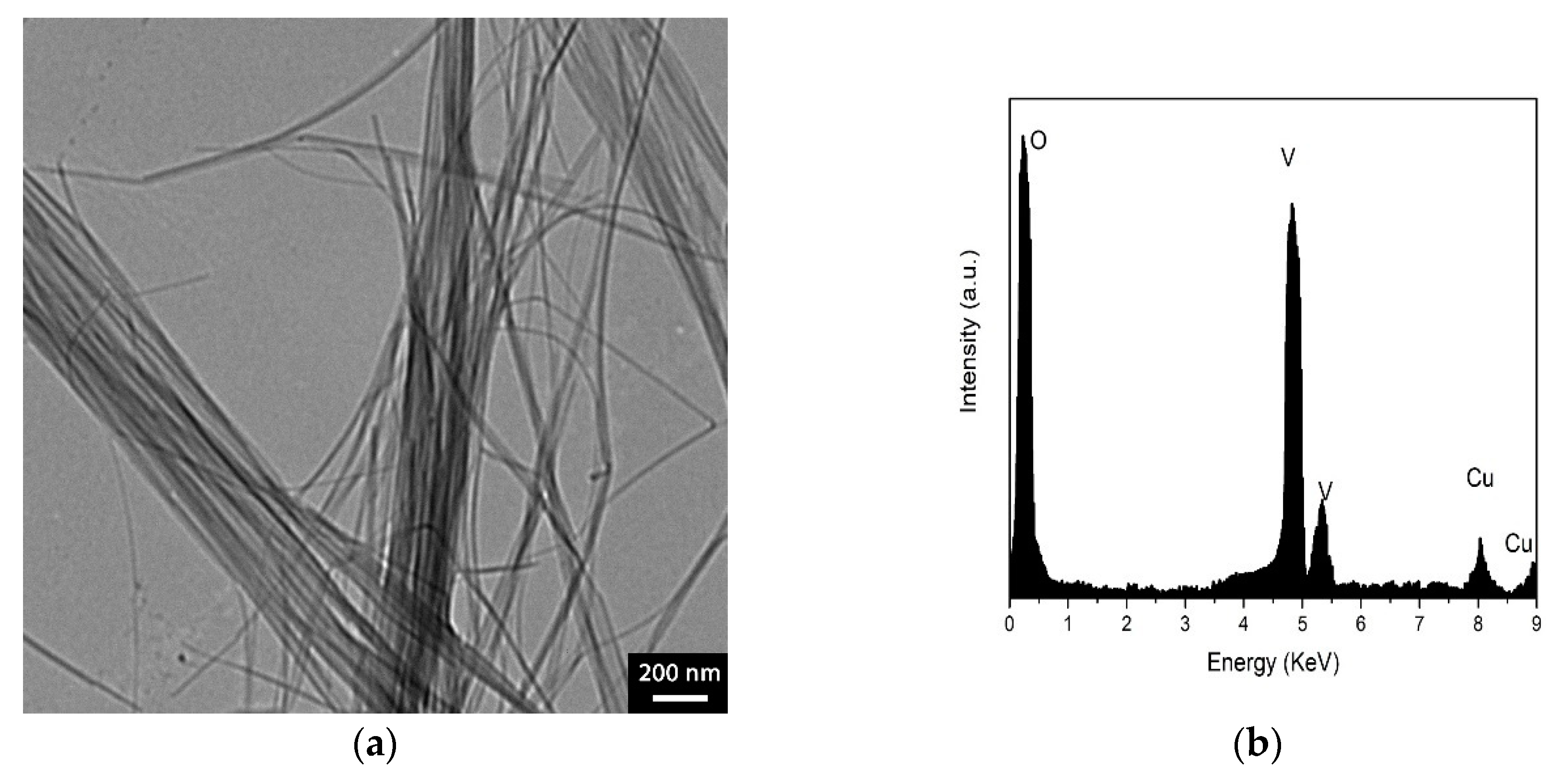
Characterization of V2O5 Nanofibers
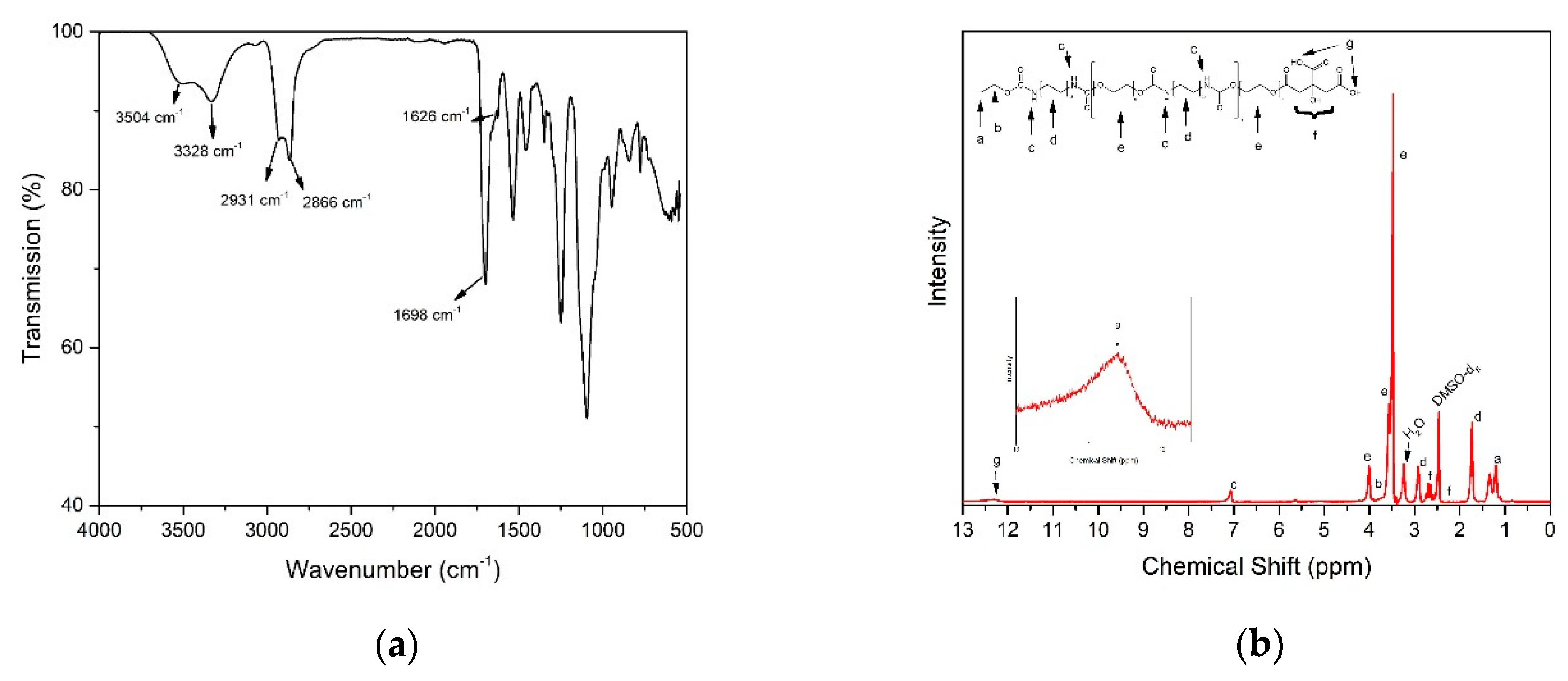
2.4. Characterization of Polymer and Composites
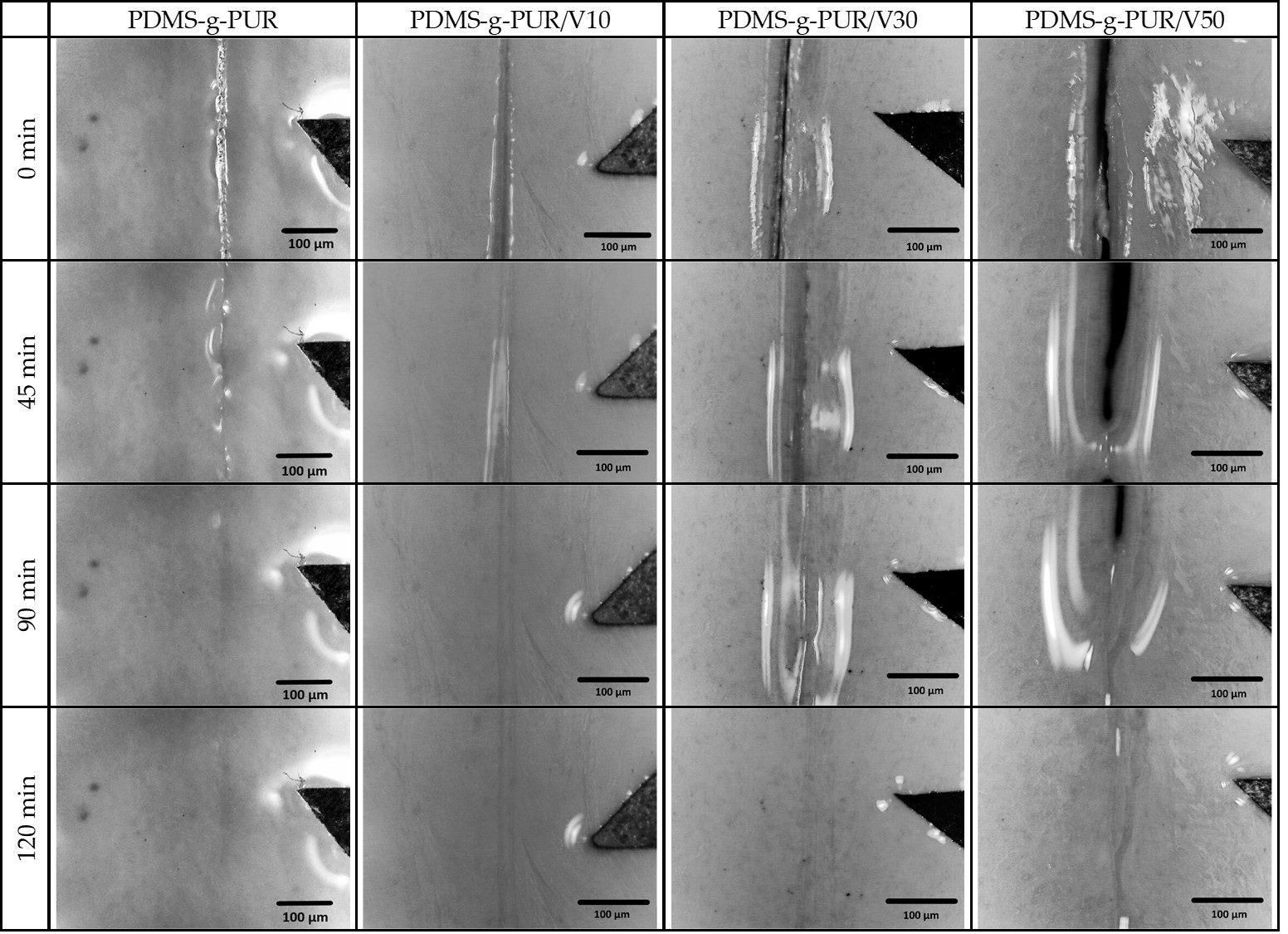
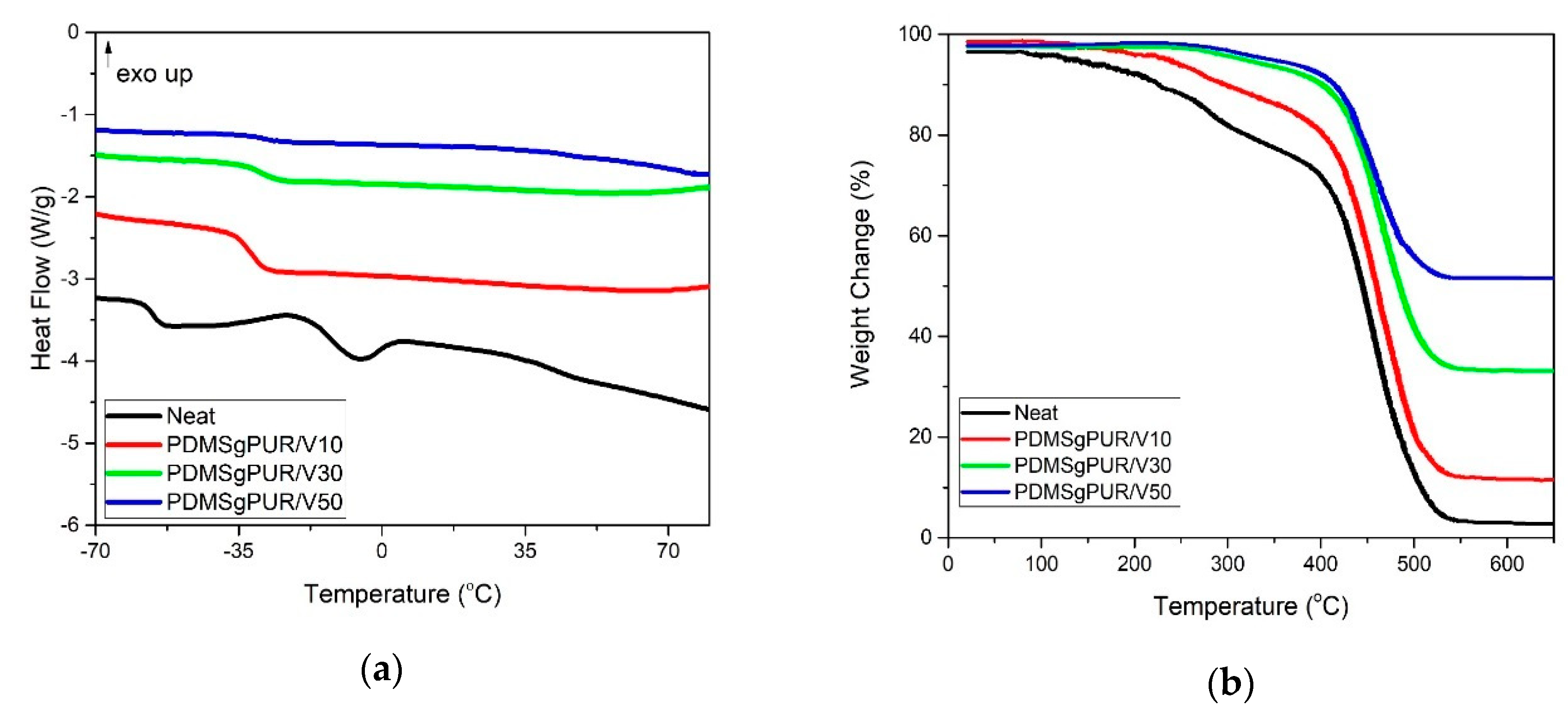
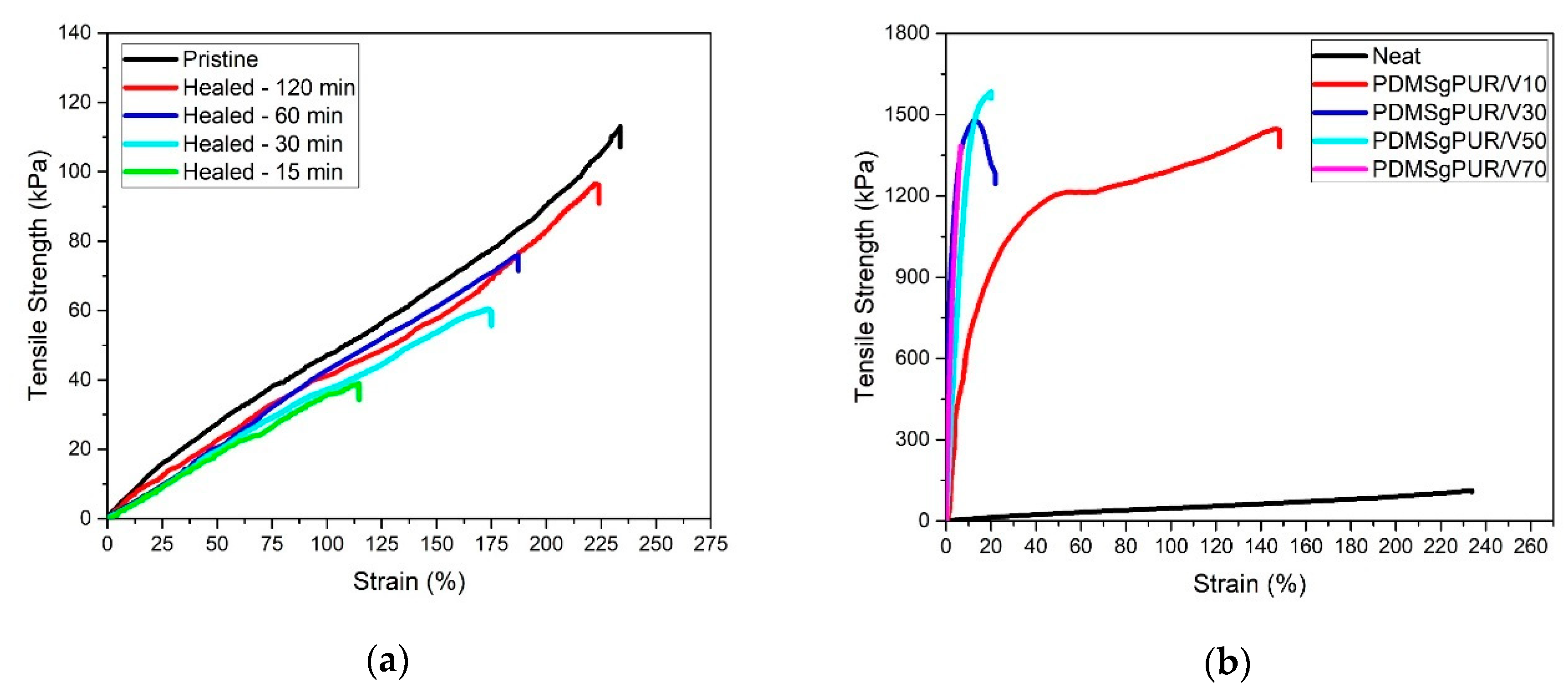
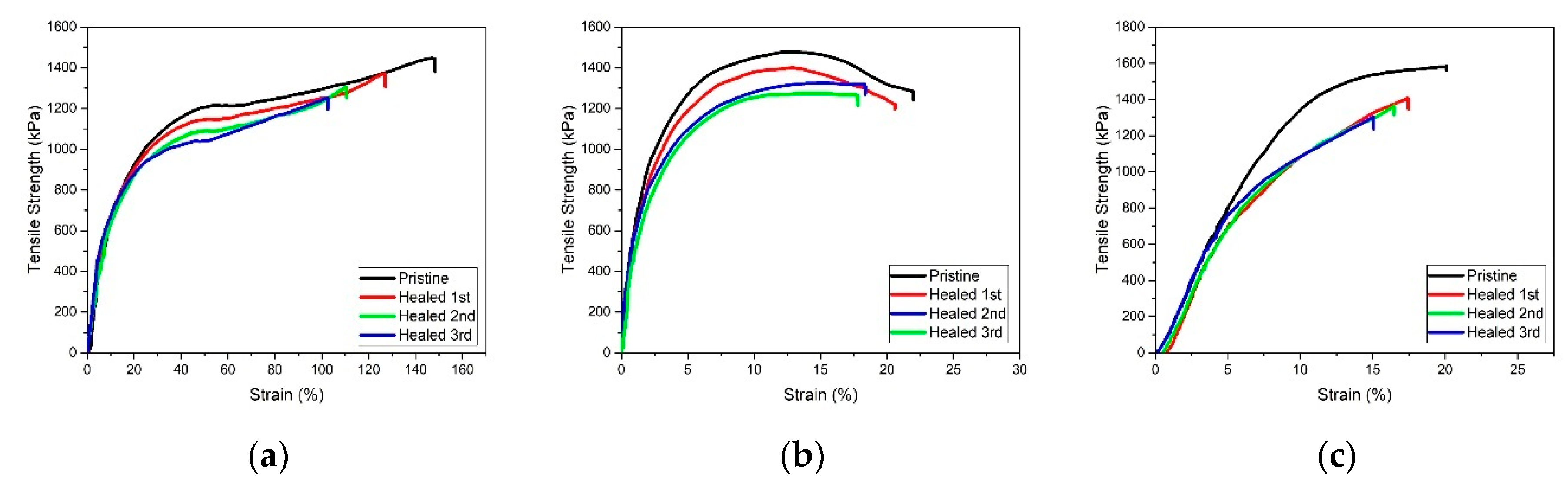
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xia, H.; Zhao, J.; Luo, G.; Xu, R.; Wu, J. A self-healing, re-moldable and biocompatible crosslinked polysiloxane elastomer. J. Mater. Chem. B 2016, 4, 982–989. [Google Scholar]
- Liu, M.; Liu, P.; Lu, G.; Xu, Z.; Yao, X. Multiphase-Assembly of Siloxane Oligomers with Improved Mechanical Strength and Water-Enhanced Healing. Angew. Chem. 2018, 130, 11412–11416. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, R.; Luo, G.; Wu, J.; Xia, H. Self-healing poly(siloxane-urethane) elastomers with remoldability, shape memory and biocompatibility. Polym. Chem. 2016, 7, 7278–7286. [Google Scholar] [CrossRef]
- Zheng, P.; McCarthy, T.J. Rediscovering Silicones: Molecularly Smooth, Low Surface Energy, Unfilled, UV/Vis-Transparent, Extremely Cross-Linked, Thermally Stable, Hard, Elastic PDMS. Langmuir 2010, 26, 18585–18590. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Andersson, H.M.; White, S.R.; Sottos, N.R.; Braun, P.V. Polydimethylsiloxane-Based Self-Healing Materials. Adv. Mater. 2006, 18, 997–1000. [Google Scholar] [CrossRef]
- Keller, M.W.; White, S.R.; Sottos, N.R. A Self-Healing Poly(Dimethyl Siloxane) Elastomer. Adv. Funct. Mater. 2007, 17, 2399–2404. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, P.; Zhang, H.; Yan, C.; Zheng, Z.; Wu, B.; Yu, Y. A Transparent, Highly Stretchable, Autonomous Self-Healing Poly(dimethyl siloxane) Elastomer. Macromol. Rapid Commun. 2017, 38, 1700110. [Google Scholar] [CrossRef]
- Kang, J.; Son, D.; Wang, G.-J.N.; Liu, Y.; Lopez, J.; Kim, Y.; Oh, J.Y.; Katsumata, T.; Mun, J.; Lee, Y.; et al. Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin. Adv. Mater. 2018, 30, 1706846. [Google Scholar] [CrossRef]
- Tee, B.C.-K.; Wang, C.; Allen, R.; Bao, Z. An electrically and mechanically self-healing composite with pressure- and flexion-sensitive properties for electronic skin applications. Nature Nanotechnol. 2012, 7, 825–832. [Google Scholar] [CrossRef]
- Roy, N.; Buhler, E.; Lehn, J.-M. The Tris-Urea Motif and Its Incorporation into Polydimethylsiloxane-Based Supramolecular Materials Presenting Self-Healing Features. Chem. Eur. J. 2013, 19, 8814–8820. [Google Scholar] [CrossRef]
- Burattini, S.; Colquhoun, H.M.; Greenland, B.W.; Hayes, W. A novel self-healing supramolecular polymer system. Faraday Discuss. 2009, 143, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Buhler, E.; Lehn, J.-M. Double dynamic self-healing polymers: Supramolecular and covalent dynamic polymers based on the bis-iminocarbohydrazide motif. Polym. Int. 2013, 63, 1400–1405. [Google Scholar] [CrossRef]
- Lu, H.; Feng, S. Supramolecular Silicone Elastomers with Healable and Hydrophobic Properties Crosslinked by “Salt-Forming Vulcanization”. J. Polym. Sci. A Polym. Chem. 2017, 55, 903–911. [Google Scholar] [CrossRef]
- Ciferri, A.; Atwood, J.L.; Steed, J.W. Supramolecular Polymers. Encycl. Supramol. Chem. 2004, 453, 1443–1452. [Google Scholar]
- Burattini, S.; Greenland, B.W.; Merino, D.H.; Weng, W.; Seppala, J.; Colquhoun, H.M.; Hayes, W.; Mackay, M.E.; Hamley, I.W.; Rowan, S.J. A Healable Supramolecular Polymer Blend Based on Aromatic π−π Stacking and Hydrogen-Bonding Interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. [Google Scholar] [CrossRef] [PubMed]
- Burattini, S.; Colquhoun, H.M.; Fox, J.D.; Friedmann, D.; Greenland, B.W.; Harris, P.J.F.; Hayes, W.; Mackay, M.E.; Rowan, S.J. A self-repairing, supramolecular polymer system: Healability as a consequence of donor–acceptor π–π stacking interactions. Chem. Commun. 2009, 0, 6717–6719. [Google Scholar] [CrossRef] [PubMed]
- Kalista, S.J.; Ward, T.C.; Oyetunji, Z. Self-Healing of Poly(Ethylene-co-Methacrylic Acid) Copolymers Following Projectile Puncture. Mech. Adv. Mater. Struct. 2007, 14, 391–397. [Google Scholar] [CrossRef]
- Zhang, A.; Yang, L.; Lin, Y.; Yan, L.; Lu, H.; Wang, L. Self-healing supramolecular elastomers based on the multi-hydrogen bonding of low-molecular polydimethylsiloxanes: Synthesis and characterization. J. Appl. Polym. Sci. 2013, 129, 2435–2442. [Google Scholar] [CrossRef]
- Stadler, R.; Freitas, L.L.; Freitas, L.D.L. Thermoplastic elastomers by hydrogen bonding 1. Rheological properties of modified polybutadiene. Colloid Polym. Sci. 1986, 264, 773–778. [Google Scholar] [CrossRef]
- Chino, K.; Ashiura, M. Themoreversible Cross-Linking Rubber Using Supramolecular Hydrogen-Bonding Networks. Macromolecules 2001, 34, 9201–9204. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.; Burtovyy, R.; Luzinov, I.; Urban, M.W. UV-induced self-repairing polydimethylsiloxane–polyurethane (PDMS–PUR) and polyethylene glycol–polyurethane (PEG–PUR) Cu-catalyzed networks. J. Mater. Chem. A 2014, 2, 15527–15534. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, W.; Tong, R.; Lu, X.; Xia, H. Thermal-healable and shape memory metallosupramolecular poly(n-butyl acrylate-co-methyl methacrylate) materials. RSC Adv. 2014, 4, 25486–25493. [Google Scholar] [CrossRef]
- Hong, G.; Lin, Y.; Chen, Y.; Xu, Y.; Zhang, H.; Weng, W.; Xia, H. Mechanoresponsive Healable Metallosupramolecular Polymers. Macromolecules 2013, 46, 8649–8656. [Google Scholar] [CrossRef]
- Deng, W.; You, Y.; Zhang, A. Supramolecular Network-Based Self-Healing Polymer Materials. In Recent Advances in Smart Self-healing Polymers and Composites; Li, G., Meng, H., Eds.; Woodhead Publishing: Cambridge, UK, 2015; pp. 181–210. [Google Scholar]
- De Espinosa, L.M.; Fiore, G.L.; Weder, C.; Foster, E.J.; Simon, Y.C. Healable supramolecular polymer solids. Prog. Polym. Sci. 2015, 49–50, 60–78. [Google Scholar] [CrossRef]
- Luan, Y.-G.; Zhang, X.-A.; Jiang, S.-L.; Chen, J.-H.; Lyu, Y.-F. Self-healing Supramolecular Polymer Composites by Hydrogen Bonding Interactions between Hyperbranched Polymer and Graphene Oxide. Chin. J. Polym. Sci. 2018, 36, 584–591. [Google Scholar] [CrossRef]
- Deflorian, F.; Rossi, S.; Scrinzi, E. Self-healing supramolecular polyurethane coatings: Preliminary study of the corrosion protective properties. Corros. Eng. Sci. Technol. 2013, 48, 147–154. [Google Scholar] [CrossRef]
- Zhang, A.; Deng, W.; Lin, Y.; Ye, J.; Dong, Y.; Lei, Y.; Chen, H. Novel supramolecular elastomer films based on linear carboxyl-terminated polydimethylsiloxane oligomers: Preparation, characterization, biocompatibility, and application in wound dressings. J. Biomater. Sci. Polym. Ed. 2014, 25, 1346–1361. [Google Scholar] [CrossRef]
- You, Y.; Zhang, A.; Lin, Y. Crosslinking mechanism of supramolecular elastomers based on linear bifunctional polydimethylsiloxane oligomers. J. Appl. Polym. Sci. 2016, 133, 43385. [Google Scholar] [CrossRef]
- Ogliani, E.; Yu, L.; Javakhishvili, I.; Skov, A.L. A thermo-reversible silicone elastomer with remotely controlled self-healing. RSC Adv. 2018, 8, 8285–8291. [Google Scholar] [CrossRef]
- Comí, M.; Lligadas, G.; Ronda, J.C.; Galià, M.; Cádiz, V. Adaptive bio-based polyurethane elastomers engineered by ionic hydrogen bonding interactions. Eur. Polym. J. 2017, 91, 408–419. [Google Scholar] [CrossRef]
- Jian, X.; Hu, Y.; Zhou, W.; Xiao, L. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2017, 29, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Bayan, R.; Karak, N. Bio-derived aliphatic hyperbranched polyurethane nanocomposites with inherent self healing tendency and surface hydrophobicity: Towards creating high performance smart materials. Compos. Part A Appl. Sci. Manuf. 2018, 110, 142–153. [Google Scholar] [CrossRef]
- Grzelak, A.W.; Boinard, P.; Liggat, J.J. The influence of diol chain extender on morphology and properties of thermally-triggered UV-stable self-healing polyurethane coatings. Prog. Org. Coat. 2018, 122, 1–9. [Google Scholar] [CrossRef]
- Wu, T.; Chen, B. Synthesis of Multiwalled Carbon Nanotube-Reinforced Polyborosiloxane Nanocomposites with Mechanically Adaptive and Self-Healing Capabilities for Flexible Conductors. ACS Appl. Mater. Interfaces 2016, 8, 24071–24078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Niu, H.; Du, X.; Zhao, S.; Yuan, Z.; Zhang, X.; Cao, R.; Yin, Y.; Zhang, C.; Zhou, T. Polymer nanocomposite enabled high-performance triboelectric nanogenerator with self-healing capability. RSC Adv. 2018, 8, 30661–30668. [Google Scholar]
- Kim, K.H.; Kim, K.H.; Huh, J.; Jo, W.H. Synthesis of thermally stable organosilicate for exfoliated poly(ethylene terephthalate) nanocomposite with superior tensile properties. Macromol. Res. 2007, 15, 178–184. [Google Scholar] [CrossRef]
- Lee, J.Y.; Buxton, G.A.; Balazs, A.C. Using nanoparticles to create self-healing composites. J. Chem. Phys. 2004, 121, 5531–5540. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Peng, Q.; Li, Y. Vanadium Pentoxide Nanobelts: Highly Selective and Stable Ethanol Sensor Materials. Adv. Mater. 2005, 17, 764–767. [Google Scholar] [CrossRef]
- Gu, G.; Schmid, M.; Chiu, P.-W.; Minett, A.; Fràysse, J.; Kim, G.-T.; Roth, S.; Kozlov, M.; Muñoz, E.; Baughman, R.H. V2O5 nanofibre sheet actuators. Nat. Mater 2003, 2, 316–319. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, K.; Gao, Y.; Luo, H.; Lu, L. Recent Progress in the Applications of Vanadium-Based Oxides on Energy Storage: From Low-Dimensional Nanomaterials Synthesis to 3D Micro/Nano-Structures and Free-Standing Electrodes Fabrication. Adv. Energy Mater. 2017, 7. [Google Scholar] [CrossRef]
- Yu, D.; Chen, C.; Xie, S.; Liu, Y.; Park, K.; Zhou, X.; Zhang, Q.; Li, J.; Cao, G. Mesoporous vanadium pentoxide nanofibers with significantly enhanced Li-ion storage properties by electrospinning. Energy Environ. Sci. 2011, 4, 858–861. [Google Scholar] [CrossRef]
- Choi, J.K.; Kim, Y.W.; Koh, J.H.; Mayes, A.M. Synthesis and characterization of nanocomposite films consisting of vanadium oxide and microphase-separated graft copolymer. Macromol. Res. 2007, 15, 553–559. [Google Scholar] [CrossRef]
- Quan, H.; Chen, D.; Xie, X.; Fan, H. Polyvinylidene fluoride/vanadium pentoxide composites with high dielectric constant and low dielectric loss. Phys. Status Solidi A 2013, 210, 2706–2709. [Google Scholar] [CrossRef]
- Burghard, Z.; Leineweber, A.; van Aken, P.A.; Dufaux, T.; Burghard, M.; Bill, J. Hydrogen-bond reinforced vanadia nanofiber paper of high stiffness. Adv. Mater. 2013, 25, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Harreld, J.H.; Wong, M.S.; Hansma, P.K.; Morse, D.E.; Stucky, G.D. Self-Healing Organosiloxane Materials Containing Reversible and Energy-Dispersive Crosslinking Domains. US Grant US6783709B2, 31 August 2004. [Google Scholar]
- Bayan, R.; Karak, N. Renewable resource derived aliphatic hyperbranched polyurethane/aluminium hydroxide–reduced graphene oxide nanocomposites as robust, thermostable material with multi-stimuli responsive shape memory features. New J. Chem. 2017, 41, 8781–8790. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition Name | V2O5 (%) | PDMS-g-PUR (%) |
|---|---|---|
| PDMS-g-PUR | - | 100 |
| PDMS-g-PUR/V10 | 10 | 90 |
| PDMS-g-PUR/V30 | 30 | 70 |
| PDMS-g-PUR/V50 | 50 | 50 |
| PDMS-g-PUR/V70 | 70 | 30 |
| 1st Healing (%) | 2nd Healing (%) | 3rd Healing (%) | |
|---|---|---|---|
| PDMS-g-PUR | 85.4 ± 1.2 | - | - |
| PDMS-g-PUR/V10 | 95.3 ± 0.4 | 90.1 ± 0.9 | 86.7 ± 0.5 |
| PDMS-g-PUR/V30 | 94.7 ± 0.5 | 89.6 ± 0.7 | 86.1 ± 0.4 |
| PDMS-g-PUR/V50 | 88.9 ± 0.9 | 86.2 ± 1.5 | 82.3 ± 1.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkem, A.S.; Capoglu, A.; Nugay, T.; Sancaktar, E.; Anac, I. Self-Healable Supramolecular Vanadium Pentoxide Reinforced Polydimethylsiloxane-Graft-Polyurethane Composites. Polymers 2019, 11, 41. https://doi.org/10.3390/polym11010041
Berkem AS, Capoglu A, Nugay T, Sancaktar E, Anac I. Self-Healable Supramolecular Vanadium Pentoxide Reinforced Polydimethylsiloxane-Graft-Polyurethane Composites. Polymers. 2019; 11(1):41. https://doi.org/10.3390/polym11010041
Chicago/Turabian StyleBerkem, Ali Sabri, Ahmet Capoglu, Turgut Nugay, Erol Sancaktar, and Ilke Anac. 2019. "Self-Healable Supramolecular Vanadium Pentoxide Reinforced Polydimethylsiloxane-Graft-Polyurethane Composites" Polymers 11, no. 1: 41. https://doi.org/10.3390/polym11010041