
Influence of Foreign Salts and Antiscalants on Calcium Carbonate Crystallization

Abstract
:1. Introduction
2. Experimental Section
2.1. Solution Preparation
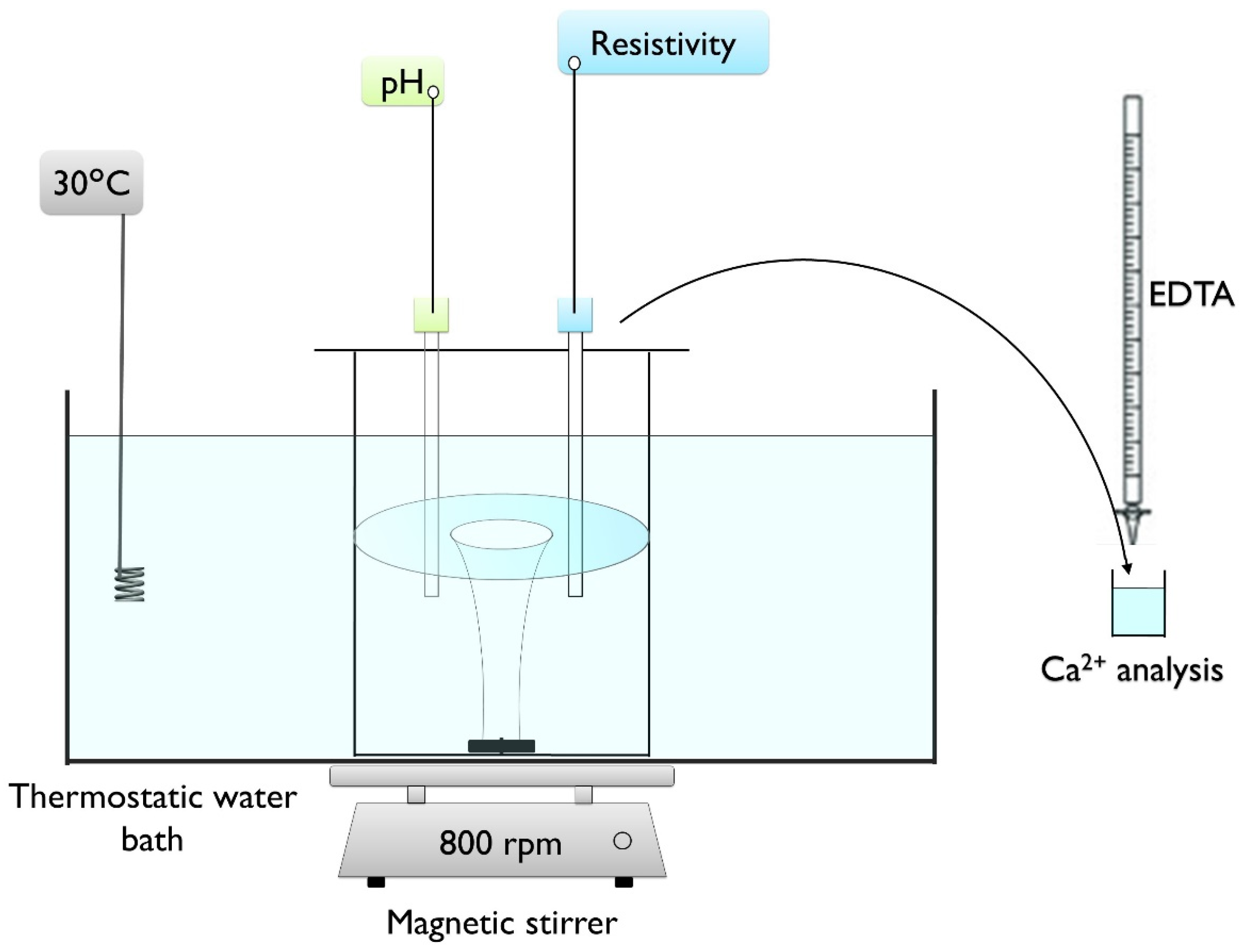
2.2. Fast Controlled Precipitation Set Up
2.3. X-ray Diffraction Method for Solid Precipitate Characterization
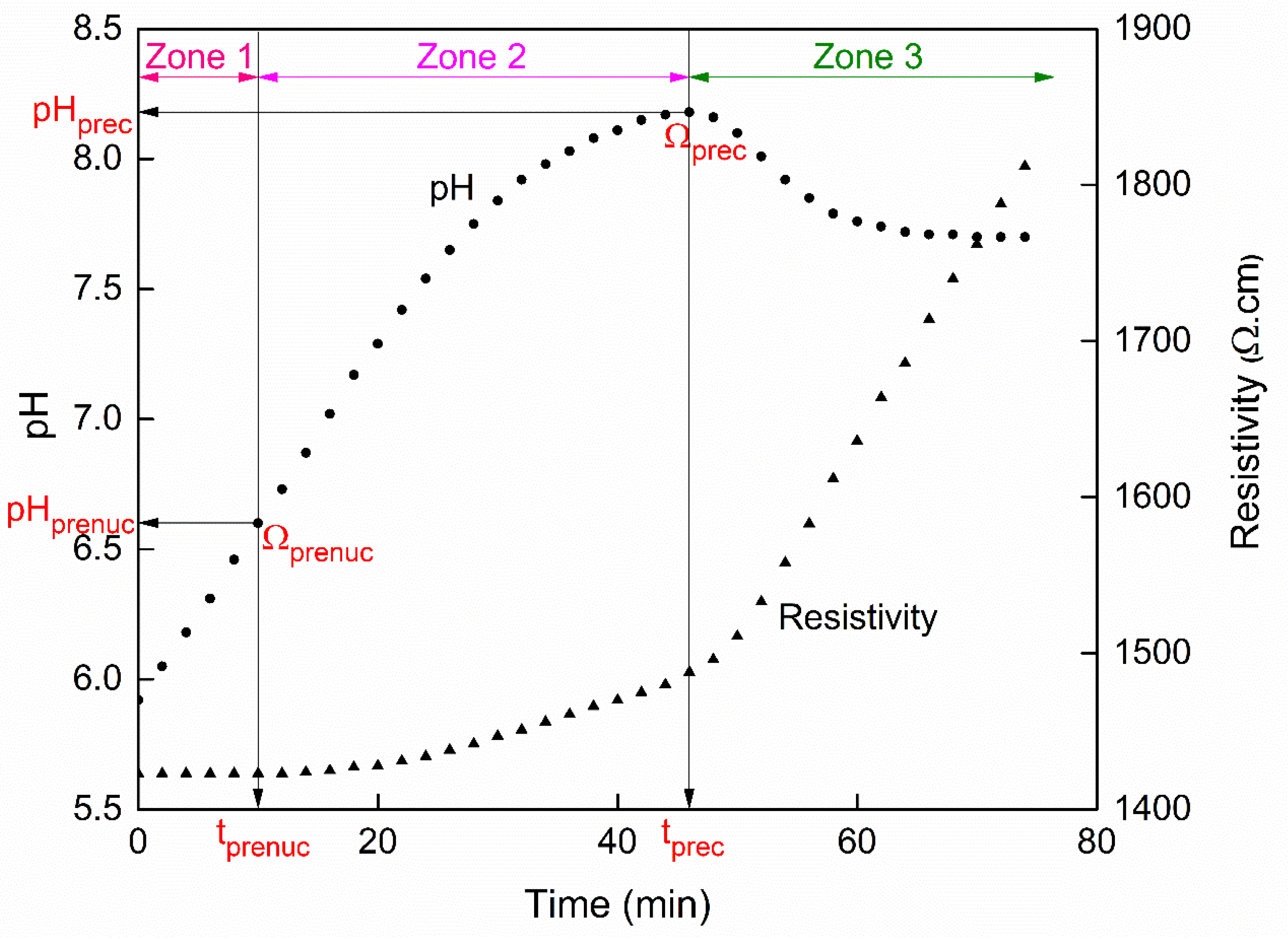
3. Results and Discussion
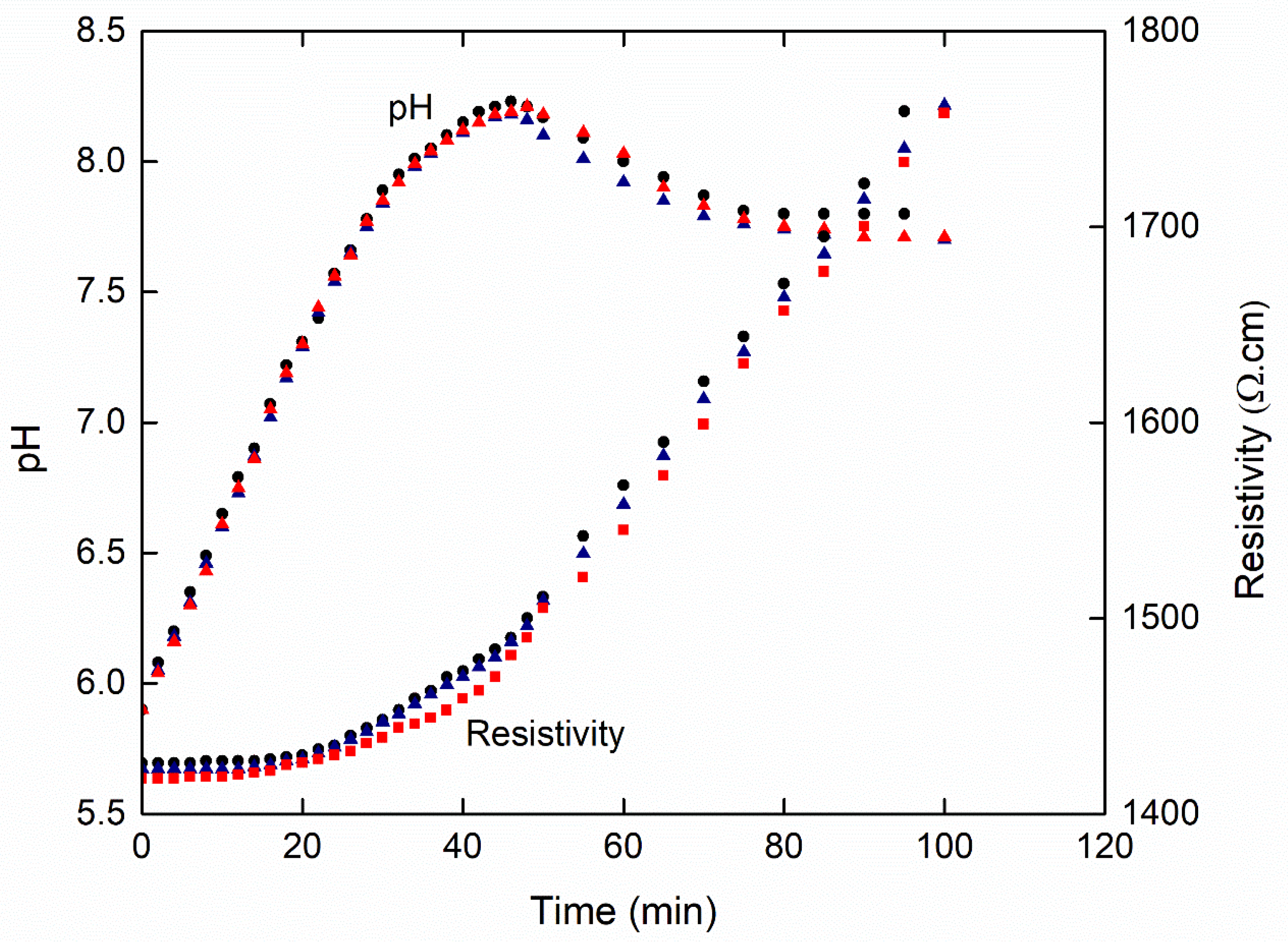
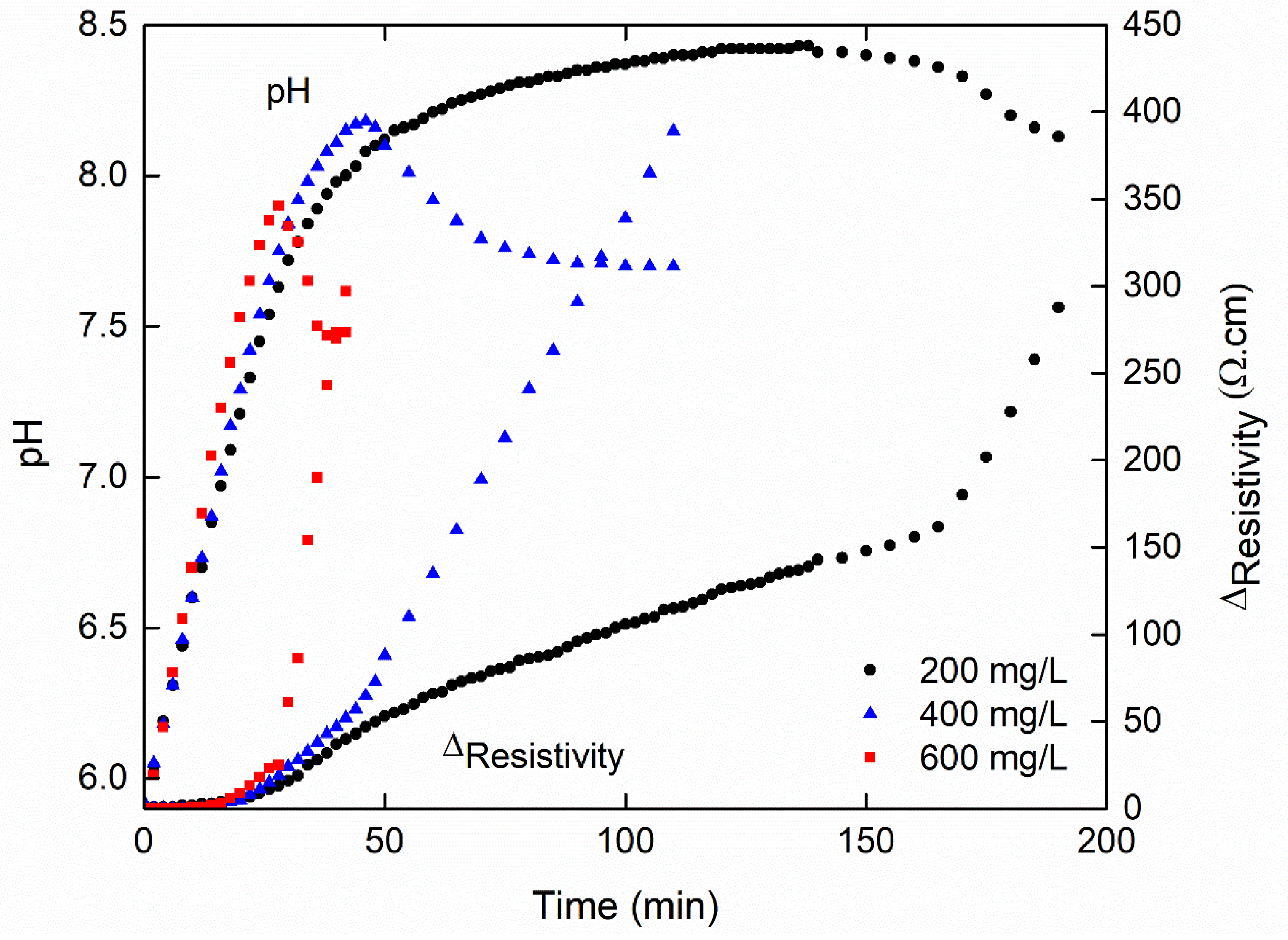
3.1. Effect of Initial Calcium Carbonate Concentration
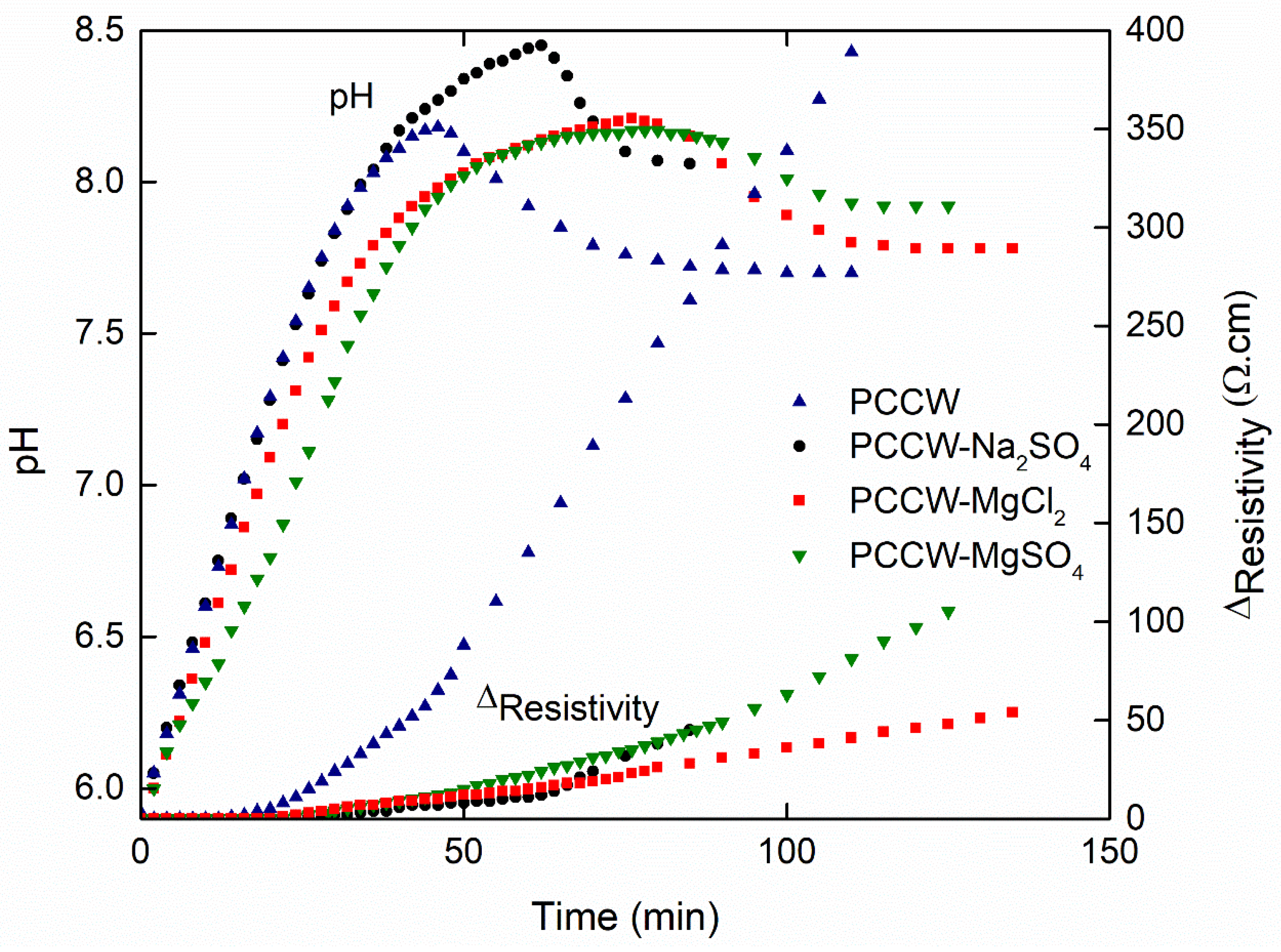
3.2. Effect of Foreign Salts
3.2.1. Influence on the Nucleation Threshold
3.2.2. Influence on the Surface Scaling
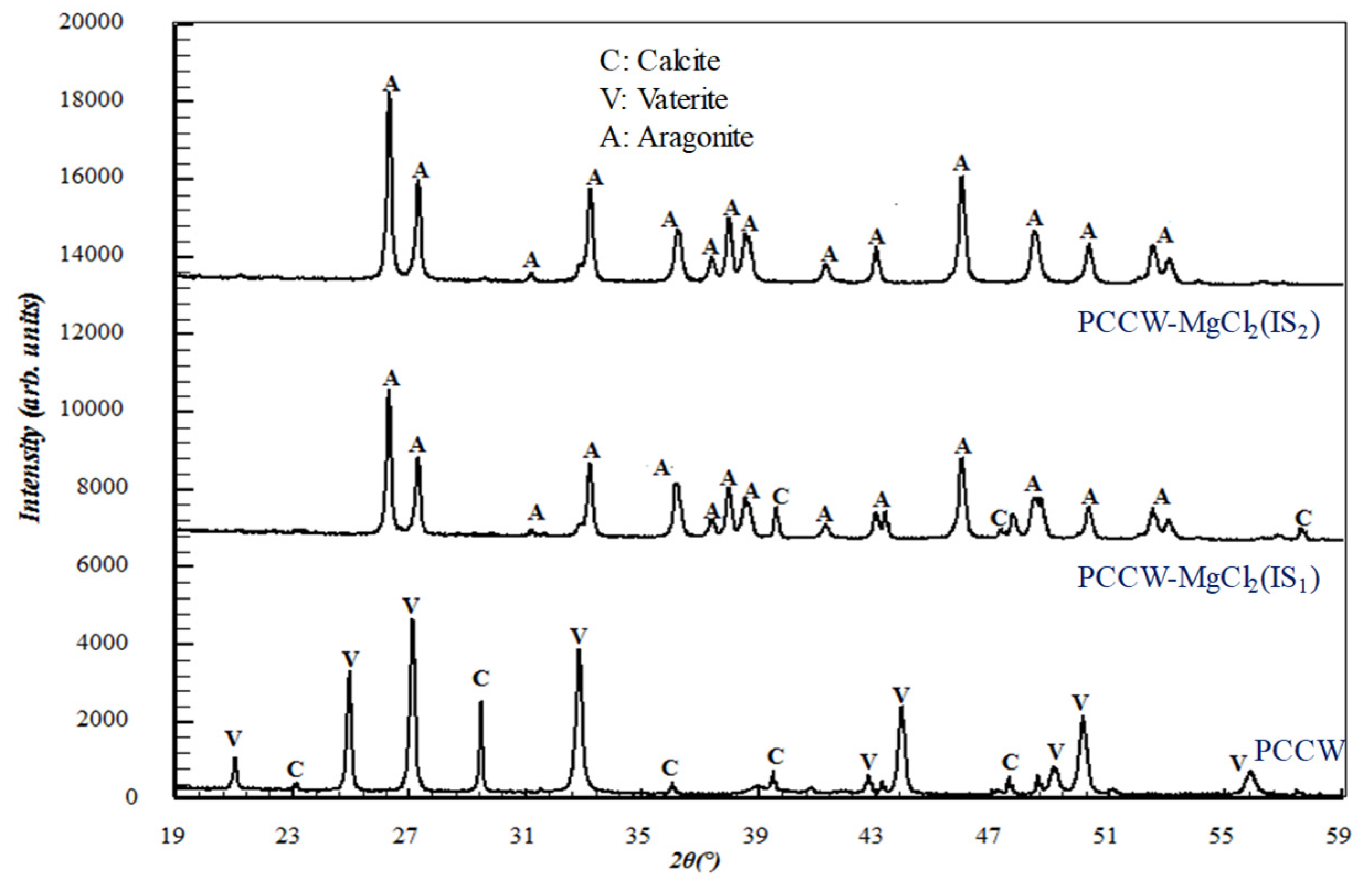
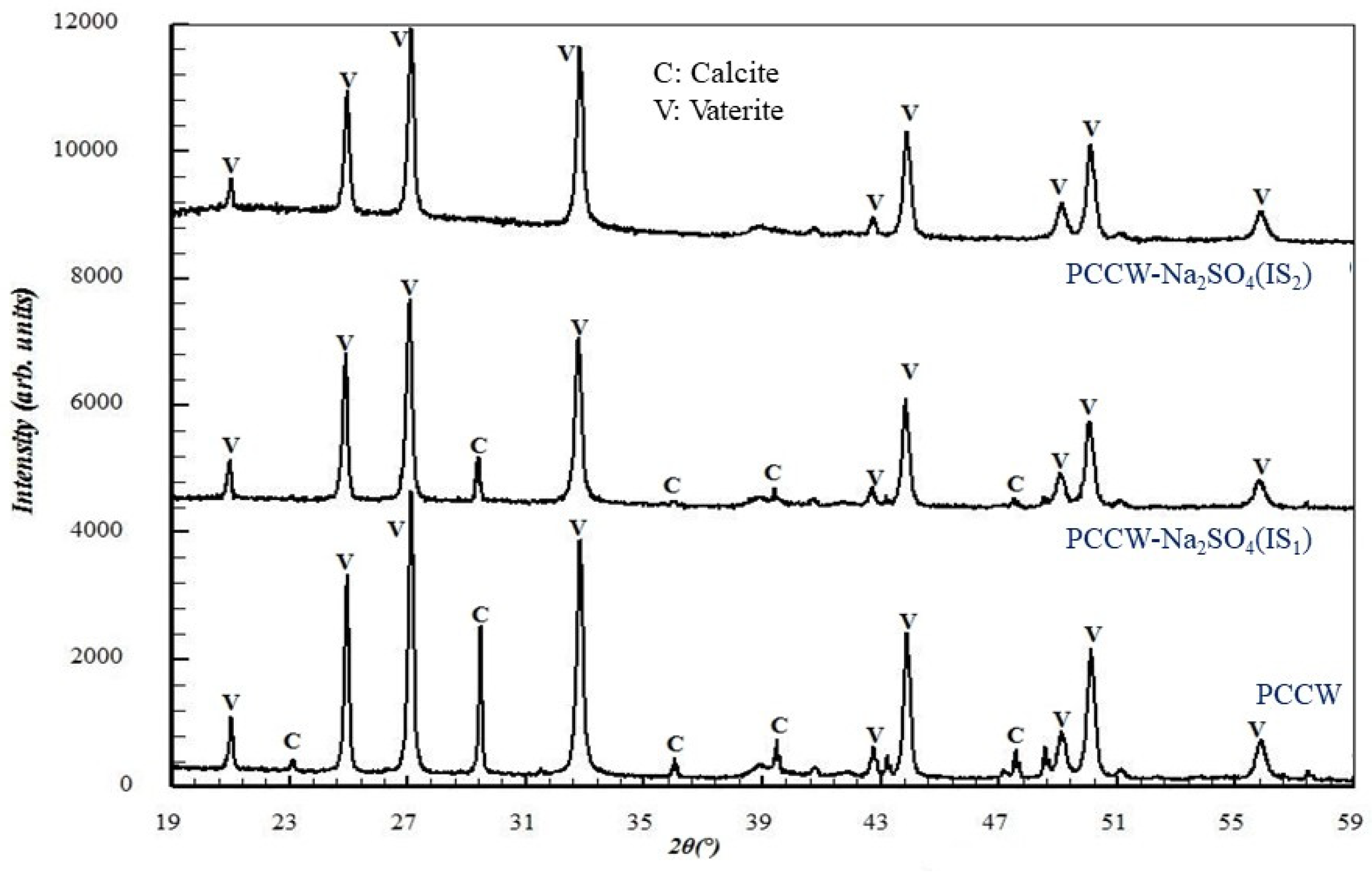
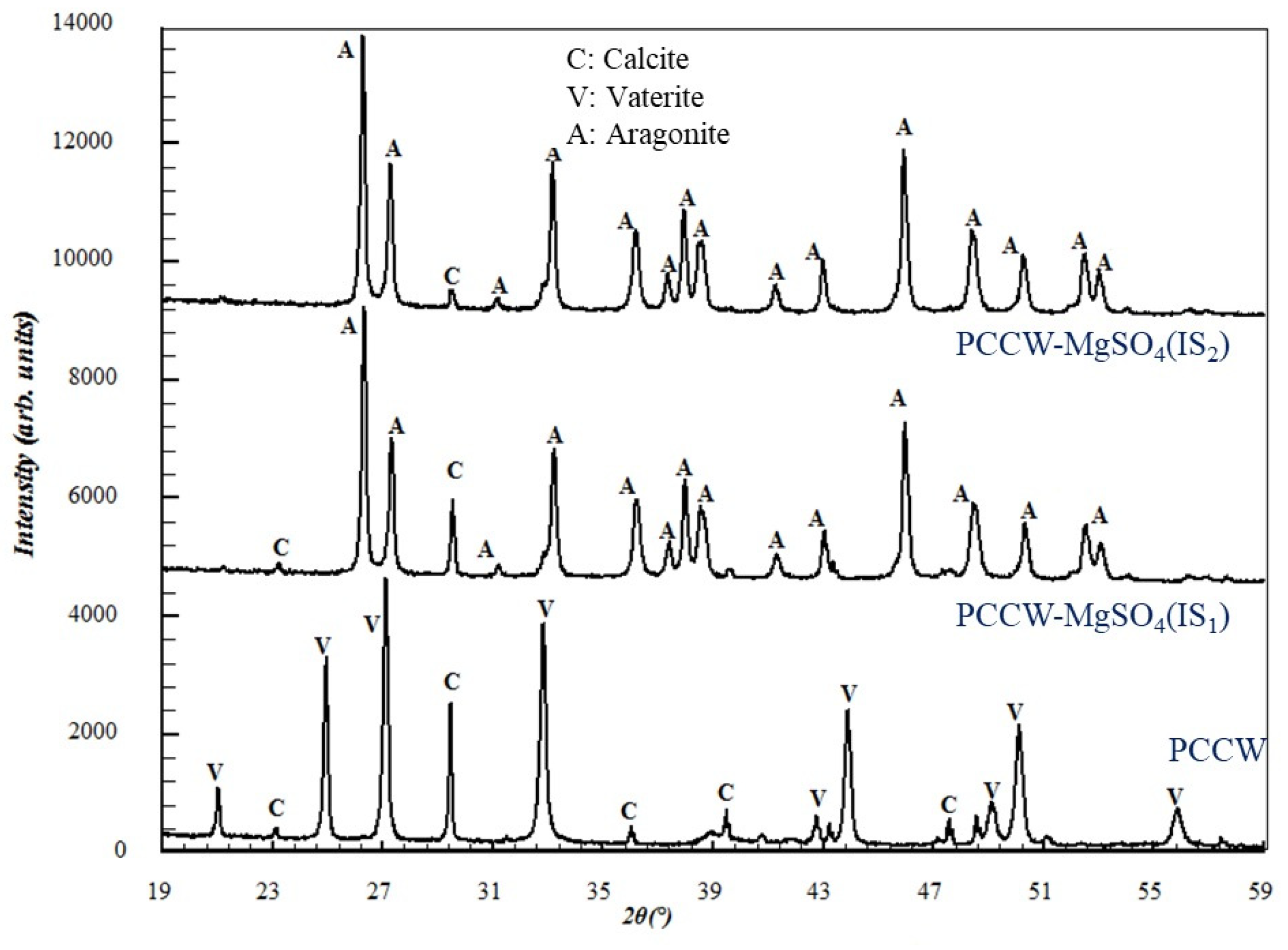
3.2.3. Influence on the Solid Precipitate Microstructure
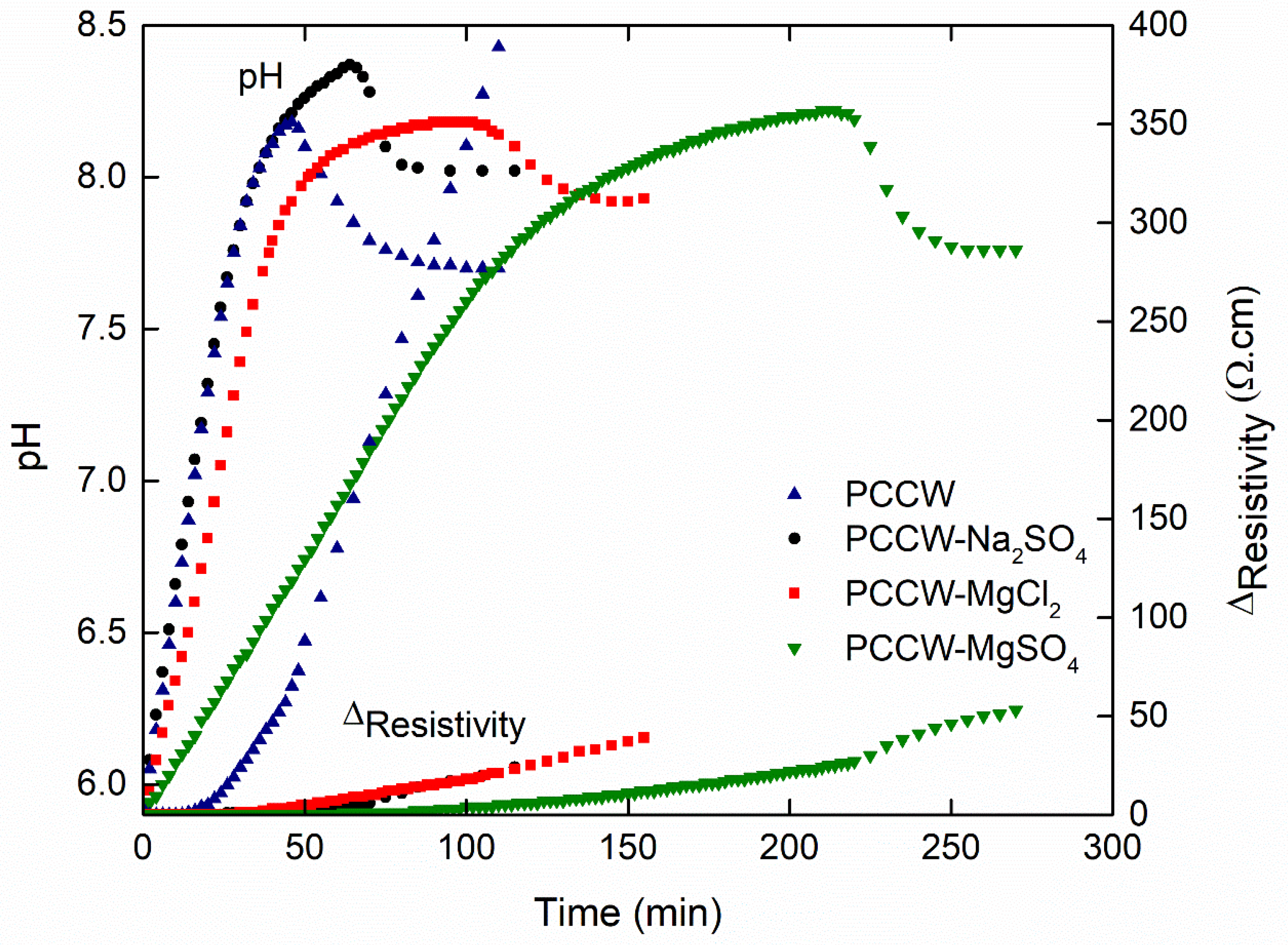
3.3. Effect of Antiscalant
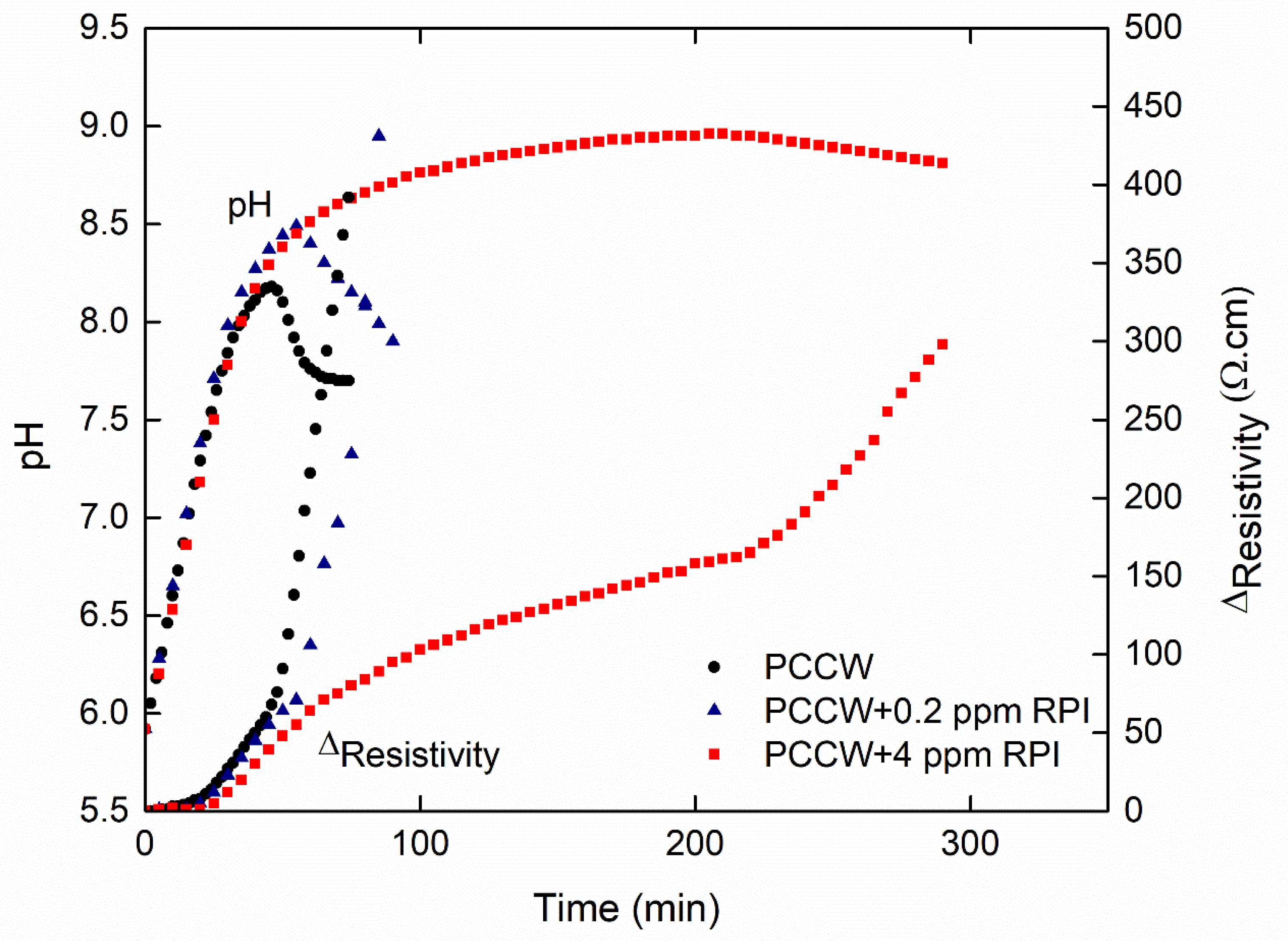
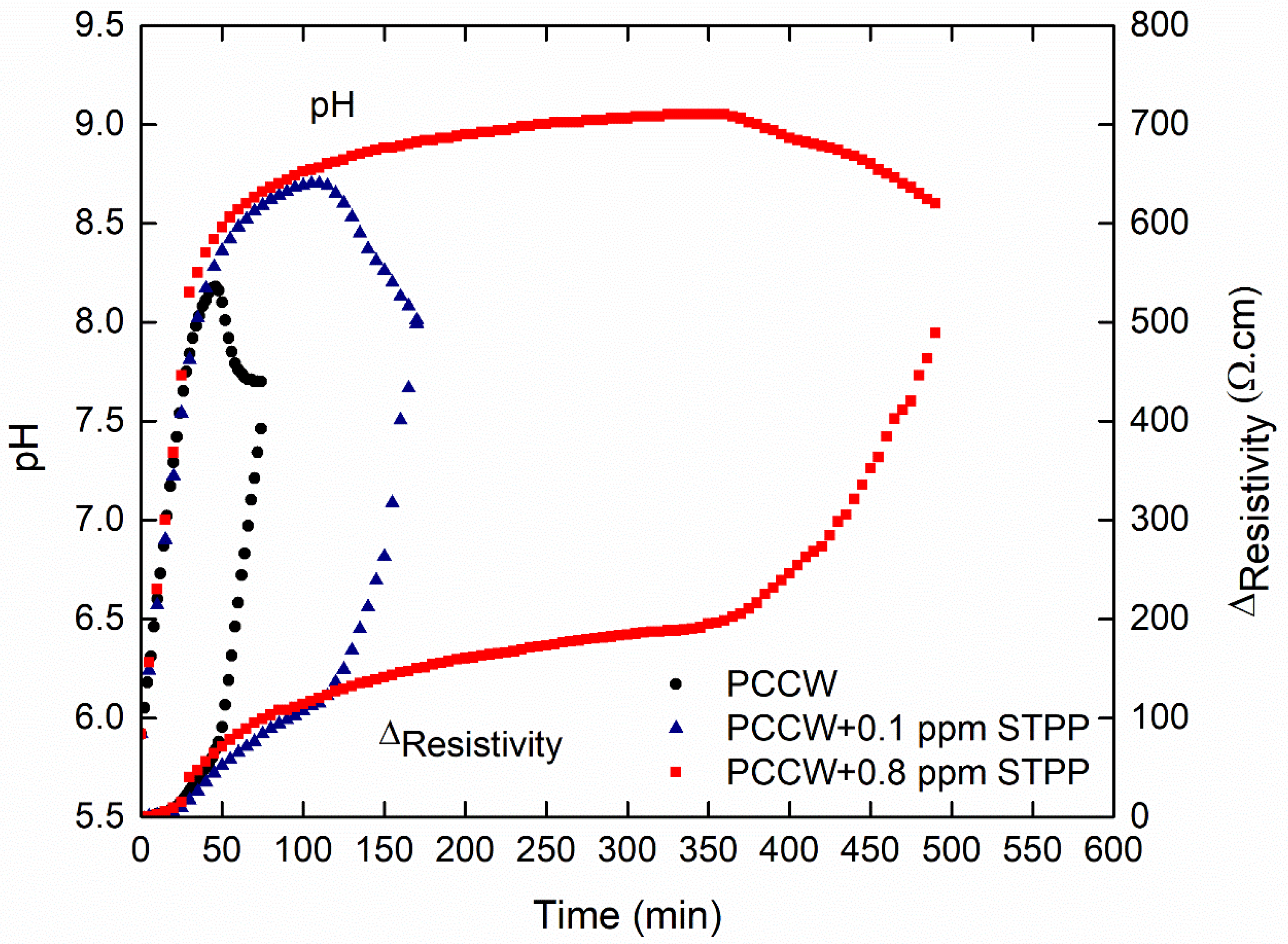
3.3.1. Influence on the Nucleation Threshold
3.3.2. Influence on the Surface Scaling
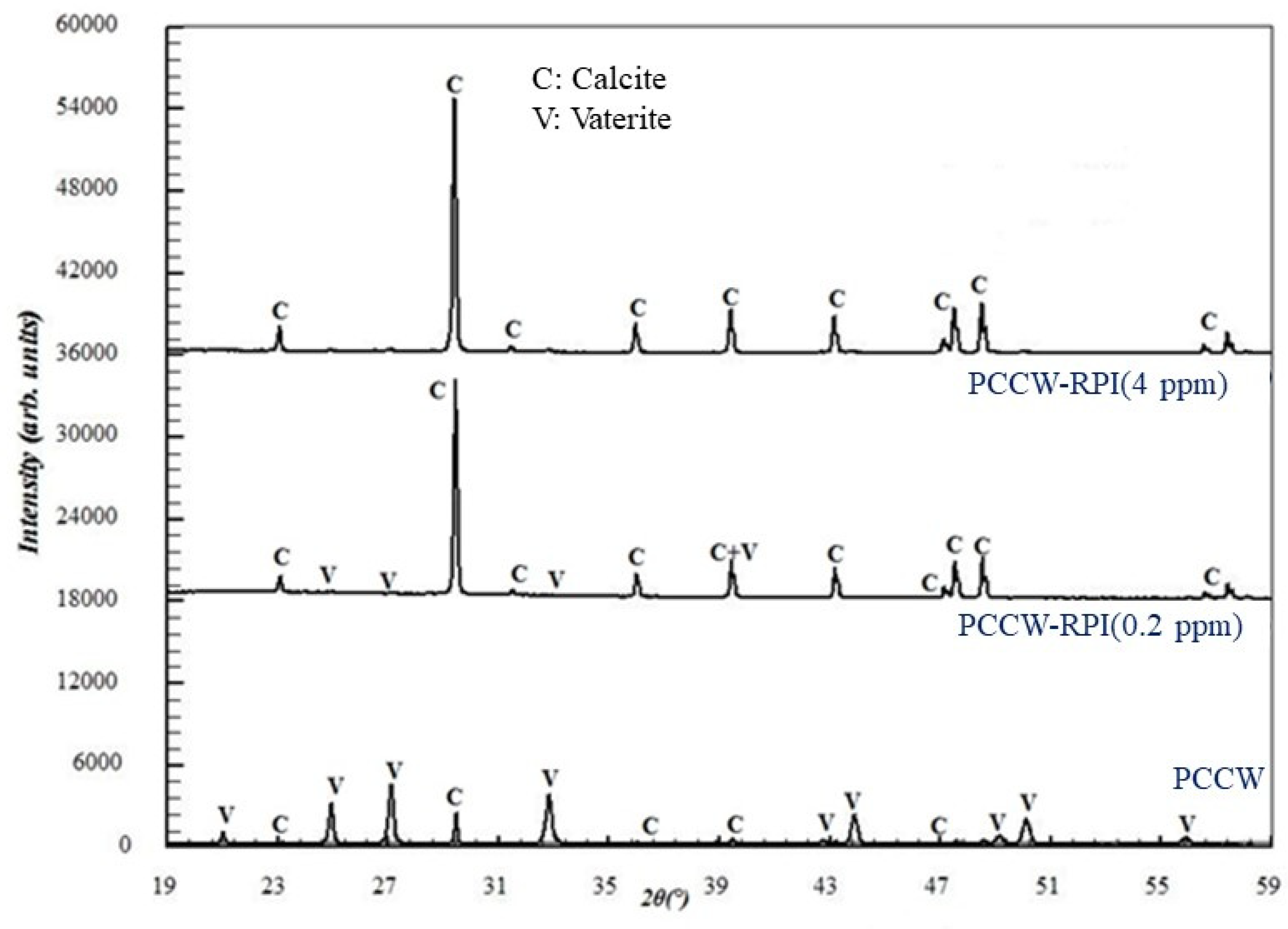
3.3.3. Influence on the Solid Precipitate Microstructure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, J.; Ruan, S.; Liu, Y.; Wang, T.; Zeng, Q.; Yan, D. Morphological characteristics of calcium carbonate crystallization in CO2 pre-cured aerated concrete. RSC Adv. 2022, 12, 14610–14620. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Yang, T.; Dong, S.; Wu, T.; Jin, W.; Wu, Z.; Wang, B.; Liang, T.; Cao, L.; Yu, L. Industrially synthesized biosafe vaterite hollow CaCO3 for controllable delivery of anticancer drugs. Mater. Today Chem. 2022, 24, 100917. [Google Scholar] [CrossRef]
- Lu, H.; Huang, Y.-C.; Hunger, J.; Gebauer, D.; Cölfen, H.; Bonn, M. Role of Water in CaCO3 Biomineralization. J. Am. Chem. Soc. 2021, 143, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Kontrec, J.; Tomašić, N.; Mlinarić, N.M.; Kralj, D.; Džakula, B.N. Effect of pH and Type of Stirring on the Spontaneous Precipitation of CaCO3 at Identical Initial Supersaturation, Ionic Strength and a(Ca2+)/a(CO32−) Ratio. Crystals 2021, 11, 1075. [Google Scholar] [CrossRef]
- Dobberschütz, S.; Nielsen, M.R.; Sand, K.K.; Civioc, R.; Bovet, N.; Stipp, S.L.S.; Andersson, M.P. The mechanisms of crystal growth inhibition by organic and inorganic inhibitors. Nat. Commun. 2018, 9, 1578. [Google Scholar] [CrossRef] [Green Version]
- Lázár, A.; Molnár, Z.; Demény, A.; Kótai, L.; Trif, L.; Béres, K.A.; Bódis, E.; Bortel, G.; Aradi, L.E.; Karlik, M.; et al. Insights into the amorphous calcium carbonate (ACC) → ikaite → calcite transformations. CrystEngComm 2023, 25, 738–750. [Google Scholar] [CrossRef]
- Dong, L.; Xu, Y.-J.; Sui, C.; Zhao, Y.; Mao, L.-B.; Gebauer, D.; Rosenberg, R.; Avaro, J.; Wu, Y.-D.; Gao, H.-L.; et al. Highly hydrated paramagnetic amorphous calcium carbonate nanoclusters as an MRI contrast agent. Nat. Commun. 2022, 13, 5088. [Google Scholar] [CrossRef]
- Raffaella, D.; Paolo, R.; Julian, D.G.; David, Q.; Denis, G. Stable prenucleation mineral clusters are liquid-like ionic polymers. Nat. Commun. 2011, 2, 590. [Google Scholar]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [Green Version]
- Ostwald, W. Studies on formation and transformation of solid materials (Studien uber die bildung und umwandlung fester korper). Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Hamdi, R.; Tlili, M.M. Influence of foreign salts on the CaCO3 pre-nucleation stage: Application of the conductometric method. CrystEngComm 2022, 24, 3256–3267. [Google Scholar] [CrossRef]
- Wolf, S.L.P.; Jähme, K.; Gebauer, D. Synergy of Mg2+ and poly(aspartic acid) in additive-controlled calcium carbonate precipitation. CrystEngComm 2015, 17, 6857–6862. [Google Scholar] [CrossRef] [Green Version]
- Song, R.-Q.; Cölfen, H. Additive controlled crystallization. CrystEngComm 2011, 13, 1249–1276. [Google Scholar] [CrossRef]
- Raj, K.S.; Devi, M.N.; Palanisamy, K.; Subramanian, V.K. Individual and synergetic effect of EDTA and NTA on polymorphism and morphology of CaCO3 crystallization process in presence of barium. J. Solid State Chem. 2021, 302, 122026. [Google Scholar]
- Sancho-Tomás, M.; Fermani, S.; Durán-Olivencia, M.A.; Otálora, F.; Gómez-Morales, J.; Falini, G.; García-Ruiz, J.M. Influence of Charged Polypeptides on Nucleation and Growth of CaCO3 Evaluated by Counterdiffusion Experiments. Cryst. Growth Des. 2013, 13, 3884–3891. [Google Scholar] [CrossRef]
- Ihli, J.; Kim, Y.-Y.; Noel, E.H.; Meldrum, F.C. The Effect of Additives on Amorphous Calcium Carbonate (ACC): Janus Behavior in Solution and the Solid State. Adv. Funct. Mater. 2013, 23, 1575–1585. [Google Scholar] [CrossRef]
- Xiang, L.; Xiang, Y.; Wang, Z.G.; Jin, Y. Influence of chemical additives on the formation of super-fine calcium carbonate. Powder Technol. 2002, 126, 129–133. [Google Scholar] [CrossRef]
- Davis, K.J.; Dove, P.M.; De Yoreo, J.J. The Role of Mg2+ as an Impurity in Calcite Growth. Science 2000, 290, 1134–1137. [Google Scholar] [CrossRef]
- Loste, E.; Wilson, R.M.; Seshadri, R.; Meldrum, F.C. The role of magnesium in stabilising amorphous calcium carbonate and controlling calcite morphologies. J. Cryst. Growth 2003, 254, 206–218. [Google Scholar] [CrossRef]
- Chong, T.H.; Sheikholeslami, R. Thermodynamics and kinetics for mixed calcium carbonate and calcium sulfate precipitation. Chem. Eng. Sci. 2001, 56, 5391–5400. [Google Scholar] [CrossRef]
- Chen, T.; Neville, A.; Yuan, M. Assessing the effect of Mg2+ on CaCO3 scale formation–bulk precipitation and surface deposition. J. Cryst. Growth 2005, 275, e1341–e1347. [Google Scholar] [CrossRef]
- Zhang, Y.; Dawe, R.A. Influence of Mg2+ on the kinetics of calcite precipitation and calcite crystal morphology. Chem. Geol. 2000, 163, 129–138. [Google Scholar] [CrossRef]
- Niedermayr, A.; Köhler, S.J.; Dietzel, M. Impacts of aqueous carbonate accumulation rate, magnesium and polyaspartic acid on calcium carbonate formation (6–40 °C). Chem. Geol. 2013, 340, 105–120. [Google Scholar] [CrossRef]
- Blue, C.R.; Giuffre, A.; Mergelsberg, S.; Han, N.; De Yoreo, J.J.; Dove, P.M. Chemical and physical controls on the transformation of amorphous calcium carbonate into crystalline CaCO3 polymorphs. Geochim. Cosmochim. Acta 2017, 196, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Blanco, J.D.; Shaw, S.; Bots, P.; Roncal-Herrero, T.; Benning, L.G. The role of Mg in the crystallization of monohydrocalcite. Geochim. Cosmochim. Acta 2014, 127, 204–220. [Google Scholar] [CrossRef]
- Zhang, A.; Xie, H.; Liu, N.; Chen, B.-L.; Ping, H.; Fu, Z.-Y.; Su, B.-L. Crystallization of calcium carbonate under the influences of casein and magnesium ions. RSC Adv. 2016, 6, 110362–110366. [Google Scholar] [CrossRef]
- Nielsen, M.R.; Sand, K.K.; Rodriguez-Blanco, J.D.; Bovet, N.; Generosi, J.; Dalby, K.N.; Stipp, S.L.S. Inhibition of Calcite Growth: Combined Effects of Mg2+ and SO42–. Cryst. Growth Des. 2016, 16, 6199–6207. [Google Scholar] [CrossRef]
- Sheikholeslami, R.; Ong, H.W.K. Kinetics and thermodynamics of calcium carbonate and calcium sulfate at salinities up to 1.5 M. Desalination 2003, 157, 217–234. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, F.; Cao, Z.; Jing, W.; Chen, Y. Crystallization of CaCO3 in the presence of sulfate and additives: Experimental and molecular dynamics simulation studies. J. Colloid Interface Sci. 2012, 377, 430–437. [Google Scholar] [CrossRef]
- Hamdi, R.; Tlili, M.M. Investigation of scale inhibitors effect on calcium carbonate nucleation process. Desalination Water Treat. 2019, 160, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Jain, T.; Sanchez, E.; Owens-Bennett, E.; Trussell, R.; Walker, S.; Liu, H. Impacts of antiscalants on the formation of calcium solids: Implication on scaling potential of desalination concentrate. Environ. Sci. Water Res. Technol. 2019, 5, 1285–1294. [Google Scholar] [CrossRef]
- Bai, S.; Naren, G.; Nakano, M.; Okaue, Y.; Yokoyama, T. Effect of polysilicic acid on the precipitation of calcium carbonate. Colloids Surf. A Physicochem. Eng. Asp. 2014, 445, 54–58. [Google Scholar] [CrossRef]
- Chhim, N.; Haddad, E.; Neveux, T.; Bouteleux, C.; Teychené, S.; Biscans, B. Performance of green antiscalants and their mixtures in controlled calcium carbonate precipitation conditions reproducing industrial cooling circuits. Water Res. 2020, 186, 116334. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, S.; Boch, R.; Leis, A.; Baldermann, A.; Domberger, G.; Schwab, C.; Dietzel, M. Green inhibitors reduce unwanted calcium carbonate precipitation: Implications for technical settings. Water Res. 2022, 208, 117850. [Google Scholar] [CrossRef]
- Gauthier, G.; Chao, Y.; Horner, O.; Alos-Ramos, O.; Hui, F.; Lédion, J.; Perrot, H. Application of the Fast Controlled Precipitation method to assess the scale-forming ability of raw river waters. Desalination 2012, 299, 89–95. [Google Scholar] [CrossRef]
- Lédion, J.; François, B.; Vienne, J. Characterization of the scaling properties of water by fast controlled precipitation test. Eur. J. Water Qual. 1997, 28, 15–35. [Google Scholar] [CrossRef]
- Rosset, R.; Sok, P.; Poindessous, G.; Amor, M.B.; Walha, K. Caractérisation de la compacité des dépôts de carbonate de calcium d’eaux géothermales du Sud tunisien par impédancemétrie. Comptes Rendus L’académie Sci. Ser. IIC Chem. 1998, 1, 751–759. [Google Scholar] [CrossRef]
- Amor, M.B.; Zgolli, D.; Tlili, M.M.; Manzola, A.S. Influence of water hardness, substrate nature and temperature on heterogeneous calcium carbonate nucleation. Desalination 2004, 166, 79–84. [Google Scholar] [CrossRef]
- Fathi, A.; Mohamed, T.; Claude, G.; Maurin, G.; Mohamed, B.A. Effect of a magnetic water treatment on homogeneous and heterogeneous precipitation of calcium carbonate. Water Res. 2006, 40, 1941–1950. [Google Scholar] [CrossRef]
- Waly, T.; Kennedy, M.D.; Witkamp, G.-J.; Amy, G.; Schippers, J.C. The role of inorganic ions in the calcium carbonate scaling of seawater reverse osmosis systems. Desalination 2012, 284, 279–287. [Google Scholar] [CrossRef]
- Mullin, J.W. Crystallization; Butterworth-Heinemann: Oxford, UK, 2001. [Google Scholar]
- Söhnel, O.; Garside, J.H. Precipitation: Basic Principles and Industrial Applications; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Chen, T.; Neville, A.; Yuan, M. Influence of on formation—Bulk precipitation and surface deposition. Chem. Eng. Sci. 2006, 61, 5318–5327. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, F.; Farfan, G.A.; Xu, H. Low-Temperature Synthesis of Disordered Dolomite and High-Magnesium Calcite in Ethanol–Water Solutions: The Solvation Effect and Implications. ACS Omega 2022, 7, 281–292. [Google Scholar] [CrossRef]
- Tlili, M.M.; Amor, M.B.; Gabrielli, C.; Joiret, S.; Maurin, G.; Rousseau, P. Study of Electrochemical Deposition of CaCO3 by In Situ Raman Spectroscopy: II. Influence of the Solution Composition. J. Electrochem. Soc. 2003, 150, C485–C493. [Google Scholar] [CrossRef]
- Zahid, A. Mineral Scale Formation and Inhibition; Plenum Press: New York, NY, USA, 1995. [Google Scholar]
- Compton, R.G.; Brown, C.A. The Inhibition of Calcite Dissolution/Precipitation: Mg2+ Cations. J. Colloid Interface Sci. 1994, 165, 445–449. [Google Scholar] [CrossRef]
- Sheng, K.; Ge, H.; Huang, X.; Zhang, Y.; Song, Y.; Ge, F.; Zhao, Y.; Meng, X. Formation and Inhibition of Calcium Carbonate Crystals under Cathodic Polarization Conditions. Crystals 2020, 10, 275. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
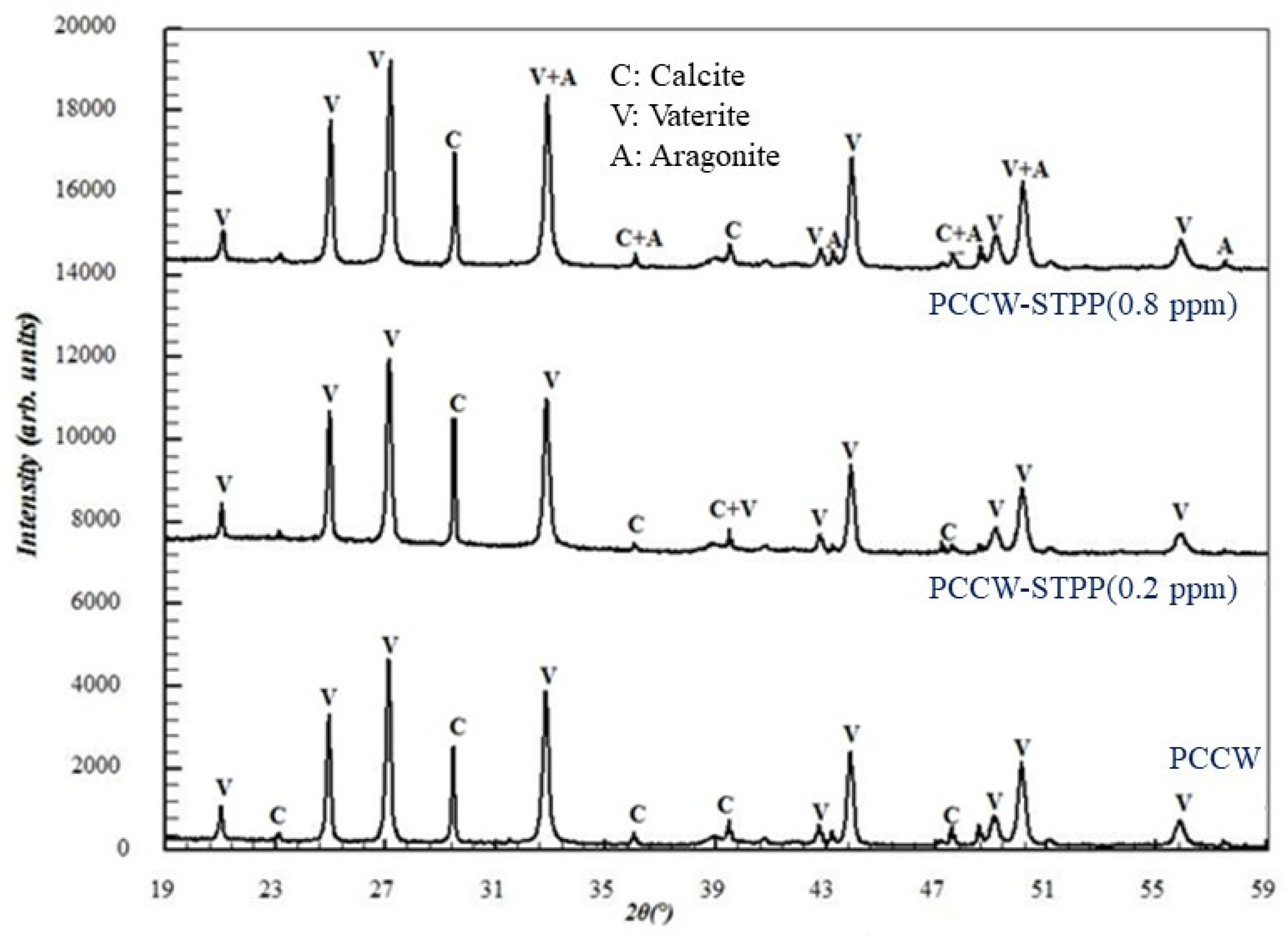{kind=link}
| [CaCO3]i (mg L−1) | tprenuc (min) | Ωprenuc | tprec (min) | Ωprec | ∆t (min) | %hete |
|---|---|---|---|---|---|---|
| 200 | 18 | 1.1 | 138 | 24 | 120 | 56 |
| 400 | 12 | 1.7 | 46 | 48 | 34 | 45 |
| 600 | 8 | 2.0 | 28 | 51 | 20 | 34 |
| IS (mol L−1) | Solution | tprenuc (min) | Ωprenuc | tprec (min) | Ωprec | τprec (%) | %hete |
|---|---|---|---|---|---|---|---|
| IS0 = 0.012 | PCCW | 12 | 1.7 | 46 | 48 | 45 | 45 |
| IS1 = 0.024 | PCCW-Na2SO4 | 20 | 5.0 | 62 | 74 | 27 | 77 |
| PCCW-MgCl2 | 20 | 3.2 | 76 | 43 | 40 | 80 | |
| PCCW-MgSO4 | 22 | 1.7 | 80 | 35 | 36 | 72 | |
| IS2 = 0.036 | PCCW-Na2SO4 | 24 | 8.7 | 64 | 55 | 26 | 72 |
| PCCW-MgCl2 | 28 | 4.4 | 102 | 35 | 38 | 79 | |
| PCCW-MgSO4 | 60 | 1.6 | 214 | 33 | 35 | 71 |
| Solution | tprenuc (min) | Ωprenuc | tprec (min) | Ωprec | τprec | %hete |
|---|---|---|---|---|---|---|
| PCCW | 12 | 1.7 | 46 | 48 | 45 | 45 |
| PCCW-RPI (0.2 ppm) | 15 | 3 | 55 | 97 | 34 | 74 |
| PCCW-RPI (4 ppm) | 20 | 5 | 210 | 286 | 29 | 79 |
| PCCW-STPP (0.1 ppm) | 15 | 2.5 | 110 | 157 | 37 | 66 |
| PCCW-STPP (0.8 ppm) | 20 | 7 | 365 | 353 | 33 | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdi, R.; Tlili, M.M. Influence of Foreign Salts and Antiscalants on Calcium Carbonate Crystallization. Crystals 2023, 13, 516. https://doi.org/10.3390/cryst13030516
Hamdi R, Tlili MM. Influence of Foreign Salts and Antiscalants on Calcium Carbonate Crystallization. Crystals. 2023; 13(3):516. https://doi.org/10.3390/cryst13030516
Chicago/Turabian StyleHamdi, Raghda, and Mohamed Mouldi Tlili. 2023. "Influence of Foreign Salts and Antiscalants on Calcium Carbonate Crystallization" Crystals 13, no. 3: 516. https://doi.org/10.3390/cryst13030516