

Structural Origin of Anisotropic Thermal Expansion of Molecular Crystals and Implication for the Density Rule Probed with Four ROY Polymorphs

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
Thermal Expansivity Measurements
3. Results
3.1. Thermal Expansivity of ROY Polymorphs
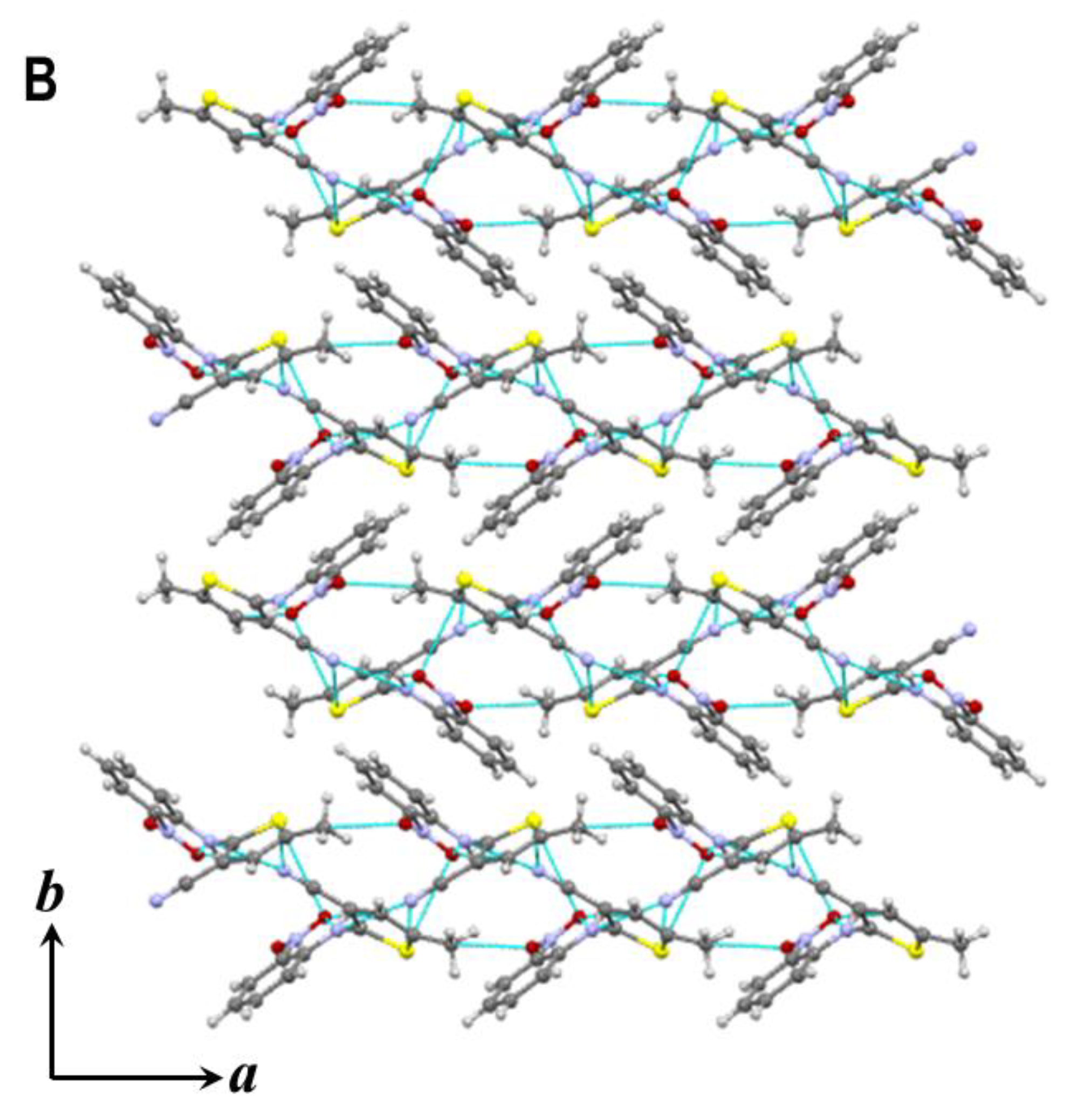
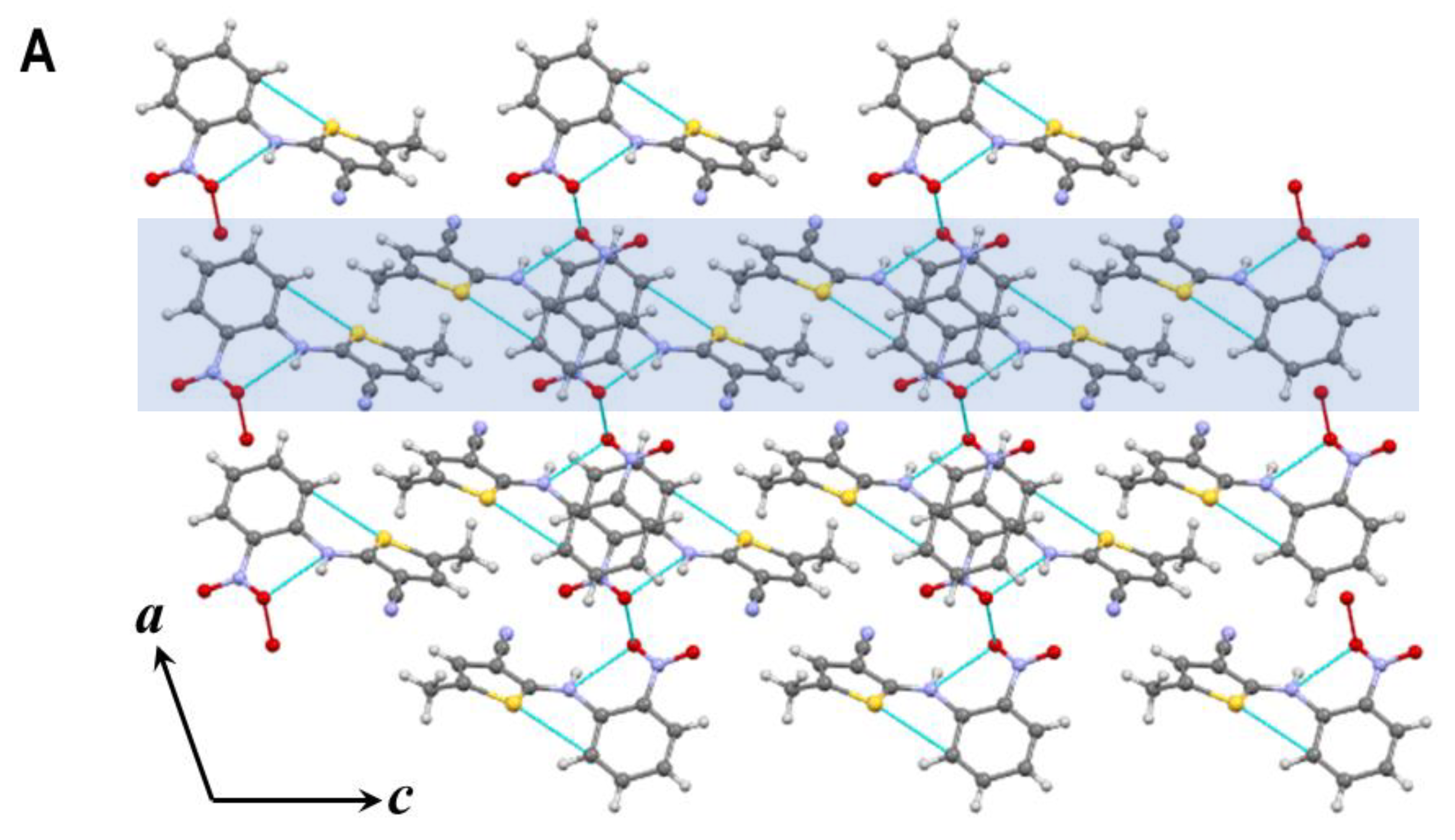
3.2. Hydrogen Bonds and Crystal Packing of Polymorphs
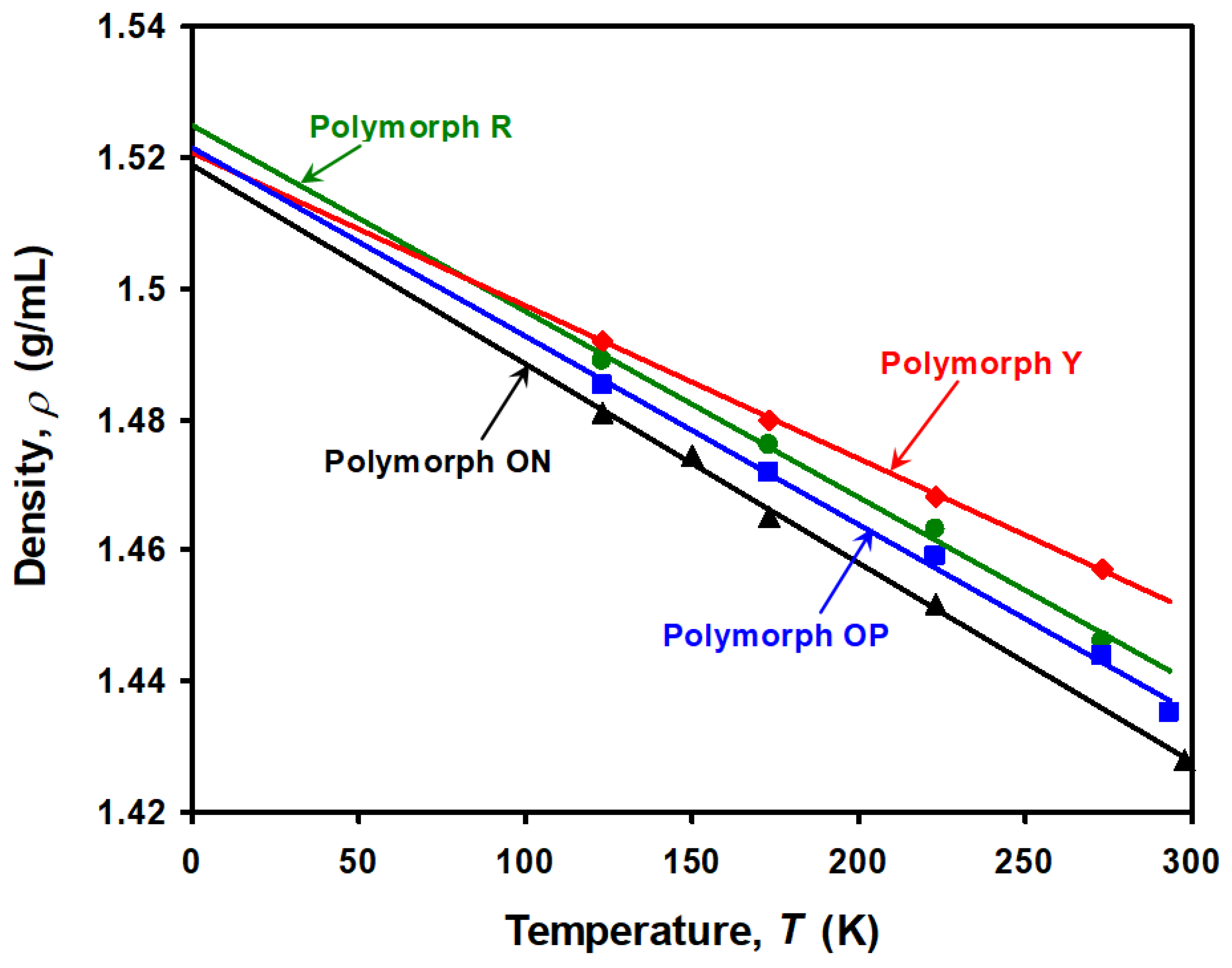
3.3. Crossover of Thermal Density Lines
4. Discussion
4.1. Structure–Expansivity Correlations
4.2. Density Crossover among Polymorphs
5. Conclusions
Supplementary Materials
CCDC Reference Numbers
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verma, A.R.; Krishna, P. Polymorphism and Polytypism in Crystals; John Wiley & Sons: New York, NY, USA, 1966. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: New York, NY, USA, 2002. [Google Scholar]
- Byrn, S.R. Solid State Chemistry of Drugs; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Brittain, H.G. (Ed.) Polymorphism in Pharmaceutical Solids; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Borka, L.; Haleblian, J.K. Crystal polymorphism of pharmaceuticals. Acta Pharm. Jugosl. 1990, 40, 71–94. [Google Scholar]
- Haleblian, J.K. Characterization of habits and crystalline modification of solids and their pharmaceutical applications. J. Pharm. Sci. 1975, 64, 1269–1288. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. 2004, 3, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Grant, D.J.W. Influence of Crystal Structure on the Tableting Properties of Sulfamerazine Polymorphs. Pharm. Res. 2001, 18, 274–280. [Google Scholar] [CrossRef]
- Taylor, R.E. Thermal Expansion of Solids; Ho, C.Y., Ed.; ASM International: Materials Park, OH, USA, 1998; Volume I-4. [Google Scholar]
- Taylor, R.E.; Denman, G.L. (Eds.) Thermal Expansion; American Institute of Physics: New York, NY, USA, 1974. [Google Scholar]
- Das, D.; Jacobs, T.; Pietraszko, A.; Barbour, L.J. Anomalous thermal expansion of an organic crystal—Implications for elucidating the mechanism of an enantiotropic phase transformation. Chem. Commun. 2011, 47, 6009–6011. [Google Scholar] [CrossRef]
- Liang, E.; Sun, Q.; Yuan, H.; Wang, J.; Zeng, G.; Gao, Q. Negative thermal expansion: Mechanisms and materials. Front. Phys. 2021, 16, 53302. [Google Scholar] [CrossRef]
- Lee, A.v.d.; Dumitrescu, D.G. Thermal expansion properties of organic crystals: A CSD study. Chem. Sci. 2021, 12, 8537–8547. [Google Scholar]
- Salvador, J.R.; Guo, F.; Hogan, T.; Kanatzidis, M.G. Zero thermal expansion in YbGaGe due to an electronic valence transition. Nature 2003, 425, 702–705. [Google Scholar] [CrossRef]
- Margadonna, S.; Prassides, K.; Fitch, A.N. Zero Thermal Expansion in a Prussian Blue Analogue. J. Am. Chem. Soc. 2004, 126, 15390–15391. [Google Scholar] [CrossRef]
- Sun, C.C. Thermal Expansion of Organic Crystals and Precision of Calculated Crystal Density: A Survey of Cambridge Crystal Database. J. Pharm. Sci. 2007, 96, 1043–1052. [Google Scholar] [CrossRef]
- Fortes, A.D.; Suard, E.; Knight, K.S. Negative Linear Compressibility and Massive Anisotropic Thermal Expansion in Methanol Monohydrate. Science 2011, 331, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Grima, J.N.; Attard, D.; Gatt, R. Unusual Thermoelastic Properties of Methanol Monohydrate. Science 2011, 331, 687–688. [Google Scholar] [CrossRef]
- White, G.K.; Choy, C.L. Thermal expansion and Grüneisen parameters of isotropic and oriented polyethylene. J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 835–846. [Google Scholar] [CrossRef]
- Burger, A.; Ramberger, R. On the Polymorphism of Pharmaceuticals and Other Molecular Crystals. I Theory of Thermodynamic Rules. Mikrochim. Acta 1979, 72, 259–271. [Google Scholar] [CrossRef]
- Burger, A.; Ramberger, R. On the Polymorphism of Pharmaceuticals and Other Molecular Crystals. II Applicability of Thermodynamic Rules. Mikrochim. Acta 1979, 72, 273–316. [Google Scholar] [CrossRef]
- Westrum, E.F.; McCullough, J.P. Thermodynamics of Crystals; Interscience Publishers: New York, NY, USA, 1963. [Google Scholar]
- Grant, D.J.W.; Higuchi, T. Solubility Behavior of Organic Compounds; John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Perumalla, S.R.; Wang, C.; Guo, Y.; Shi, L.; Sun, C.C. Robust bulk preparation and characterization of sulfamethazine and saccharine salt and cocrystal polymorphs. Cryst. Eng. Commun. 2019, 21, 2089–2096. [Google Scholar] [CrossRef]
- Yu, L. Inferring Thermodynamic Stability Relationship of Polymorphs from Melting Data. J. Pharm. Sci. 1995, 94, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Stephenson, G.A.; Mitchell, C.A.; Bunnell, C.A.; Snorek, S.V.; Bowyer, J.B.; Borchardt, T.B.; Stowell, J.G.; Byrn, S.R. Thermochemistry and Conformational Polymorphism of a Hexamorphic Crystal System. J. Am. Chem. Soc. 2000, 122, 585–591. [Google Scholar] [CrossRef]
- Yu, L.; Huang, J.; Jones, K.J. Measuring Free-Energy Difference between Crystal Polymorphs through Eutectic Melting. J. Phys. Chem. B 2005, 109, 19915–19922. [Google Scholar] [CrossRef]
- Chen, S.; Guzei, I.A.; Yu, L. New polymorphs of ROY and new record for coexisting polymorphs of solved structures. J. Am. Chem. Soc. 2005, 127, 9881–9885. [Google Scholar] [CrossRef]
- Yao, Z.S.; Guan, H.; Shiota, Y.; He, C.T.; Wang, X.L.; Wu, S.Q.; Zheng, X.; Su, S.Q.; Yoshizawa, K.; Kong, X.; et al. Giant anisotropic thermal expansion actuated by thermodynamically assisted reorientation of imidazoliums in a single crystal. Nat. Commun. 2019, 10, 4805. [Google Scholar] [CrossRef] [PubMed]
- Janiak, A.; Esterhuysen, C.; Barbour, L.J. A thermo-responsive structural switch and colossal anisotropic thermal expansion in a chiral organic solid. Chem. Commun. 2018, 54, 3727–3730. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Saha, B.K. Steric guided anomalous thermal expansion in a dimorphic organic system. CrystEngComm 2014, 16, 2340–2343. [Google Scholar] [CrossRef]
- Ding, X.; Crawford, A.W.; Derrick, W.P.; Unruh, D.K.; Groeneman, R.H.; Hutchins, K.M. Thermal Expansion Properties and Mechanochemical Synthesis of Stoichiometric Cocrystals Containing Tetrabromobenzene as a Hydrogen- and Halogen-Bond Donor. Chem. Eur. J. 2021, 27, 16329–16333. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C-H…O Hydrogen Bond: Structural Implications and Supramolecular Design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C. Materials science tetrahedron--a useful tool for pharmaceutical research and development. J. Pharm. Sci. 2009, 98, 1671–1687. [Google Scholar] [CrossRef]
- Chen, X.; Stowell, J.G.; Morris, K.R.; Byrn, S.R. Quantitative study of solid-state acid–base reactions between polymorphs of flufenamic acid and magnesium oxide using X-ray powder diffraction. J. Pharm. Biomed. Anal. 2010, 51, 866–874. [Google Scholar] [CrossRef]
- Chattoraj, S.; Shi, L.; Sun, C.C. Understanding the relationship between crystal structure, plasticity and compaction behaviour of theophylline, methyl gallate, and their 1:1 co-crystal. Cryst. Eng. Comm. 2010, 12, 2466–2472. [Google Scholar] [CrossRef]
- Wang, K.; Mishra, M.K.; Sun, C.C. Exceptionally Elastic Single-Component Pharmaceutical Crystals. Chem. Mater. 2019, 31, 1794–1799. [Google Scholar] [CrossRef]
- Hu, S.; Mishra, M.K.; Sun, C.C. Twistable Pharmaceutical Crystal Exhibiting Exceptional Plasticity and Tabletability. Chem. Mater. 2019, 31, 3818–3822. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, C.C.; Liu, L.; Wang, C.; Shi, P.; Xu, J.; Wu, S.; Gong, J. Structural Origins of Elastic and 2D Plastic Flexibility of Molecular Crystals Investigated with Two Polymorphs of Conformationally Rigid Coumarin. Chem. Mater. 2021, 33, 1053–1060. [Google Scholar] [CrossRef]
- Brand, H.E.A.; Fortes, A.D.; Wood, I.G.; Knight, K.S.; Vocadlo, L. The thermal expansion and crystal structure of mirabilite (Na2SO4.10D2O) from 4.2 to 300 K, determined by time-of-flight neutron powder diffraction. Phys. Chem. Miner. 2009, 36, 29–46. [Google Scholar] [CrossRef]
- Fortes, A.D.; Wood, I.G.; Knight, K.S. The crystal structure and thermal expansion tensor of MgSO4–11D2O (meridianiite) determined by neutron powder diffraction. Phys. Chem. Miner. 2008, 35, 207–221. [Google Scholar] [CrossRef]
- Das, D.; Jacobs, T.; Barbour, L.J. Exceptionally large positive and negative anisotropic thermal expansion of an organic crystalline material. Nat. Mater. 2010, 9, 36–39. [Google Scholar] [CrossRef]
- Amoros, J.L.; Canut, M.L.; Neira, E. Thermal Expansion of β-Succinic Acid and |alpha-Adipic Acid in Relation to Their Crystal Structures. Proc. Royal Soc. Lond. Ser. A 1965, 285, 370–381. [Google Scholar]
- Stephenson, G.A. Anisotropic lattice contraction in pharmaceuticals: The influence of cryo-crystallography on calculated powder diffraction patterns. J. Pharm. Sci. 2006, 95, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, C.C. Computational Techniques for Predicting Mechanical Properties of Organic Crystals: A Systematic Evaluation. Mol. Pharm. 2019, 16, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Korcok, J.L.; Katz, M.J.; Leznoff, D.B. Impact of Metallophilicity on “Colossal” Positive and Negative Thermal Expansion in a Series of Isostructural Dicyanometallate Coordination Polymers. J. Am. Chem. Soc. 2009, 131, 4866–4871. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Glukhov, I.V.; Antipin, M.Y.; Lyssenko, K.A. “Higher density does not mean higher stability” mystery of paracetamol finally unraveled. Chem. Commun. 2010, 46, 3469–3471. [Google Scholar] [CrossRef]
- Van Wylen, G.J.; Sonntag, R.E. Fundamentals of Classical Thermodynamics, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1985. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorph | Temperature (K) | Crystal System | Axial Expansivity a × 10−6 (K−1) | Angular Expansivity b × 10−6 (K−1) | ||||
|---|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | |||
| Y | 123 | Monoclinic | 13.80 | 133.72 | 34.41 | Invariant | 22.78 | Invariant |
| 173 | 13.79 | 132.98 | 34.34 | 22.74 | ||||
| 223 | 13.79 | 132.19 | 34.29 | 22.71 | ||||
| 298 | 13.76 | 130.75 | 34.20 | 22.69 | ||||
| R | 123 | Triclinic | 135.44 | 78.90 | 14.32 | −8.306 | 25.20 | −73.37 |
| 173 | 134.64 | 78.62 | 14.31 | −8.308 | 25.17 | −73.59 | ||
| 223 | 133.79 | 78.33 | 14.30 | −8.311 | 25.14 | −73.86 | ||
| 273 | 132.73 | 77.97 | 14.29 | −8.316 | 25.10 | −74.18 | ||
| OP | 123 | Monoclinic | 174.66 | 13.023 | 18.14 | Invariant | 30.95 | Invariant |
| 173 | 173.31 | 13.010 | 18.13 | 30.92 | ||||
| 223 | 171.85 | 13.003 | 18.11 | 30.87 | ||||
| 273 | 170.19 | 12.997 | 18.09 | 30.81 | ||||
| ON | 123 | Monoclinic | 131.73 | 51.67 | 20.13 | Invariant | 72.80 | Invariant |
| 173 | 130.94 | 51.49 | 20.10 | 72.53 | ||||
| 223 | 130.19 | 51.34 | 20.09 | 72.25 | ||||
| 298 | 128.76 | 51.22 | 20.07 | 71.89 | ||||
| Polymorph | Hydrogen Bond | Bond Type | Bond Length (Ǻ) | Bond Expansivity × 10−6 (K−1) * |
|---|---|---|---|---|
| Y | N2…H–N1 | Intermolecular | 3.067 | 88.03 |
| S1…H–N2 | Intermolecular | 3.304 | 96.85 | |
| O2…H–C9 | Intermolecular | 3.184 | 75.37 | |
| O2…H–N1 | Intramolecular | 2.620 | 7.63 | |
| R | O2…H–O2 | Intermolecular | 2.936 | 66.07 |
| O2…H–N1 | Intramolecular | 2.634 | −6.07 | |
| S1…H–C2 | Intramolecular | 3.145 | 39.43 | |
| OP | O1…H–O1 | Intermolecular | 2.943 | 102.62 |
| O2…H–N1 | Intramolecular | 2.654 | −18.84 | |
| S1…H–C2 | Intramolecular | 3.239 | 30.26 | |
| ON | O2…H–N1 | Intramolecular | 2.616 | −3.27 |
| S1…H–C2 | Intramolecular | 3.356 | −59.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chattoraj, S.; Sun, C.C. Structural Origin of Anisotropic Thermal Expansion of Molecular Crystals and Implication for the Density Rule Probed with Four ROY Polymorphs. Crystals 2023, 13, 270. https://doi.org/10.3390/cryst13020270
Chattoraj S, Sun CC. Structural Origin of Anisotropic Thermal Expansion of Molecular Crystals and Implication for the Density Rule Probed with Four ROY Polymorphs. Crystals. 2023; 13(2):270. https://doi.org/10.3390/cryst13020270
Chicago/Turabian StyleChattoraj, Sayantan, and Changquan Calvin Sun. 2023. "Structural Origin of Anisotropic Thermal Expansion of Molecular Crystals and Implication for the Density Rule Probed with Four ROY Polymorphs" Crystals 13, no. 2: 270. https://doi.org/10.3390/cryst13020270