
Quantum Chemical GA-MLR, Cluster Model, and Conceptual DFT Descriptors Studies on the Binding Interaction of Estrogen Receptor Alpha with Endocrine Disrupting Chemicals

Abstract
:1. Introduction
2. Computational Details
2.1. Dataset
2.2. Quantum Chemical Descriptors
2.3. GA-MLR Method
2.4. Molecular Docking
2.5. Quantum Chemical Cluster Model Approach
2.6. Density Functional Theory Calculations
3. Results and Discussion
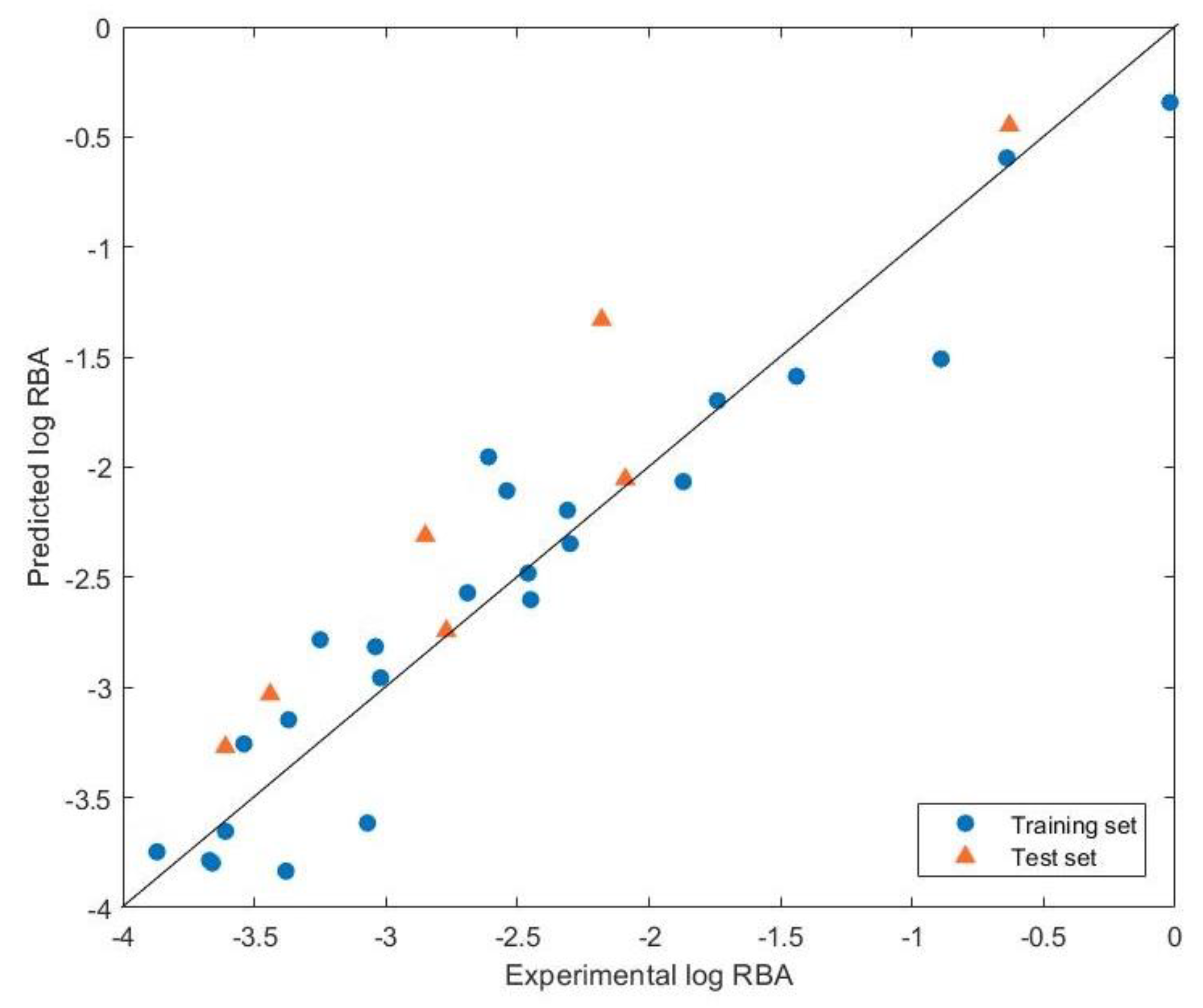
3.1. Quantum Chemical GA-MLR Model
Dipole Moment + 0.0067 (±0.0012) APSA
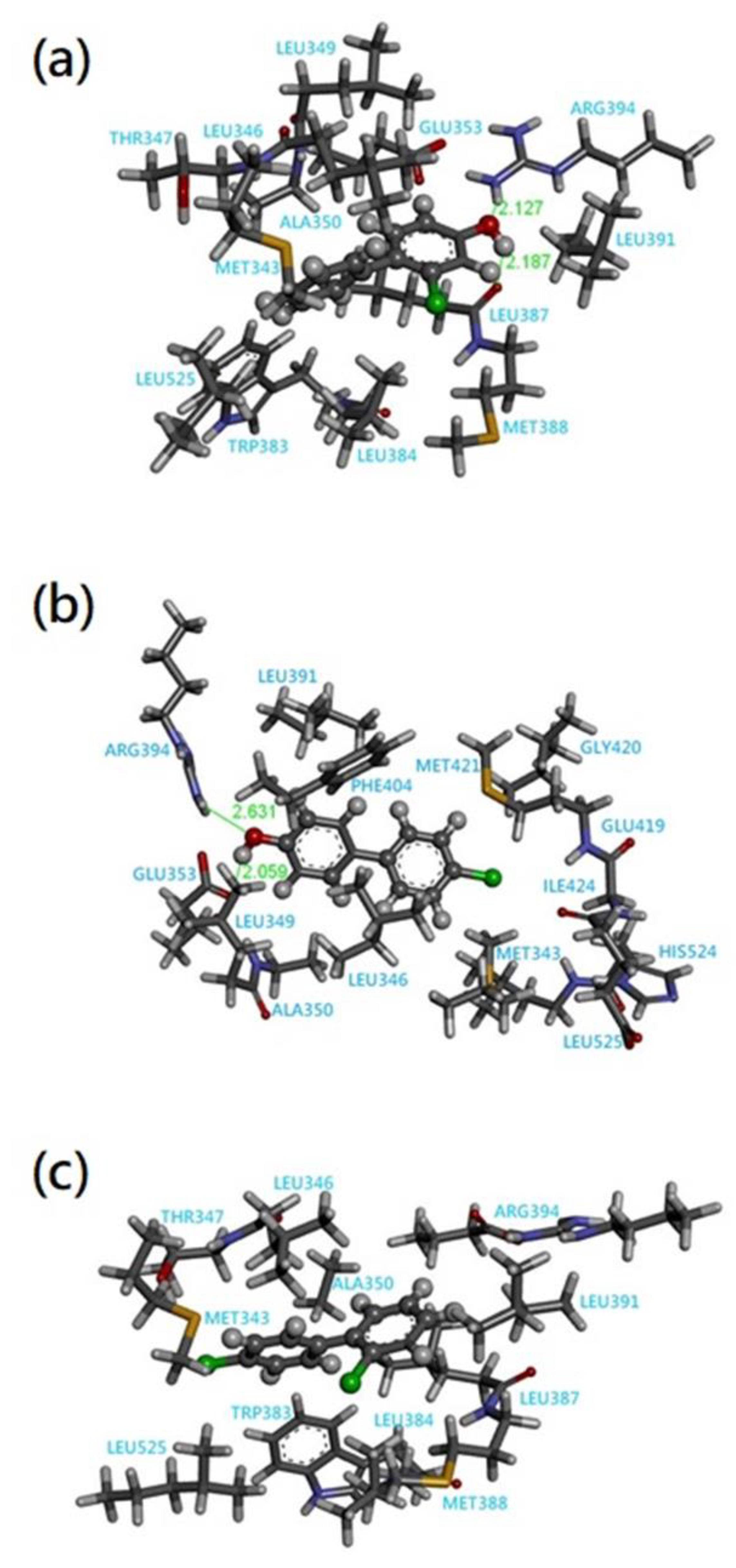
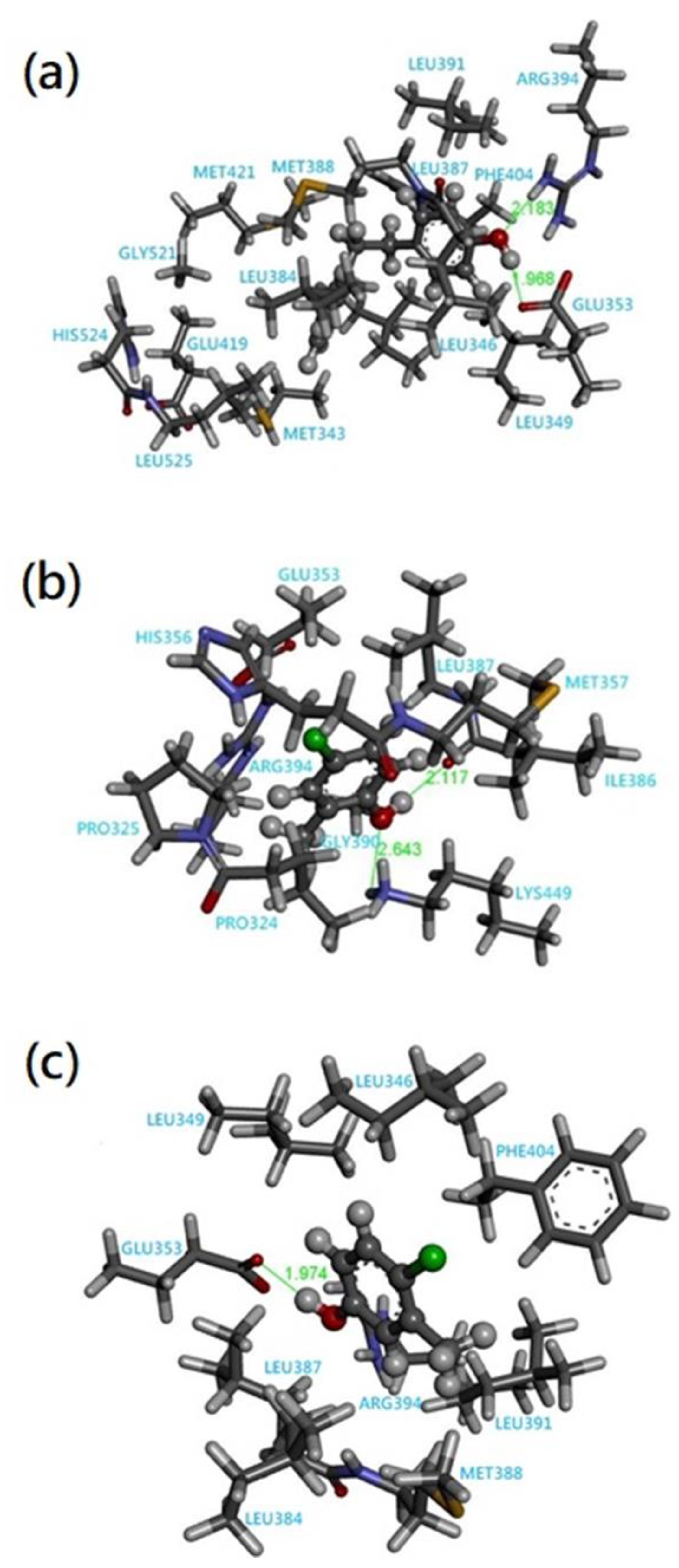
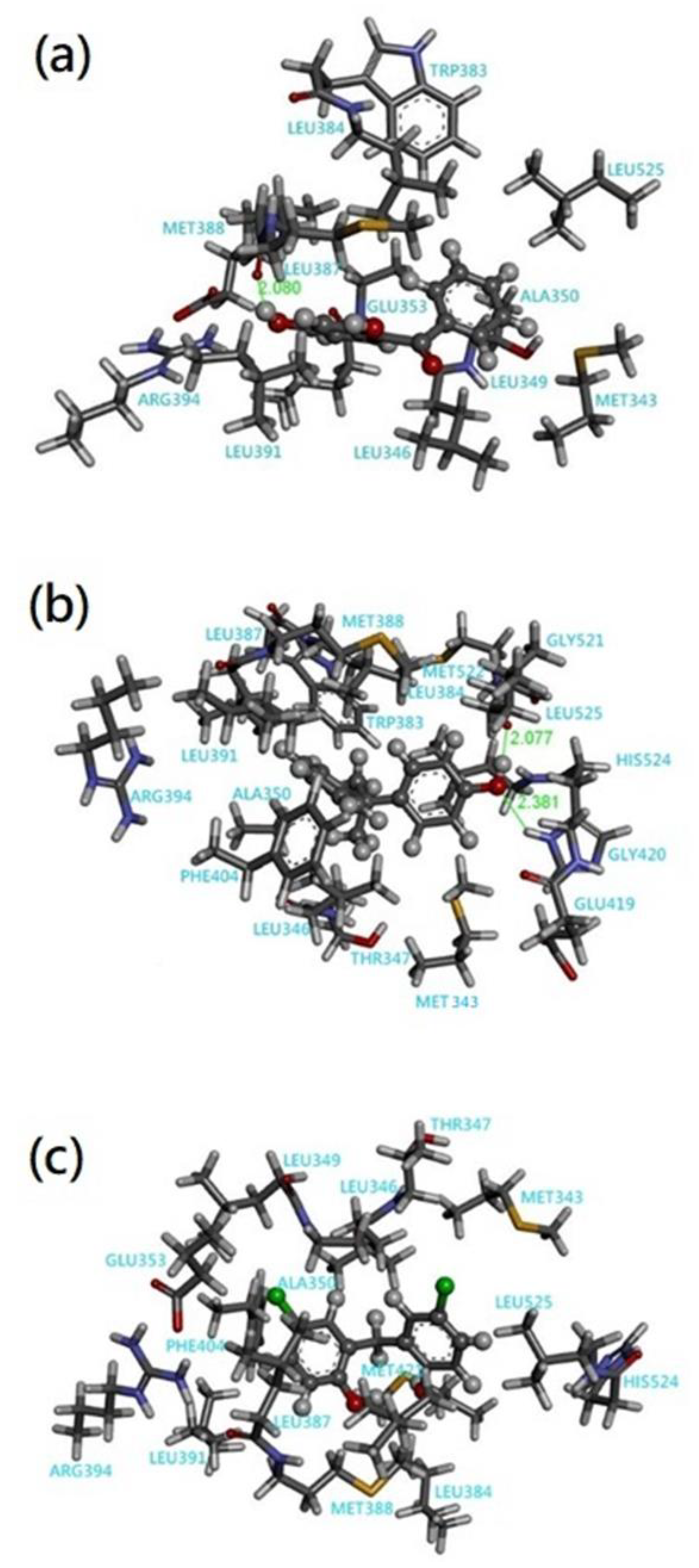
3.2. Quantum Chemical Cluster Model
3.3. Conceptual Density Functional Theory
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EFSA Scientific Committee. Scientific Opinion on the hazard assessment of endocrine disruptors: Scientific criteria for identification of endocrine disruptors and appropriateness of existing test methods for assessing effects mediated by these substances on human health and the environment. EFSA J. 2013, 11, 3132. [Google Scholar] [CrossRef]
- Sellami, A.; Montes, M.; Lagarde, N. Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods. Int. J. Mol. Sci. 2021, 22, 2846. [Google Scholar] [CrossRef]
- Lee, S.; Barron, M.G. Structure-Based Understanding of Binding Affinity and Mode of Estrogen Receptor alpha Agonists and Antagonists. PLoS ONE 2017, 12, e0169607. [Google Scholar] [CrossRef] [Green Version]
- Toporova, L.; Balaguer, P. Nuclear receptors are the major targets of endocrine disrupting chemicals. Mol. Cell. Endocrinol. 2020, 502, 110665. [Google Scholar] [CrossRef]
- Uzzaman, M.; Mahmud, T. Structural modification of aspirin to design a new potential cyclooxygenase (COX-2) inhibitors. Silico Pharmacol. 2020, 8, 1. [Google Scholar] [CrossRef]
- Uzzaman, M.; Hasan, M.K.; Mahmud, S.; Yousuf, A.; Islam, S.; Uddin, M.N.; Barua, A. Physicochemical, spectral, molecular docking and ADMET studies of Bisphenol analogues; A computational approach. Inform. Med. Unlocked 2021, 25, 100706. [Google Scholar] [CrossRef]
- Matsushima, A. A Novel Action of Endocrine-Disrupting Chemicals on Wildlife; DDT and Its Derivatives Have Remained in the Environment. Int. J. Mol. Sci. 2018, 19, 1377. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, S.; Zhang, J.; Wen, Y.; Zhang, C.; Liu, W. Distinct mechanisms of endocrine disruption of DDT-related pesticides toward estrogen receptor α and estrogen-related receptor γ. Environ. Toxicol. Chem. 2012, 31, 2597–2605. [Google Scholar] [CrossRef]
- Landeros-Martinez, L.L.; Glossman-Mitnik, D.; Flores-Holguin, N. Interaction of Tamoxifen Analogs With the Pocket Site of Some Hormone Receptors. A Molecular Docking and Density Functional Theory Study. Front. Chem. 2018, 6, 293. [Google Scholar] [CrossRef]
- Żwierełło, W.; Maruszewska, A.; Skórka-Majewicz, M.; Goschorska, M.; Baranowska-Bosiacka, I.; Dec, K.; Styburski, D.; Nowakowska, A.; Gutowska, I. The influence of polyphenols on metabolic disorders caused by compounds released from plastics—Review. Chemosphere 2020, 240, 124901. [Google Scholar] [CrossRef]
- Manzetti, S.; van der Spoel, E.R.; van der Spoel, D. Chemical Properties, Environmental Fate, and Degradation of Seven Classes of Pollutants. Chem. Res. Toxicol. 2014, 27, 713–737. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xu, L.; Fang, H.; Hong, H.; Perkins, R.; Harris, S.; Bearden, E.D.; Shi, L.; Tong, W. The EDKB: An established knowledge base for endocrine disrupting chemicals. BMC Bioinform. 2010, 11 (Suppl. 6), S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkoç, S.; Tüzün, B.; Özalp, A.; Kökbudak, Z. Investigation of structural, electronical and in vitro cytotoxic activity properties of some heterocyclic compounds. J. Mol. Struct. 2021, 1246, 131127. [Google Scholar] [CrossRef]
- Tsuneda, T.; Song, J.W.; Suzuki, S.; Hirao, K. On Koopmans′ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Levy, M. Comment on “Significance of the highest occupied Kohn-Sham eigenvalue”. Phys. Rev. B 1997, 56, 16021. [Google Scholar] [CrossRef]
- Casida, M.E. Correlated optimized effective-potential treatment of the derivative discontinuity and of the highest occupied Kohn-Sham eigenvalue: A Janak-type theorem for the optimized effective-potential model. Phys. Rev. B 1999, 59, 4694–4698. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demircioğlu, Z.; Büyükgüngör, O.; Ersanlı, C.C. Chemical Activity Studies with Density Functional Theory. SETSCI Conf. Index. Syst. 2018, 3, 695–697. [Google Scholar]
- Pathak, S.K.; Srivastava, R.; Sachan, A.K.; Prasad, O.; Sinha, L.; Asiri, A.M.; Karabacak, M. Experimental (FT-IR, FT-Raman, UV and NMR) and quantum chemical studies on molecular structure, spectroscopic analysis, NLO, NBO and reactivity descriptors of 3,5-Difluoroaniline. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 135, 283–295. [Google Scholar] [CrossRef]
- Aiken, L.S.; West, S.G.; Pitts, S.C. Multiple Linear Regression. In Handbook of Psychology; Weiner, I.B., Ed.; Wiley: Hoboken, NJ, USA, 2003; pp. 481–507. [Google Scholar] [CrossRef]
- Ambure, P.; Gajewicz-Skretna, A.; Cordeiro, M.; Roy, K. New Workflow for QSAR Model Development from Small Data Sets: Small Dataset Curator and Small Dataset Modeler. Integration of Data Curation, Exhaustive Double Cross-Validation, and a Set of Optimal Model Selection Techniques. J. Chem. Inf. Model. 2019, 59, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Alam, S.; Khan, F. QSAR, docking, ADMET, and system pharmacology studies on tormentic acid derivatives for anticancer activity. J. Biomol. Struct. Dyn. 2018, 36, 2373–2390. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Ambure, P.; Kar, S.; Ojha, P.K. Is it possible to improve the quality of predictions from an “intelligent” use of multiple QSAR/QSPR/QSTR models? J. Chemom. 2018, 32, e2992. [Google Scholar] [CrossRef]
- Olasupo, S.B.; Uzairu, A.; Adamu, G.S.; Uba, S. Computational Modeling and Pharmacokinetics/ADMET Study of Some Arylpiperazine Derivatives as Novel Antipsychotic Agents Targeting Depression. Chem. Afr. 2020, 3, 979–988. [Google Scholar] [CrossRef]
- Jaworska, J.S.; Comber, M.; Auer, C.; Van Leeuwen, C.J. Summary of a workshop on regulatory acceptance of (Q)SARs for human health and environmental endpoints. Environ. Health Perspect. 2003, 111, 1358–1360. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01. Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA suite of programs: An versatile platform for cheminformatics and drug design projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar] [CrossRef]
- Mekenyan, O.G.; Veith, G.D. Relationships between descriptors for hydrophobicity and soft electrophilicity in predicting toxicity. SAR QSAR Environ. Res. 1993, 1, 335–344. [Google Scholar] [CrossRef]
- Veith, G.D.; Mekenyan, O.G. A QSAR Approach for Estimating the Aquatic Toxicity of Soft Electrophiles [QSAR for Soft Electrophiles]. Quant. Struct.-Act. Relatsh. 1993, 12, 349–356. [Google Scholar] [CrossRef]
- Bary, G.; Ghani, L.; Jamil, M.I.; Arslan, M.; Ahmed, W.; Ahmad, A.; Sajid, M.; Ahmad, R.; Huang, D. Designing small organic non-fullerene acceptor molecules with diflorobenzene or quinoline core and dithiophene donor moiety through density functional theory. Sci. Rep. 2021, 11, 19683. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, D.; Liu, C. Evaluation of the effectiveness of AM1 geometry used in calculating O–H bond dissociation enthalpy. J. Mol. Struct. THEOCHEM 2002, 618, 181–189. [Google Scholar] [CrossRef]
- Zheng, G.; Irle, S.; Morokuma, K. Performance of the DFTB method in comparison to DFT and semiempirical methods for geometries and energies of C20–C86 fullerene isomers. Chem. Phys. Lett. 2005, 412, 210–216. [Google Scholar] [CrossRef]
- Derosa, P.A. A combined semiempirical-DFT study of oligomers within the finite-chain approximation, evolution from oligomers to polymers. J. Comput. Chem. 2009, 30, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Villar, R.; Gil, M.J.; García, J.I.; Martínez-Merino, V. Are AM1 ligand-protein binding enthalpies good enough for use in the rational design of new drugs? J. Comput. Chem. 2005, 26, 1347–1358. [Google Scholar] [CrossRef]
- Derosa, P.; Koraboina, K.; Sanders, M. A combined model to study conductive properties of polymers with atomic resolution. In Proceedings of the AIChE Annual Meeting, Conference Proceedings, San Francisco, CA, USA, 12–17 November 2006. [Google Scholar]
- Celik, L.; Lund, J.D.D.; Schiøtt, B. Exploring Interactions of Endocrine-Disrupting Compounds with Different Conformations of the Human Estrogen Receptor α Ligand Binding Domain: A Molecular Docking Study. Chem. Res. Toxicol. 2008, 21, 2195–2206. [Google Scholar] [CrossRef]
- Ye, H.; Dudley, S.Z.; Shaw, I.C. Intimate estrogen receptor-α/ligand relationships signal biological activity. Toxicology 2018, 408, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ekena, K.; Weis, K.E.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Identification of amino acids in the hormone binding domain of the human estrogen receptor important in estrogen binding. J. Biol. Chem. 1996, 271, 20053–20059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekena, K.; Weis, K.E.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Different residues of the human estrogen receptor are involved in the recognition of structurally diverse estrogens and antiestrogens. J. Biol. Chem. 1997, 272, 5069–5075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Liu, X.; Liu, X.-C.; Pan, W.-X.; Fu, J.-J.; Zhang, A.-Q. The Effect of Structural Diversity on Ligand Specificity and Resulting Signaling Differences of Estrogen Receptor α. Chem. Res. Toxicol. 2019, 32, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Radek, J.T.; Meyers, M.J.; Nettles, K.W.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Agard, D.A.; Greene, G.L. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 2002, 9, 359–364. [Google Scholar] [CrossRef]
- Pearson, R.G.; Songstad, J. Application of the Principle of Hard and Soft Acids and Bases to Organic Chemistry. J. Am. Chem. Soc. 1967, 89, 1827–1836. [Google Scholar] [CrossRef]
- Chakraborty, D.; Chattaraj, P.K. Conceptual density functional theory based electronic structure principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; De Proft, F.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual density functional theory: Status, prospects, issues. Theor. Chem. Acc. 2020, 139. [Google Scholar] [CrossRef]
- Geerlings, P.; Proft, F.D.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Lopachin, R.M.; Gavin, T.; Decaprio, A.; Barber, D.S. Application of the Hard and Soft, Acids and Bases (HSAB) theory to toxicant—Target interactions. Chem. Res. Toxicol. 2012, 25, 239–251. [Google Scholar] [CrossRef]
- Rokhina, E.V.; Suri, R.P. Application of density functional theory (DFT) to study the properties and degradation of natural estrogen hormones with chemical oxidizers. Sci. Total. Environ. 2012, 417–418, 280–290. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; De Proft, F.; Ayers, P.W.; Geerlings, P. On the applicability of local softness and hardness. Phys. Chem. Chem. Phys. 2010, 12, 1072–1080. [Google Scholar] [CrossRef] [Green Version]
- Faver, J.; Kenneth, M.; Merz, J. Utility of the Hard/Soft Acid-Base Principle via the Fukui Function in Biological Systems. J. Chem. Theory Comput. 2010, 6, 548–559. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Compound | HOMO (eV) | LUMO (eV) | μ+ (a.u.) | μ− (a.u.) | Dipole Moment (D) | APSA (Å2) |
|---|---|---|---|---|---|---|---|
| PCB | 2,3,4,5-Tetrachloro-4′-biphenylol | −7.502 | −0.430 | −0.070 | −0.182 | 4.753 | 399.6 |
| 2,5-Dichloro-4′-biphenylol | −7.407 | −0.083 | −0.088 | −0.186 | 2.142 | 353.9 | |
| 2-Chloro-4-biphenylol | −7.693 | 0.384 | −0.077 | −0.188 | 1.065 | 342.0 | |
| 4-Chloro-4′-biphenylol | −7.141 | 0.012 | −0.082 | −0.179 | 3.506 | 383.3 | |
| 4-Hydroxybiphenyl | −7.487 | 0.550 | −0.069 | −0.180 | 1.777 | 322.3 | |
| 3-Phenylphenol | −7.556 | 0.628 | −0.066 | −0.180 | 1.772 | 325.9 | |
| 2,4′-Dichlorobiphenyl | −8.203 | 0.275 | −0.083 | −0.203 | 3.964 | 404.9 | |
| Phenol | 4-n-Octylphenol | −7.270 | 0.860 | −0.061 | −0.173 | 1.811 | 452.9 |
| 2-sec-Butylphenol | −7.369 | 0.894 | −0.061 | −0.174 | 1.513 | 320.0 | |
| 4-sec-Butylphenol | −7.285 | 0.824 | −0.062 | −0.173 | 1.837 | 305.3 | |
| 4-tert-Butylphenol | −7.292 | 0.847 | −0.062 | −0.173 | 1.850 | 293.8 | |
| 4-Chloro-3-methylphenol | −7.454 | 0.657 | −0.069 | −0.180 | 3.190 | 249.0 | |
| 4-Phenethylphenol | −7.288 | 0.831 | −0.060 | −0.173 | 1.768 | 389.2 | |
| 3-Ethylphenol | −7.446 | 0.897 | −0.061 | −0.176 | 1.434 | 264.1 | |
| a,a-Dimethyl-b-ethylallenolicacid | −6.976 | −0.207 | −0.087 | −0.176 | 1.487 | 370.8 | |
| 4-Chloro-2-methylphenol | −7.439 | 0.638 | −0.069 | −0.180 | 3.670 | 258.8 | |
| 2-Cholor-4-methylphenol | −7.476 | 0.576 | −0.071 | −0.181 | 4.155 | 258.3 | |
| Heptylp-hydroxybenzoate | −7.793 | −0.079 | −0.092 | −0.195 | 4.521 | 447.5 | |
| 2-Ethylhexyl-4-hydroxybenzoate | −7.793 | −0.082 | −0.092 | −0.195 | 1.723 | 430.2 | |
| Benzyl4-hydroxybenzoate | −7.805 | −0.113 | −0.093 | −0.196 | 1.833 | 366.5 | |
| DDT | o,p′-DDT | −8.134 | −0.246 | −0.099 | −0.211 | 3.389 | 491.9 |
| 2,4-Dihydroxybenzophenone | −7.558 | −0.726 | −0.107 | −0.195 | 7.199 | 291.3 | |
| Phenolphthalein | −7.497 | −0.520 | −0.101 | −0.192 | 9.135 | 363.6 | |
| Phenol red | −7.631 | −0.332 | −0.097 | −0.194 | 9.753 | 355.4 | |
| 4,4′-Sulfonyldiphenol | −7.766 | −0.261 | −0.082 | −0.196 | 8.662 | 258.9 | |
| 4,4′-Dihydoxy-benzophenone | −7.708 | −0.662 | −0.108 | −0.204 | 4.990 | 272.9 | |
| 2,2′-Methylenebis(4-chlorophenol) | −7.468 | 0.165 | −0.082 | −0.189 | 3.955 | 374.5 | |
| Bis(4-hydroxyphenyl)methane | −7.145 | 0.758 | −0.061 | −0.174 | 1.332 | 303.5 | |
| Monohydroxy methoxychlor | −7.335 | 0.049 | −0.087 | −0.188 | 2.570 | 443.8 | |
| Monohydroxy methoxychlor olefin | −7.057 | −0.150 | −0.067 | −0.173 | 4.977 | 429.8 | |
| p-Cumylphenol | −7.259 | 0.712 | −0.061 | −0.173 | 1.612 | 376.0 |
| Category | Compound | Expt. | Pred. | Δ (Expt. − Pred.) |
|---|---|---|---|---|
| PCB | 2,3,4,5−Tetrachloro−4′−biphenylol | −0.64 | −0.60 | −0.04 |
| 2,5−Dichloro−4′−biphenylol | −1.44 | −1.59 | 0.15 | |
| 2−Chloro−4−biphenylol | −2.77 | −2.74 | −0.03 | |
| 4−Chloro−4′−biphenylol | −2.18 | −1.33 | −0.85 | |
| 4−Hydroxybiphenyl | −3.04 | −2.81 | −0.23 | |
| 3−Phenylphenol | −3.44 | −3.03 | −0.41 | |
| 2,4′−Dichlorobiphenyl | −3.61 | −3.65 | 0.04 | |
| Phenol | 4−n−Octylphenol | −2.31 | −2.20 | −0.11 |
| 2−sec−Butylphenol | −3.54 | −3.26 | −0.28 | |
| 4−sec−Butylphenol | −3.37 | −3.15 | −0.22 | |
| 4−tert−Butylphenol | −3.61 | −3.27 | −0.34 | |
| 4−Chloro−3−methylphenol | −3.38 | −3.83 | 0.45 | |
| 4−Phenethylphenol | −2.69 | −2.57 | −0.12 | |
| 3−Ethylphenol | −3.87 | −3.75 | −0.12 | |
| a,a−Dimethyl−b−ethylallenolicacid | −0.02 | −0.34 | 0.32 | |
| 4−Chloro−2−methylphenol | −3.67 | −3.79 | 0.12 | |
| 2−Cholor−4−methylphenol | −3.66 | −3.80 | 0.14 | |
| Heptylp−hydroxybenzoate | −2.09 | −2.05 | −0.04 | |
| 2−Ethylhexyl−4−hydroxybenzoate | −1.74 | −1.70 | −0.04 | |
| Benzyl4−hydroxybenzoate | −2.54 | −2.11 | −0.43 | |
| DDT | o,p′−DDT | −2.85 | −2.31 | −0.54 |
| 2,4−Dihydroxybenzophenone | −2.61 | −1.95 | −0.66 | |
| Phenolphthalein | −1.87 | −2.07 | 0.20 | |
| Phenol red | −3.25 | −2.78 | −0.47 | |
| 4,4′−Sulfonyldiphenol | −3.07 | −3.62 | 0.55 | |
| 4,4′−Dihydoxy−benzophenone | −2.46 | −2.48 | 0.02 | |
| 2,2′−Methylenebis(4−chlorophenol) | −2.45 | −2.60 | 0.15 | |
| Bis(4−hydroxyphenyl)methane | −3.02 | −2.96 | −0.06 | |
| Monohydroxy methoxychlor | −0.89 | −1.51 | 0.62 | |
| Monohydroxy methoxychlor olefin | −0.63 | −0.45 | −0.18 | |
| p−Cumylphenol | −2.30 | −2.35 | 0.05 |
| Descriptor | VIF | t−Value |
|---|---|---|
| LUMO | 3.152 | −10.740 |
| μ− | 2.561 | 6.595 |
| Dipole Moment | 2.076 | −4.273 |
| APSA | 1.162 | 5.496 |
| Category | Compound | ρ+Max(H) (a.u.) | ρ+Max (a.u.) | f+Max | Sites for Nucleophilic Attack | s+Max (a.u.) | f−Max | Site for Electrophilic Attack | s−Max (a.u.) | ΔE (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|---|
| PCB | 2−Chloro−4−biphenylol | 0.369 | 0.369 | 0.129 | Cl1 | 0.582 | 0.125 | O1 | 0.567 | −16.93 |
| 4−Chloro−4′−biphenylol | 0.366 | 0.366 | 0.107 | Cl1 | 0.553 | 0.095 | O2 | 0.492 | −24.33 | |
| 2,4′−Dichlorobiphenyl | 0.167 | 0.167 | 0.097 | C5 | 0.407 | 0.241 | Cl2 | 1.010 | −9.84 | |
| Phenol | 4−Chloro−3−methylphenol | 0.367 | 0.367 | 0.115 | C15 | 0.519 | 0.191 | O1 | 0.860 | −16.94 |
| 4−Phenethylphenol | 0.363 | 0.363 | 0.118 | C5 | 0.522 | 0.124 | Cl1 | 0.548 | −22.19 | |
| 4−Chloro−2−methylphenol | 0.366 | 0.366 | 0.111 | C4 | 0.504 | 0.185 | Cl1 | 0.837 | −10.19 | |
| DDT | 2,4−Dihydroxybenzophenone | 0.371 | 0371 | 0.151 | O2 | 0.860 | 0.132 | C10 | 0.748 | −24.37 |
| p−Cumylphenol | 0.364 | 0.364 | 0.070 | C14 | 0.312 | 0.120 | O1 | 0.537 | −26.62 | |
| 2,2′−Methylenebis(4−chlorophenol) | 0.371 | 0.371 | 0.099 | C13 | 0.461 | 0.118 | Cl2 | 0.548 | −21.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, S.-C.; Hsi, H.-C.; Chang, C.-M. Quantum Chemical GA-MLR, Cluster Model, and Conceptual DFT Descriptors Studies on the Binding Interaction of Estrogen Receptor Alpha with Endocrine Disrupting Chemicals. Crystals 2023, 13, 228. https://doi.org/10.3390/cryst13020228
Chi S-C, Hsi H-C, Chang C-M. Quantum Chemical GA-MLR, Cluster Model, and Conceptual DFT Descriptors Studies on the Binding Interaction of Estrogen Receptor Alpha with Endocrine Disrupting Chemicals. Crystals. 2023; 13(2):228. https://doi.org/10.3390/cryst13020228
Chicago/Turabian StyleChi, Shu-Chun, Hsing-Cheng Hsi, and Chia-Ming Chang. 2023. "Quantum Chemical GA-MLR, Cluster Model, and Conceptual DFT Descriptors Studies on the Binding Interaction of Estrogen Receptor Alpha with Endocrine Disrupting Chemicals" Crystals 13, no. 2: 228. https://doi.org/10.3390/cryst13020228