
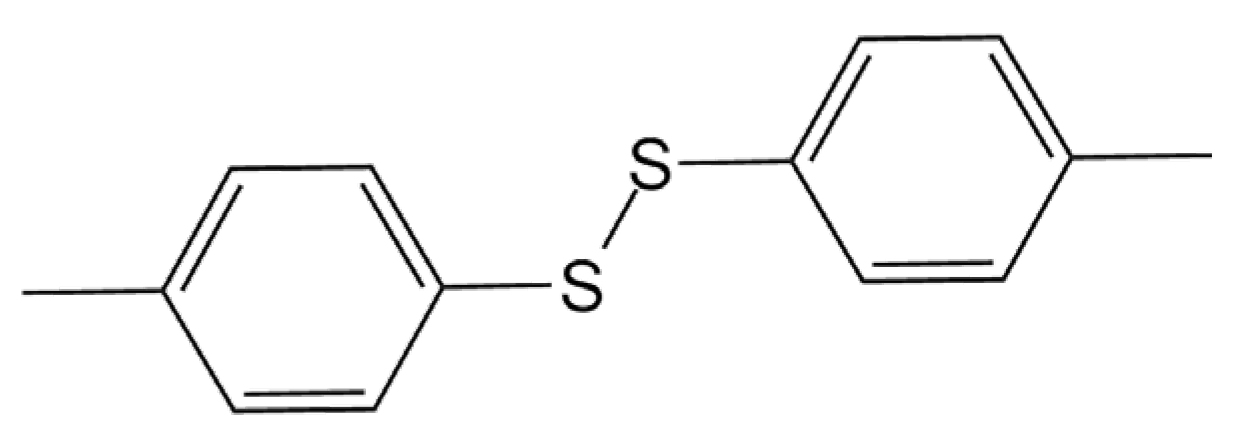
Computational Investigation of the Stability of Di-p-Tolyl Disulfide “Hidden” and “Conventional” Polymorphs at High Pressures


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Details
2.1. FF Method Calculations
2.2. Periodic DFT Calculations
2.3. Equations of States
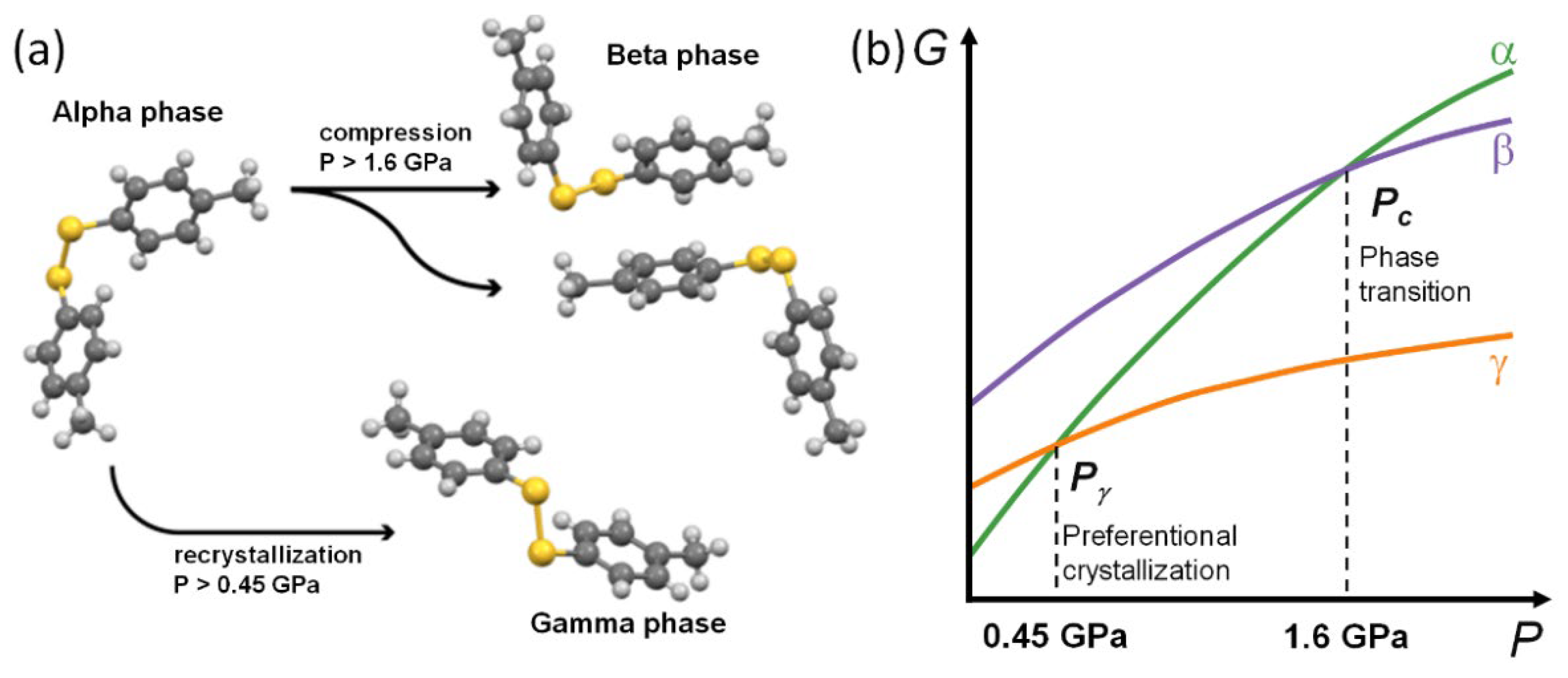
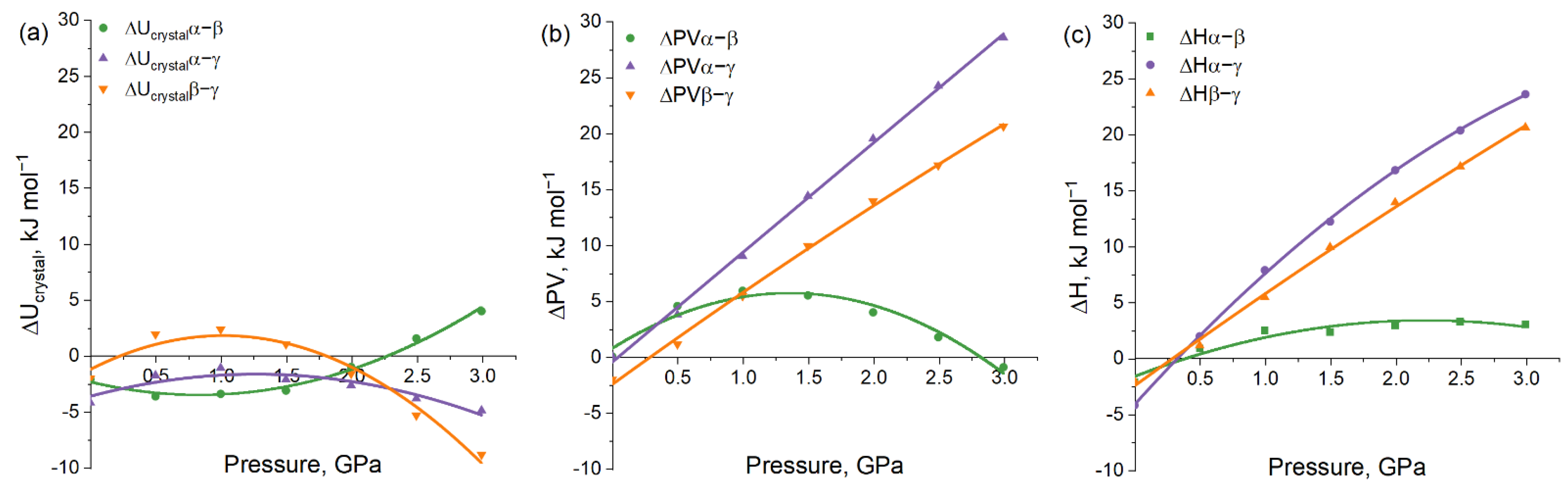
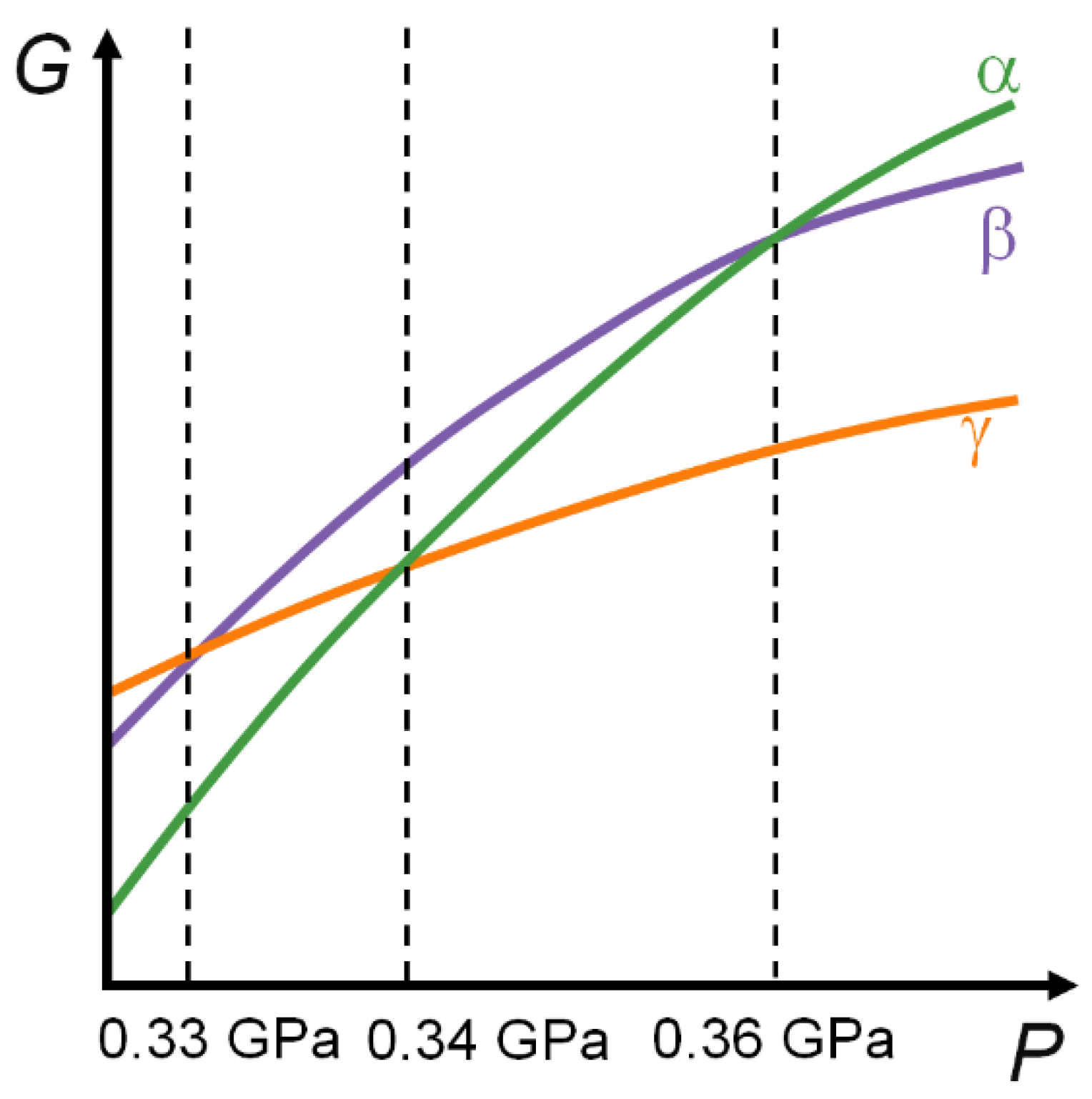
3. Results and Discussion
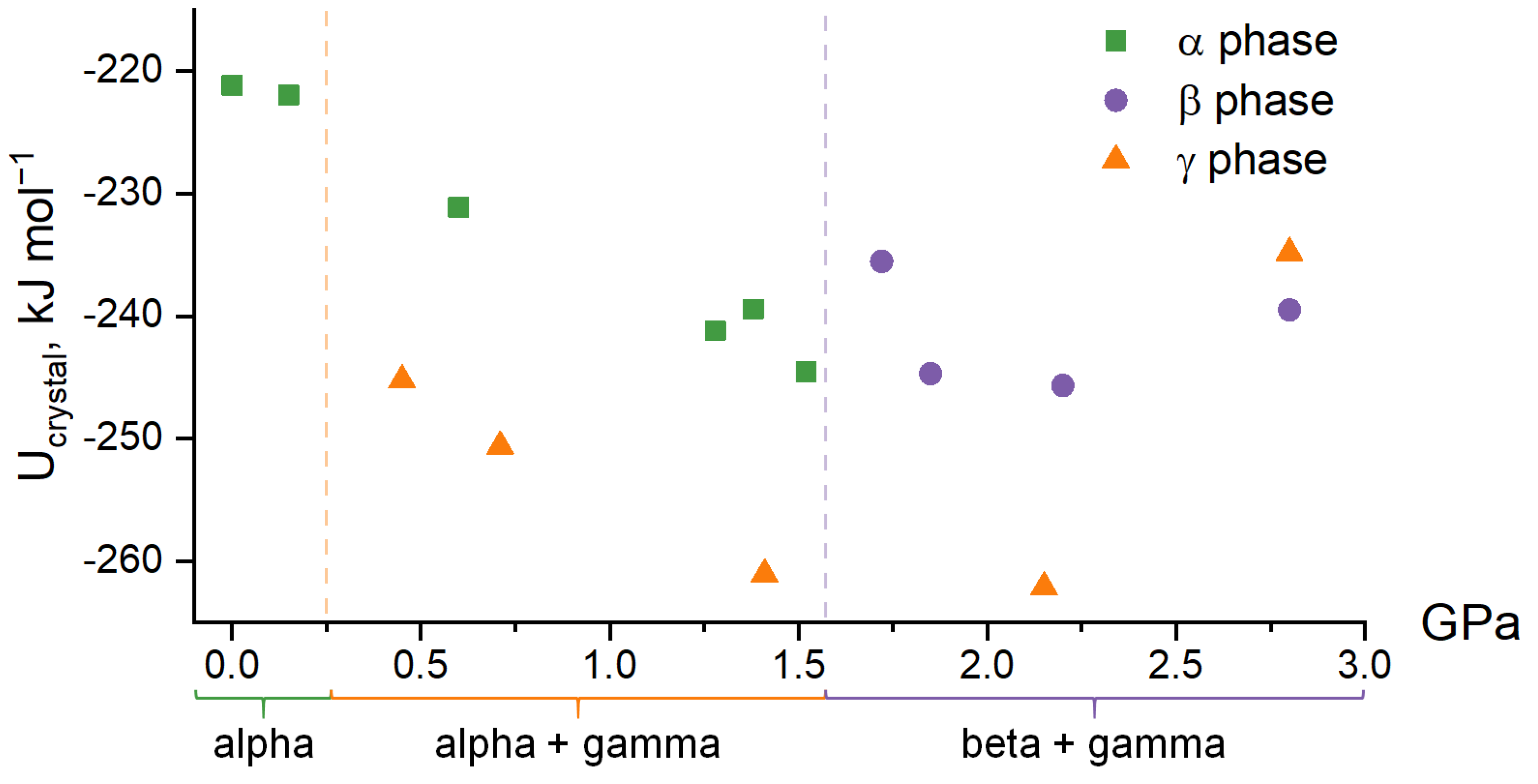
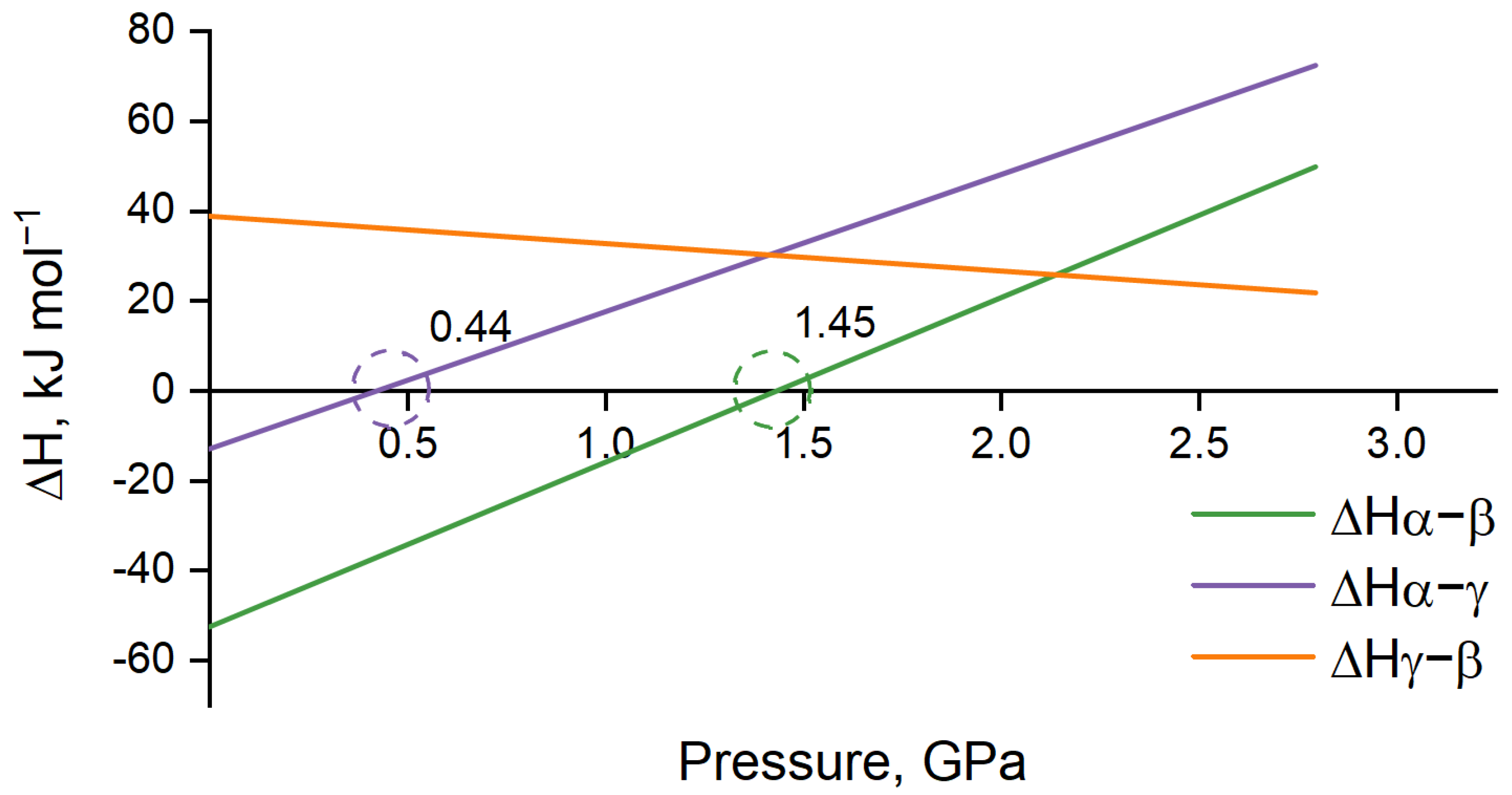
3.1. Force Field Calculations
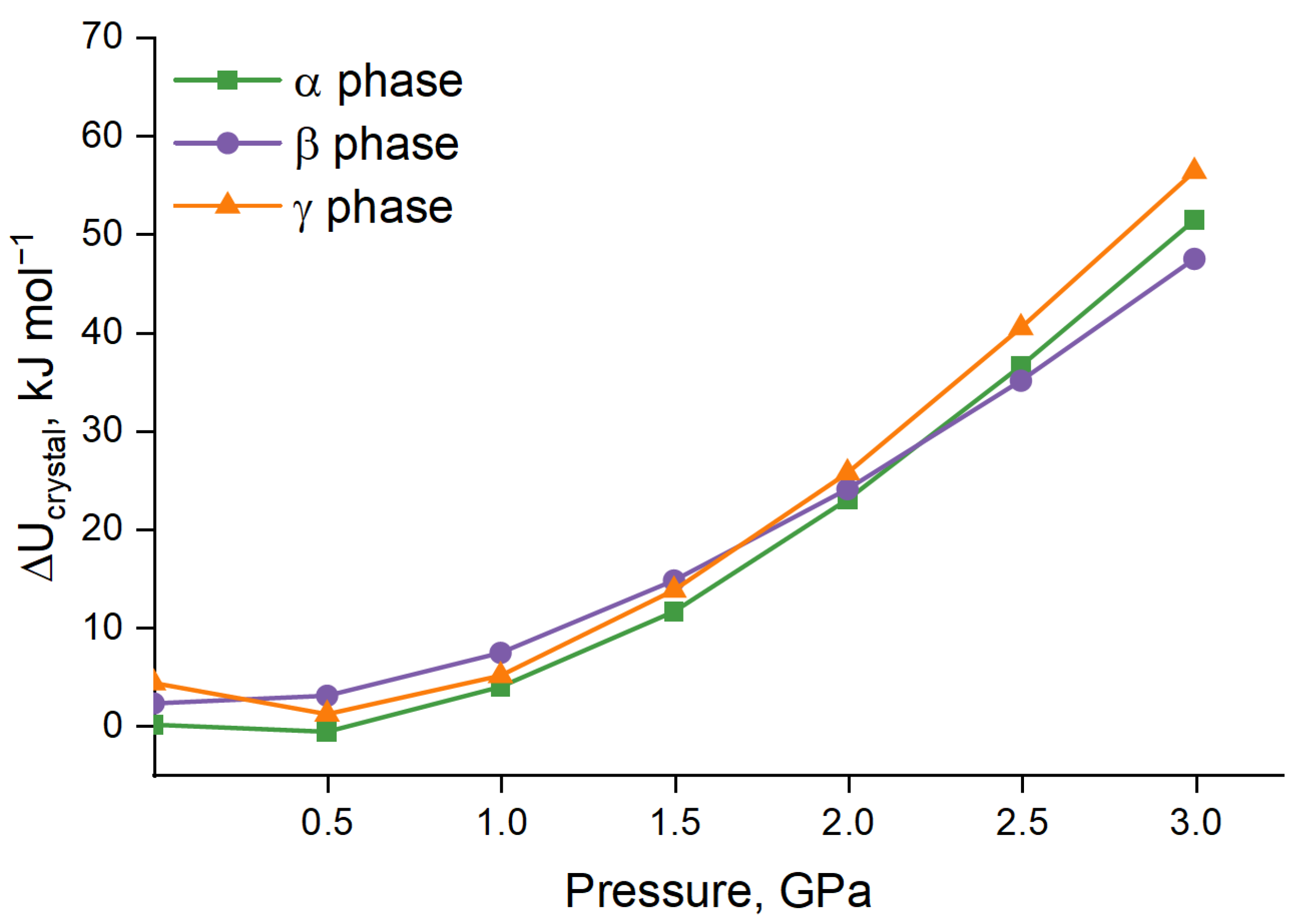
3.2. Periodic DFT Calculations with EoS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Errandonea, D. Pressure-Induced Phase Transformations. Crystals 2020, 10, 595. [Google Scholar] [CrossRef]
- Thiel, A.M.; Damgaard-Møller, E.; Overgaard, J. High-Pressure Crystallography as a Guide in the Design of Single-Molecule Magnets. Inorg. Chem. 2020, 59, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Moggach, S.A.; Oswald, I.D.H. Crystallography Under High Pressures. In 21st Century Challenges in Chemical Crystallography I. Structure and Bonding; Springer: Cham, Switzerland, 2020; pp. 141–198. ISBN 9783030647421. [Google Scholar]
- Görbitz, C.H. Crystal structures of amino acids: From bond lengths in glycine to metal complexes and high-pressure polymorphs. Crystallogr. Rev. 2015, 21, 160–212. [Google Scholar] [CrossRef]
- Skakunova, K.D.; Rychkov, D.A. Low Temperature and High-Pressure Study of Bending L-Leucinium Hydrogen Maleate Crystals. Crystals 2021, 11, 1575. [Google Scholar] [CrossRef]
- Sharma, S.M.; Garg, N. Material Studies at High Pressure. In Materials under Extreme Conditions; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–47. ISBN 9780128014424. [Google Scholar]
- Colmenero, F. Organic acids under pressure: Elastic properties, negative mechanical phenomena and pressure induced phase transitions in the lactic, maleic, succinic and citric acids. Mater. Adv. 2020, 1, 1399–1426. [Google Scholar] [CrossRef]
- Colmenero, F.; Lunelli, B. Fluorine-substituted cyclobutenes in the solid state: Crystal structures, vibrational spectra and mechanical and thermodynamic properties. J. Phys. Chem. Solids 2022, 160, 110337. [Google Scholar] [CrossRef]
- Ratajczyk, P.; Katrusiak, A.; Bogdanowicz, K.A.; Przybył, W.; Krysiak, P.; Kwak, A.; Iwan, A. Mechanical strain, thermal and pressure effects on the absorption edge of an organic charge-transfer polymer for flexible photovoltaics and sensors. Mater. Adv. 2022, 3, 2697–2705. [Google Scholar] [CrossRef]
- Konar, S.; Hobday, C.L.; Bull, C.L.; Funnell, N.P.; Chan, Q.F.; Fong, A.; Atceken, N.; Pulham, C.R. High-Pressure Structural Behavior of para-Xylene. Cryst. Growth Des. 2022, 22, 3862–3869. [Google Scholar] [CrossRef]
- Wilson, C.J.G.; Cervenka, T.; Wood, P.A.; Parsons, S. Behavior of Occupied and Void Space in Molecular Crystal Structures at High Pressure. Cryst. Growth Des. 2022, 22, 2328–2341. [Google Scholar] [CrossRef]
- Neumann, M.A.; van de Streek, J.; Fabbiani, F.P.A.; Hidber, P.; Grassmann, O. Combined crystal structure prediction and high-pressure crystallization in rational pharmaceutical polymorph screening. Nat. Commun. 2015, 6, 7793. [Google Scholar] [CrossRef]
- Mazurek, A.; Szeleszczuk, Ł.; Pisklak, D.M. Can We Predict the Pressure Induced Phase Transition of Urea? Application of Quantum Molecular Dynamics. Molecules 2020, 25, 1584. [Google Scholar] [CrossRef] [PubMed]
- Paliwoda, D.; Dziubek, K.F.; Katrusiak, A. Imidazole Hidden Polar Phase. Cryst. Growth Des. 2012, 12, 4302–4305. [Google Scholar] [CrossRef]
- Anioła, M.; Katrusiak, A. Conformational Conversion of 4,4′-Bipyridinium in a Hidden High-Pressure Phase. Cryst. Growth Des. 2015, 15, 764–770. [Google Scholar] [CrossRef]
- Patyk-Kaźmierczak, E.; Kaźmierczak, M. A new high-pressure benzocaine polymorph—Towards understanding the molecular aggregation in crystals of an important active pharmaceutical ingredient (API). Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 56–64. [Google Scholar] [CrossRef]
- Fedorov, A.Y.; Rychkov, D.A.; Losev, E.A.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V. Effect of pressure on two polymorphs of tolazamide: Why no interconversion? CrystEngComm 2017, 19, 2243–2252. [Google Scholar] [CrossRef]
- Sobczak, S.; Katrusiak, A. Colossal Strain Release by Conformational Energy Up-Conversion in a Compressed Molecular Crystal. J. Phys. Chem. C 2017, 121, 2539–2545. [Google Scholar] [CrossRef]
- Rychkov, D.A. A Short Review of Current Computational Concepts for High-Pressure Phase Transition Studies in Molecular Crystals. Crystals 2020, 10, 81. [Google Scholar] [CrossRef]
- Munday, L.B.; Chung, P.W.; Rice, B.M.; Solares, S.D. Simulations of High-Pressure Phases in RDX. J. Phys. Chem. B 2011, 115, 4378–4386. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Stare, J.; Boldyreva, E.V. Pressure-driven phase transition mechanisms revealed by quantum chemistry: L-serine polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 6671–6676. [Google Scholar] [CrossRef]
- Wood, P.A.; Francis, D.; Marshall, W.G.; Moggach, S.A.; Parsons, S.; Pidcock, E.; Rohl, A.L. A study of the high-pressure polymorphs of L-serine using ab initio structures and PIXEL calculations. CrystEngComm 2008, 10, 1154. [Google Scholar] [CrossRef]
- Thomas, S.P.; Spackman, M.A. The Polymorphs of ROY: A Computational Study of Lattice Energies and Conformational Energy Differences. Aust. J. Chem. 2018, 71, 279. [Google Scholar] [CrossRef]
- Fedorov, A.Y.; Rychkov, D.A. Comparison of different computational approaches for unveiling the high-pressure behavior of organic crystals at a molecular level. Case study of tolazamide polymorphs. J. Struct. Chem. 2020, 61, 1356–1366. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Gonzalez-Platas, J.; Alvaro, M.; Nestola, F.; Angel, R. EosFit7-GUI: A new graphical user interface for equation of state calculations, analyses and teaching. J. Appl. Crystallogr. 2016, 49, 1377–1382. [Google Scholar] [CrossRef]
- Angel, R.J.; Alvaro, M.; Gonzalez-Platas, J. EosFit7c and a Fortran module (library) for equation of state calculations. Z. für Krist.-Cryst. Mater. 2014, 229, 405–419. [Google Scholar] [CrossRef]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: New York, NY, USA, 2002; Volume 14, ISBN 978-0-19-923656-5. [Google Scholar]
- Hunter, S.; Sutinen, T.; Parker, S.F.; Morrison, C.A.; Williamson, D.M.; Thompson, S.; Gould, P.J.; Pulham, C.R. Experimental and DFT-D Studies of the Molecular Organic Energetic Material RDX. J. Phys. Chem. C 2013, 117, 8062–8071. [Google Scholar] [CrossRef]
- Kolesnik, E.N.; Goryainov, S.V.; Boldyreva, E.V. Different behavior of L- and DL-serine crystals at high pressures: Phase transitions in L-serine and stability of the DL-serine structure. Dokl. Phys. Chem. 2005, 404, 169–172. [Google Scholar] [CrossRef]
- Moggach, S.A.; Allan, D.R.; Morrison, C.A.; Parsons, S.; Sawyer, L. Effect of pressure on the crystal structure of L-serine-I and the crystal structure of L-serine-II at 5.4 GPa. Acta Crystallogr. Sect. B Struct. Sci. 2005, 61, 58–68. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnova, V.Y.; Iurchenkova, A.A.; Rychkov, D.A. Computational Investigation of the Stability of Di-p-Tolyl Disulfide “Hidden” and “Conventional” Polymorphs at High Pressures. Crystals 2022, 12, 1157. https://doi.org/10.3390/cryst12081157
Smirnova VY, Iurchenkova AA, Rychkov DA. Computational Investigation of the Stability of Di-p-Tolyl Disulfide “Hidden” and “Conventional” Polymorphs at High Pressures. Crystals. 2022; 12(8):1157. https://doi.org/10.3390/cryst12081157
Chicago/Turabian StyleSmirnova, Valeriya Yu., Anna A. Iurchenkova, and Denis A. Rychkov. 2022. "Computational Investigation of the Stability of Di-p-Tolyl Disulfide “Hidden” and “Conventional” Polymorphs at High Pressures" Crystals 12, no. 8: 1157. https://doi.org/10.3390/cryst12081157