Clicking Azides and Alkynes with Poly(pyrazolyl)borate-Copper(I) Catalysts: An Experimental and Computational Study

 , , ,
, , ,  , ,
, ,  and
and
Abstract
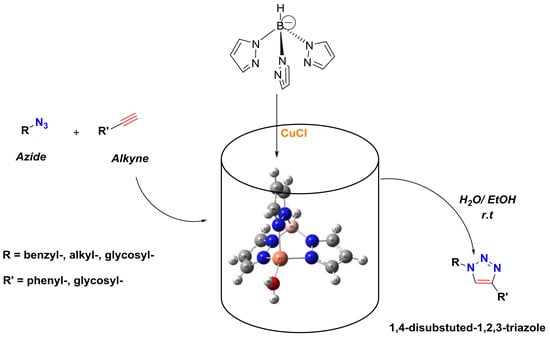
:
1. Introduction
2. Results and Discussion
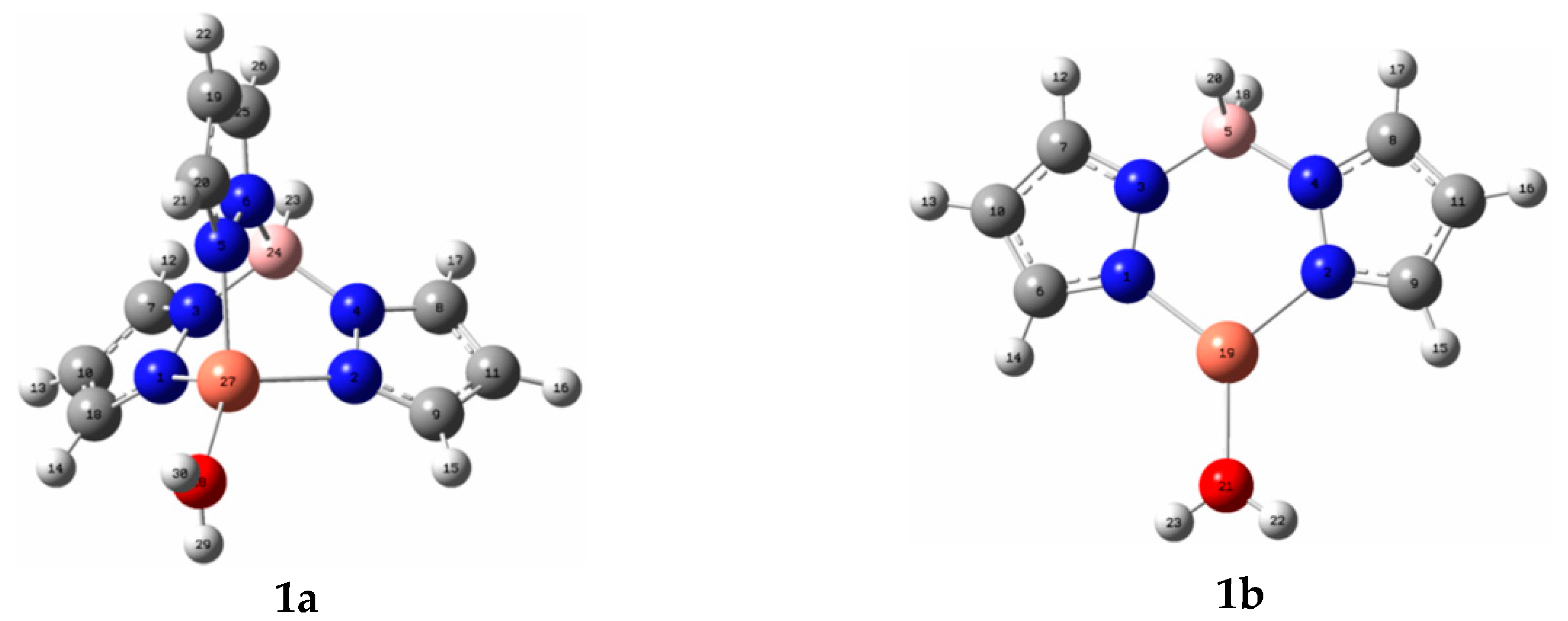
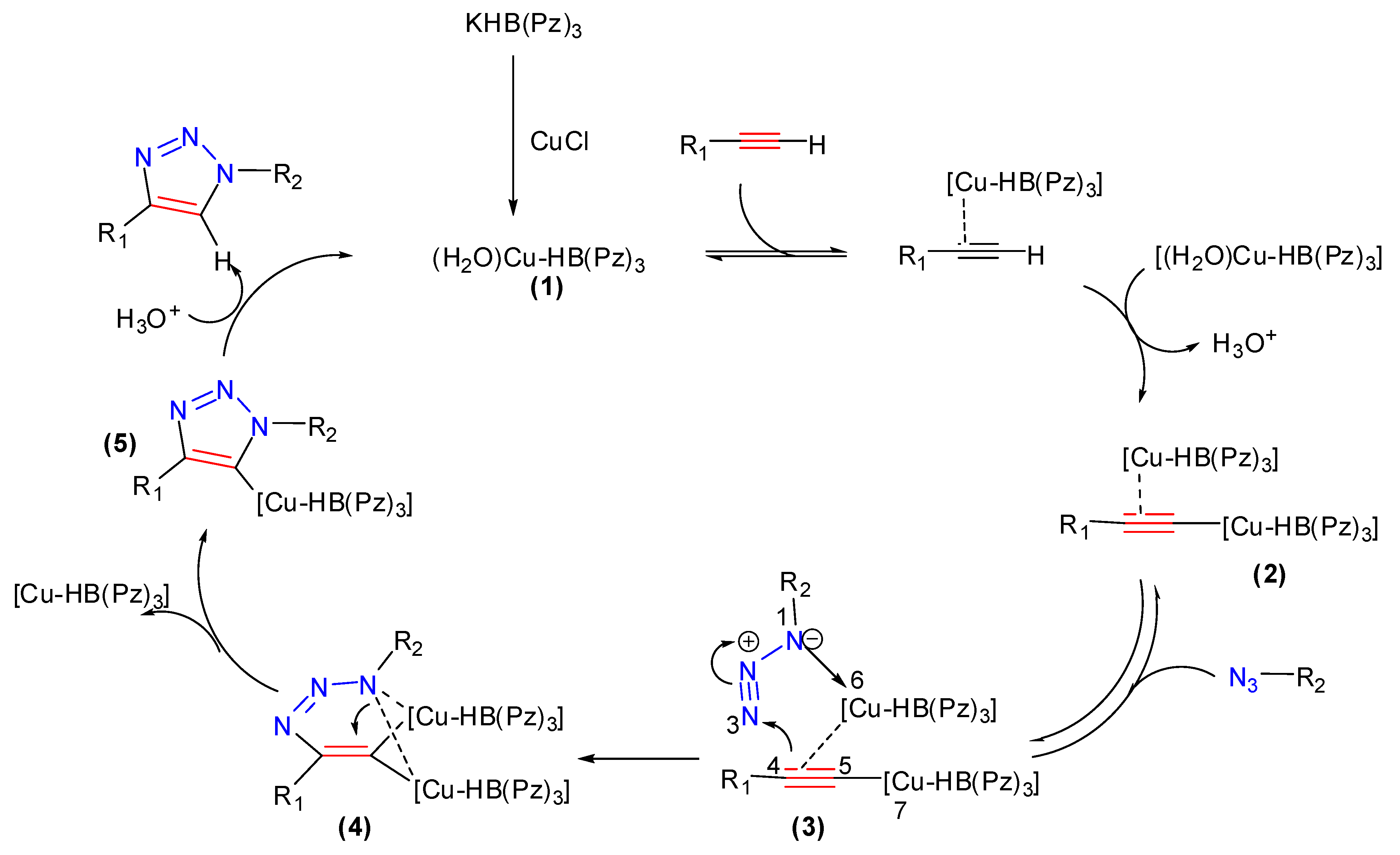
2.1. Poly(pyrazolyl)borate Copper(I)-Catalyzed Alkyne–Azide 32CA Reaction
2.2. Global and Local Conceptual DFT (CDFT) Reactivity Index Analysis
3. Materials and Methods
3.1. Reagents and Physical Measurements
3.2. Computational Methods
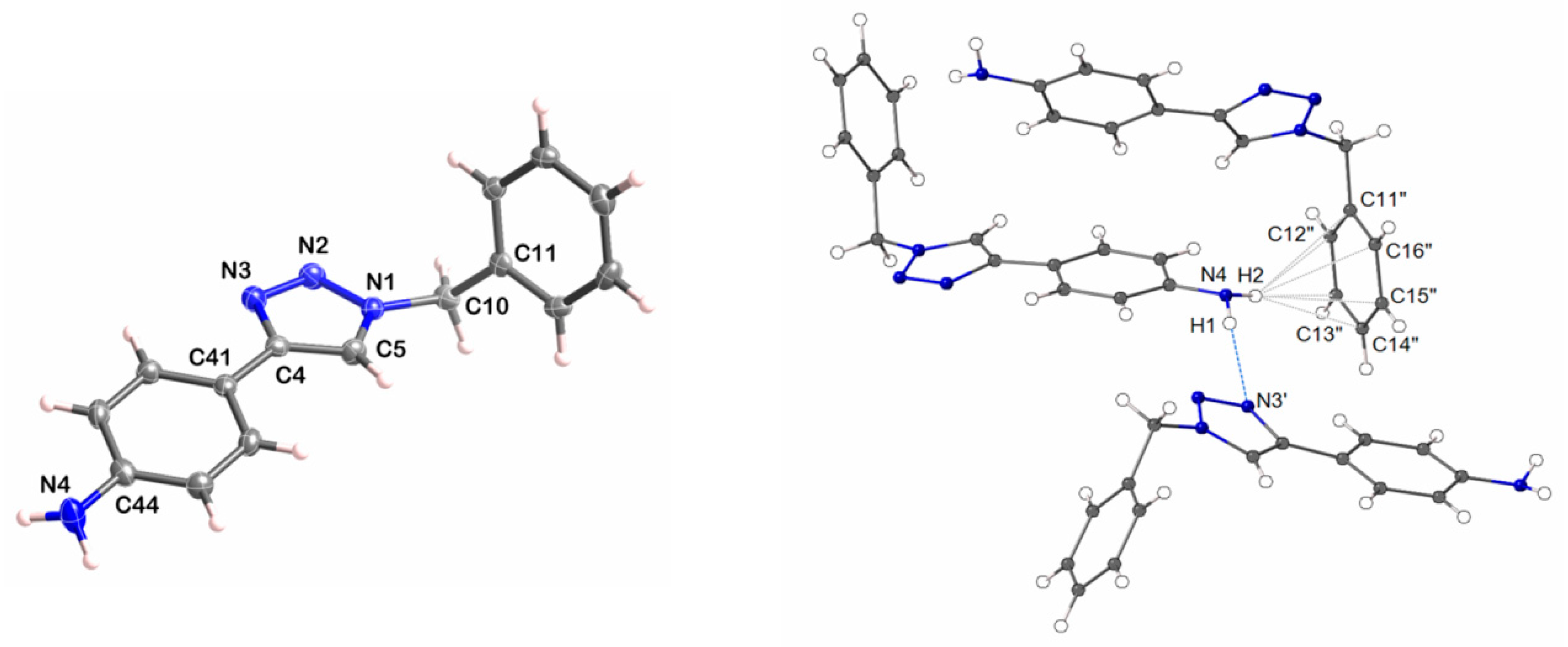
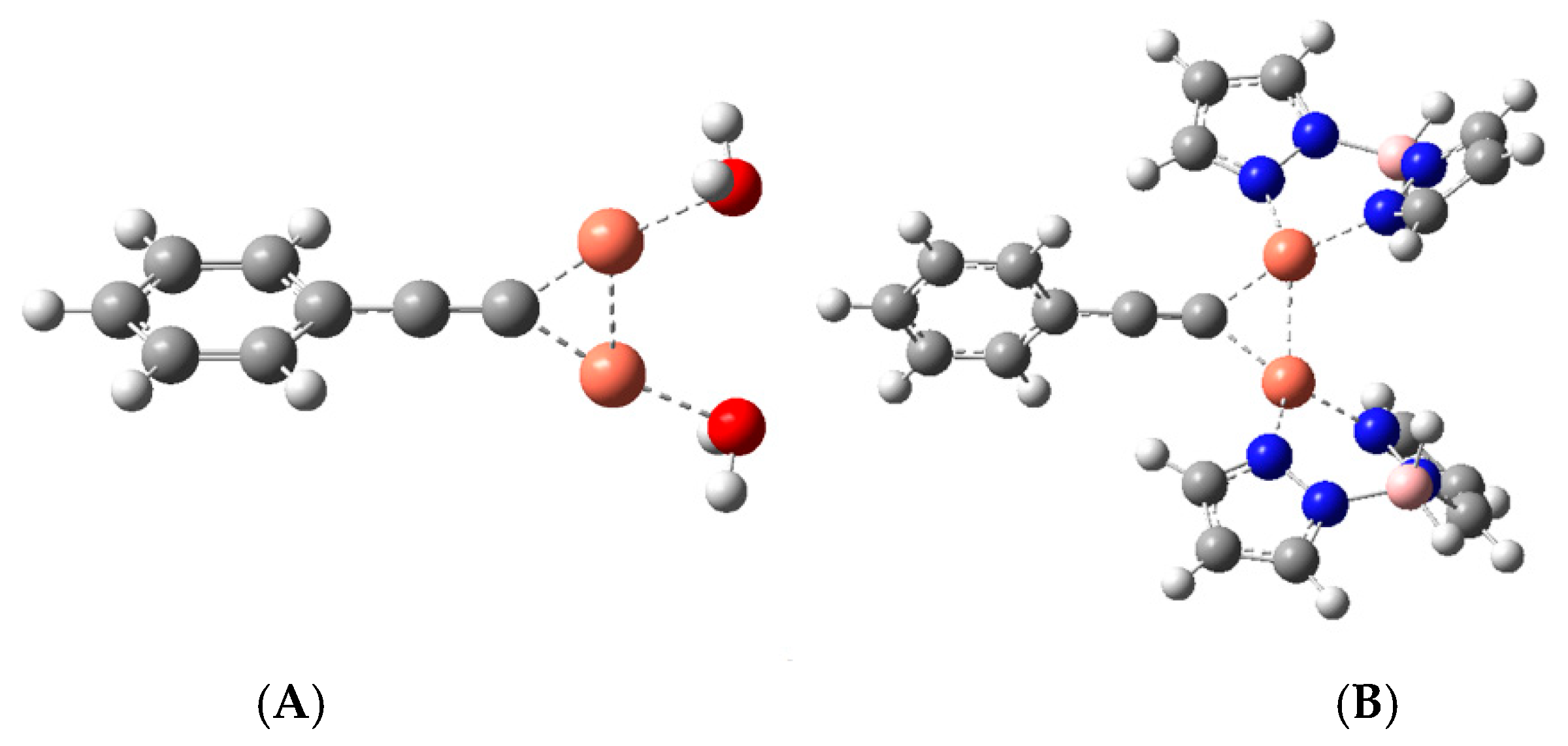
3.3. X-ray Crystallography
3.4. Protection of Sugars
3.4.1. Preparation of 1,2:3,4-di-O-Isopropylidene-α-D-galactopyranose
3.4.2. Preparation of O-Methyl-2,3-O-isopropylidene-D-ribofuranoside
3.5. General Procedure for the Tosylation of Sugars
3.5.1. 3,4-di-O-Isopropylidene-6-O-p-toluenesulfonyl-α-D-galactopyranose
3.5.2. O-Methyl-2,3-O-isopropylidene-5-O-p-toluenesulfonyl-D-ribofuranoside
3.6. General Procedure for the Preparation of Sugar Azides
3.6.1. 6-Azido-1,2: 3,4-di-O-isopropylidene-D–galactopyranose
3.6.2. 5-Azido-1-O-methyl-2,3-O-isopropylidene-D-ribofuranoside
3.7. General Procedure for the Synthesis of 1,2,3-Triazoles
3.7.1. Synthesis of 1-Benzyl-4-phenyl-1H-1,2,3-triazole (3a)
3.7.2. Synthesis of 1-Benzyl-4-(4-fluorophenyl)-1H-1,2,3-triazole (3b)
3.7.3. Synthesis of 1-Benzyl-4-p-tolyl-1H-1,2,3-triazole (3c)
3.7.4. Synthesis of Methyl 4-(1-Benzyl-1H-1,2,3-triazol-4-yl)benzoate (3d)
3.7.5. Synthesis of 4-(1-Benzyl-1H-1,2,3-triazol-4-yl)benzaldehyde (3e)
3.7.6. Synthesis of 1-Benzyl-4-(4-phenoxyphenyl)-1H-1,2,3-triazole (3f)
3.7.7. Synthesis of 1-Phenethyl-4-phenyl-1H-1,2,3-triazole (3g)
3.7.8. Synthesis of 4-(1-Benzyl-1H-1,2,3-triazol-4-yl)benzenamine (3h)
3.7.9. Synthesis of 1-Benzyl-4-(((2,2,7,7-tetramethyltetrahydro-5H-bis([1,3]dioxolo)[4,5-b:4′,5′-d]pyran-5-yl)methoxy)methyl)-1H-1,2,3-triazole (3i)
3.7.10. Synthesis of 4-Phenyl-1-((2,2,7,7-tetramethyltetrahydro-5H-bis([1,3]dioxolo)[4,5-b:4′,5′-d]pyran-5-yl)methyl)-1H-1,2,3-triazole (3j)
3.7.11. Synthesis of 1-((6-Methoxy-2,2-dimethyltetrahydrofuro [3,4-d][1,3]dioxol-4-yl)methyl)-4-(((2,2,7,7-tetramethyltetrahydro-5H-bis([1,3]dioxolo)[4,5-b:4′,5′-d]pyran-5-yl)methoxy)methyl)-1H-1,2,3-triazole (3k)
3.7.12. Synthesis of 4-Phenyl-1-(12-(4-phenyl-1H-1,2,3-triazol-1-yl)dodecyl)-1H-1,2,3-triazole (3l)
3.7.13. Synthesis of 1,12-bis(4-(((2,2,7,7-Tetramethyltetrahydro-5H-bis([1,3]dioxolo)[4,5-b:4’,5’-d]pyran-5-yl)methoxy)methyl)-1H-1,2,3-triazol-1-yl)dodecane (3m)
3.7.14. Synthesis of 1,12-bis(4-(((2,2,7,7-Tetramethyltetrahydro-5H-bis([1,3]dioxolo)[4,5-b:4’,5’-d]pyran-5-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)dodecane (3n)
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Presolski, S.I.; Díaz Díaz, D.; Fokin, V.V.; Finn, M.G. Ligand-Accelerated Cu-Catalyzed Azide−Alkyne Cycloaddition: A Mechanistic Report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Presolski, S.I.; Gardinier, S.; Lim, Y.H.; Finn, M.G. Benzimidazole and Related Ligands for Cu-Catalyzed Azide−Alkyne Cycloaddition. J. Am. Chem. Soc. 2007, 129, 12696–12704. [Google Scholar] [CrossRef] [PubMed]
- Bahsis, L.; Ben El Ayouchia, H.; Anane, H.; Triki, S.; Julve, M.; Stiriba, S.E. Copper(II)-dipicolinate-mediated clickable azide–alkyne cycloaddition in water as solvent. J. Coord. Chem. 2018, 71, 633–643. [Google Scholar] [CrossRef]
- Candelon, N.; Lastécouères, D.; Diallo, A.K.; Aranzaes, J.R.; Astruc, D.; Vincent, J.M. A highly active and reusable copper(I)-tren catalyst for the ‘click’ 1,3-dipolar cycloaddition of azides and alkynes. Chem. Commun. 2008, 6, 741–743. [Google Scholar] [CrossRef]
- Özçubukçu, S.; Ozkal, E.; Jimeno, C.; Pericàs, M.A. A Highly Active Catalyst for Huisgen 1,3-Dipolar Cycloadditions Based on the Tris(triazolyl)methanol−Cu(I) Structure. Org. Lett. 2009, 11, 4680–4683. [Google Scholar] [CrossRef]
- Cano, I.; Nicasio, M.C.; Pérez, P.J. Copper(I) complexes as catalysts for the synthesis of N-sulfonyl-1,2,3-triazoles from N-sulfonylazides and alkynes. Org. Biomol. Chem. 2010, 8, 536–538. [Google Scholar] [CrossRef]
- Trofimenko, S. Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Janiak, C.; Scharmann, T.G. Supramolecular C H⋯O, C H⋯N and C H⋯Cl interactions in metal compounds with multi-topic poly(pyrazolyl)borate ligands. Polyhedron 2003, 22, 1123–1133. [Google Scholar] [CrossRef]
- Kealey, S.; White, A.J.P.; Gee, A.D.; Long, N.J. Evaluation of [12C/11C]Carbon Monoxide Binding to Copper(I) Tris(pyrazolyl)borate Complexes. Eur. J. Inorg. Chem. 2014, 2014, 1896–1905. [Google Scholar] [CrossRef]
- Fujisawa, K.; Ono, T.; Ishikawa, Y.; Amir, N.; Miyashita, Y.; Okamoto, K.; Lehnert, N. Structural and Electronic Differences of Copper(I) Complexes with Tris(pyrazolyl)methane and Hydrotris(pyrazolyl)borate Ligands. Inorg. Chem. 2006, 45, 1698–1713. [Google Scholar] [CrossRef]
- Conry, R.R.; Ji, G.; Tipton, A.A. Synthesis and Characterization of Copper(I) Complexes with a Fairly Bulky Tris(pyrazolyl)hydroborate Ligand. Probing the Flexibility of the Metal-Containing Pocket Formed by the Ligand. Inorg. Chem. 1999, 38, 906–913. [Google Scholar] [CrossRef]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P.J. Trispyrazolylborate coinage metals complexes: Structural features and catalytic transformations. Coord. Chem. Rev. 2019, 390, 171–189. [Google Scholar] [CrossRef]
- Yap, G.P.A.; Jove, F.; Urbano, J.; Alvarez, E.; Trofimenko, S.; Díaz-Requejo, M.M.; Pérez, P.J. Unusual Polybrominated Polypyrazolylborates and Their Copper(I) Complexes: Synthesis, Characterization, and Catalytic Activity. Inorg. Chem. 2007, 46, 780–787. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Lobbia, G.G.; Papini, G. Synthesis and Properties of Poly(pyrazolyl)borate and Related Boron-Centered Scorpionate Ligands. Part A: Pyrazole-Based Systems. MROC 2010, 7, 84–124. [Google Scholar] [CrossRef]
- Díaz-Requejo, M.M.; Pérez, P.J. Coinage Metal Catalyzed C−H Bond Functionalization of Hydrocarbons. Chem. Rev. 2008, 108, 3379–3394. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Churchill, M.R.; DeBoer, B.G.; Rotella, F.J.; Abu Salah, O.M.; Bruce, M.I. Determination of the crystal structure and molecular geometry of [hydrotris(1-pyrazolyl) borato] copper(I) carbonyl. Unique structural investigation of a copper-carbonyl linkage. Inorg. Chem. 1975, 14, 2051–2056. [Google Scholar] [CrossRef]
- Fujisawa, K.; Kobayashi, T.; Fujita, K.; Kitajima, N.; Moro-oka, Y.; Miyashita, Y.; Yamada, Y.; Okamoto, K. Mononuclear Copper(II) Hydroxo Complex: Structural Effect of a 3-Position of Tris(pyrazolyl)borates. BCSJ 2000, 73, 1797–1804. [Google Scholar] [CrossRef]
- Harmand, L.; Lescure, M.H.; Candelon, N.; Duttine, M.; Lastécouères, D.; Vincent, J.M. Huisgen click cycloadditions from a copper(II)-tren precatalyst without external sacrificial reductant. Tetrahedron Lett. 2012, 53, 1417–1420. [Google Scholar] [CrossRef]
- Gonda, Z.; Novák, Z. Highly active copper-catalysts for azide-alkyne cycloaddition. Dalton Trans. 2009, 39, 726–729. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Worrell, B.T.; Malik, J.A.; Fokin, V.V. Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide–Alkyne Cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef]
- Nolte, C.; Mayer, P.; Straub, B.F. Isolation of a Copper(I) Triazolide: A “Click” Intermediate. Angew. Chem. Int. Ed. 2007, 46, 2101–2103. [Google Scholar] [CrossRef]
- Calvo-Losada, S.; Pino, M.S.; Quirante, J.J. On the regioselectivity of the mononuclear copper-catalyzed cycloaddition of azide and alkynes (CuAAC). A quantum chemical topological study. J. Mol. Model. 2014, 20, 2187. [Google Scholar] [CrossRef]
- Ben El Ayouchia, H.; Bahsis, L.; Anane, H.; Domingo, L.R.; Stiriba, S.E. Understanding the mechanism and regioselectivity of the copper(I) catalyzed [3 + 2] cycloaddition reaction between azide and alkyne: A systematic DFT study. RSC Adv. 2018, 8, 7670–7678. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Roy, L.E.; Hay, P.J.; Martin, R.L. Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. [Google Scholar] [CrossRef]
- Frisch, M.J.; Hratchian, H.P.; Nielsen, A.B. Gaussian 09; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. In Horizons of Quantum Chemistry; Fukui, K., Pullman, B., Eds.; Springer: Amstelredam, The Netherlands, 1980; pp. 5–15. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a | 1b | Exp a | Exp b | |
|---|---|---|---|---|
| Bond Lengths (Å) | ||||
| Cu–O | 2.090 | 2.008 | - | 1.918 |
| Cu–N1 | 2.109 | 1.966 | 2.039 | 2.004 |
| Cu–N5 | 2.075 | 1.966 | 2.059 | 2.061 |
| Cu–N2 | 2.109 | - | 2.039 | 2.094 |
| N1–N3 | 1.390 | 1.386 | 1.378 | |
| N2–N4 | 1.390 | 1.386 | 1.353 | |
| N5–N6 | 1.390 | - | 1.350 | - |
| N3–B | 1.553 | 1.578 | 1.526 | - |
| N4–B | 1.555 | 1.578 | 1.558 | - |
| N6–B | 1.553 | - | 1.540 | - |
| Bond Angles (°) | ||||
| O–Cu–N1 | 119.447 | 127.425 | - | 131.9 |
| O–Cu–N2 | 128.412 | 127.592 | - | 114.5 |
| O–Cu–N5 | 119.596 | - | - | 121.7 |
| N1–Cu–N2 | 93.974 | 104.981 | 90.4 | 92.7 |
| N1–Cu–N5 | 93.149 | 92.4 | 92.1 | |
| N2–Cu–N5 | 93.982 | - | 90.8 | 95.4 |

| Entry | Ligand | Solvent | Time (h) | Yield (%) |
|---|---|---|---|---|
| 1 | - | Toluene | 48 | - |
| 2 | - | H2O/EtOH | 48 | 50 |
| 3 | HB(Pz)3− | CH3CN | 24 | 89 |
| 4 | H2B(Pz)2− | CH3CN | 24 | 78 |
| 5 | HB(Pz)3− | CH3OH | 24 | 92 |
| 6 | H2B(Pz)2− | CH3OH | 24 | 83 |
| 7 | HB(Pz)3− | EtOH | 24 | 91 |
| 8 | H2B(Pz)2− | EtOH | 24 | 80 |
| 9 | HB(Pz)3− | H2O | 24 | 92 |
| 10 | H2B(Pz)2− | H2O | 24 | 83 |
| 11 | HB(Pz)3− | H2O/EtOH | 24 | 95 |
| 12 | H2B(Pz)2− | H2O/EtOH | 24 | 84 |

| Entry | Alkyne | Azide | Product | Ligand | Yield a (%) | TON b |
|---|---|---|---|---|---|---|
| 1 |  |  | 3a | HB(pz)3− | 92 | 18.4 |
| H2B(pz)2− | 84 | 16.8 | ||||
| 2 |  | | 3b | HB(pz)3− | 93 | 18.4 |
| H2B(pz)2− | 89 | 17.8 | ||||
| 3 |  | | 3c | HB(pz)3− | 89 | 17.8 |
| H2B(pz)2− | 87 | 17.4 | ||||
| 4 |  | | 3d | HB(pz)3− | 76 | 15.2 |
| H2B(pz)2− | 70 | 14 | ||||
| 5 |  | | 3e | HB(pz)3− | 92 | 18.4 |
| H2B(pz)2− | 88 | 17.6 | ||||
| 6 |  | | 3f | HB(pz)3− | 89 | 17.8 |
| H2B(pz)2− | 86 | 17.2 | ||||
| 7 | |  | 3g | HB(pz)3− | 88 | 17.6 |
| H2B(pz)2− | 82 | 16.4 | ||||
| 8 |  | | 3h | HB(pz)3− | 73 | 14.6 |
| H2B(pz)2− | 70 | 14 | ||||
| 9 |  | | 3i | HB(pz)3− | 83 | 16.6 |
| H2B(pz)2− | 80 | 16 | ||||
| 10 | |  | 3j | HB(pz)3− | 89 | 17.8 |
| H2B(pz)2− | 85 | 17 | ||||
| 11 | |  | 3k | HB(Pz)3− | 80 | 16 |
| H2B(Pz)2− | 78 | 78 | ||||
| 12 | | N3–(CH2)12–N3 | 3l | HB(Pz)3− | 90 | 18 |
| H2B(Pz)2− | 77 | 15.4 | ||||
| 13 | | N3–(CH2)12–N3 | 3m | HB(pz)3− | 80 | 16 |
| H2B(pz)2− | 78 | 15.6 | ||||
| 14 |  | N3–(CH2)12–N3 | 3n | HB(pz)3− | 85 | 17 |

| Entry a | Cu Loading (mol %) | Solvent | T (°C) | Time (h) | Yield (%) | References |
|---|---|---|---|---|---|---|
| Cu(I)-HB(pz)3− | 5 | EtOH/H2O | 25 | 24 | 92 | This work |
| Cu(I)-H2B(pz)2− | 5 | EtOH/H2O | 25 | 24 | 84 | |
| Cu(II)-tren | 0.2 | n-Octane | 25 | 24 | 84 | [22] |
| Cu(I)-tren | 0.05 | Toluene | 60 | 24 | 86 | [7] |
| Cu(I)-TBTM | 0.5 | H2O | 25 | 4 | 94 | [8] |
| Cu(I)-TBTA | 1 | t-BuOH/H2O | 25 | 24 | 84 | [19] |
| C3H7COOCu(PPh3)2 | 0.15 | Dichloromethane | 28 | 3 | 99 | [23] |
| Species | µ (eV) | η (eV) | ω (eV) | N (eV) |
|---|---|---|---|---|
| Dinuclear Cu(I)-acetylide | 1.68 | 1.09 | 1.29 | 10.25 |
| Phenylazide | −3.62 | 5.17 | 1.27 | 2.92 |
| Phenyl acetylene | −3.53 | 5.51 | 1.13 | 2.83 |
| Dinuclear Cu(I)-acetylide | 0.38 | 3.47 | 0.02 | 7.76 |

| Number of the Atom | |||
|---|---|---|---|
| Phenylazide | N1 N2 N3 | 0.29 0.18 −0.09 | |
| Simplest dinuclear Cu(I)-acetylide (A) | C4 C5 Cu | 0.28 −0.07 0.25 | |
| Dinuclear Cu(I)-acetylide stabilized by bis(pyrazolyl)borate (B) | C4 C5 Cu | 0.28 −0.12 0.30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahsis, L.; Ben El Ayouchia, H.; Anane, H.; Ramirez de Arellano, C.; Bentama, A.; El Hadrami, E.M.; Julve, M.; Domingo, L.R.; Stiriba, S.-E. Clicking Azides and Alkynes with Poly(pyrazolyl)borate-Copper(I) Catalysts: An Experimental and Computational Study. Catalysts 2019, 9, 687. https://doi.org/10.3390/catal9080687
Bahsis L, Ben El Ayouchia H, Anane H, Ramirez de Arellano C, Bentama A, El Hadrami EM, Julve M, Domingo LR, Stiriba S-E. Clicking Azides and Alkynes with Poly(pyrazolyl)borate-Copper(I) Catalysts: An Experimental and Computational Study. Catalysts. 2019; 9(8):687. https://doi.org/10.3390/catal9080687
Chicago/Turabian StyleBahsis, Lahoucine, Hicham Ben El Ayouchia, Hafid Anane, Carmen Ramirez de Arellano, Abdeslem Bentama, El Mestafa El Hadrami, Miguel Julve, Luis R. Domingo, and Salah-Eddine Stiriba. 2019. "Clicking Azides and Alkynes with Poly(pyrazolyl)borate-Copper(I) Catalysts: An Experimental and Computational Study" Catalysts 9, no. 8: 687. https://doi.org/10.3390/catal9080687