Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis


Abstract
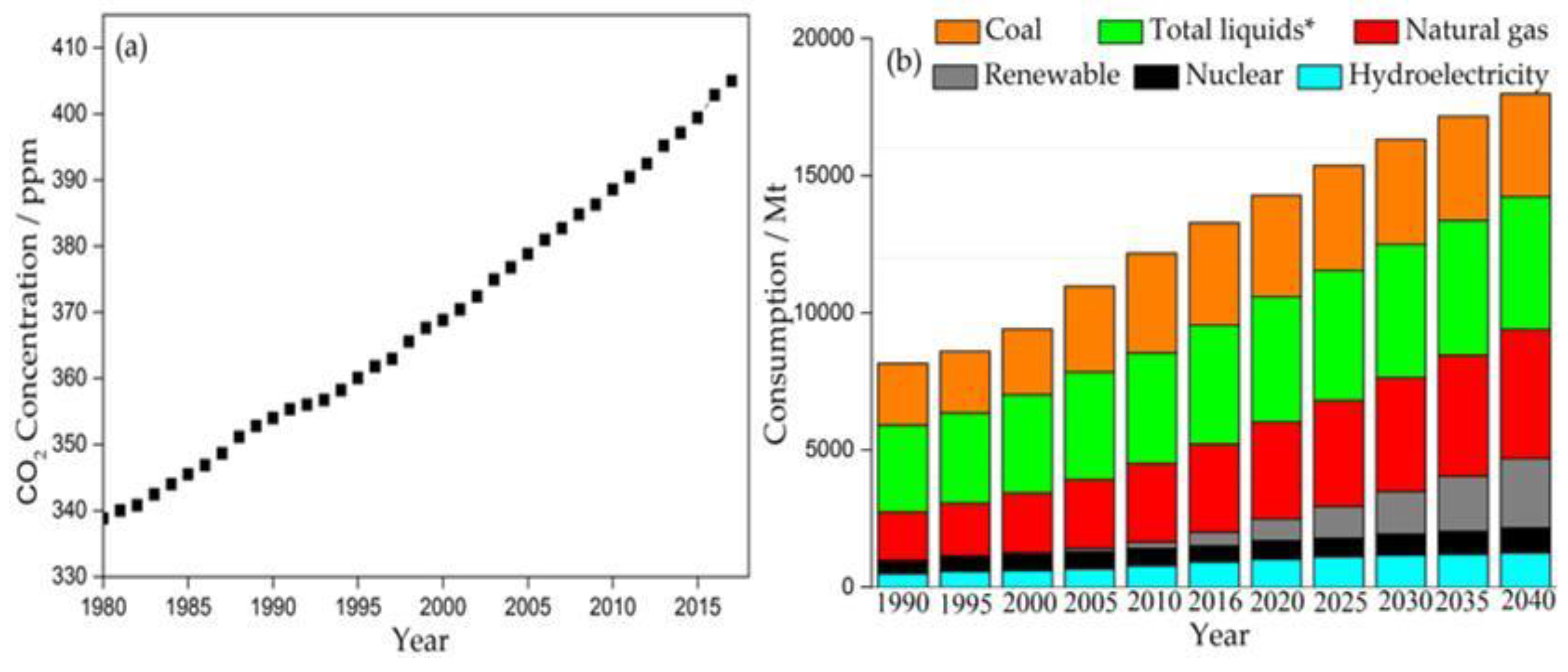
:1. Introduction
2. Heterogeneous Catalysis
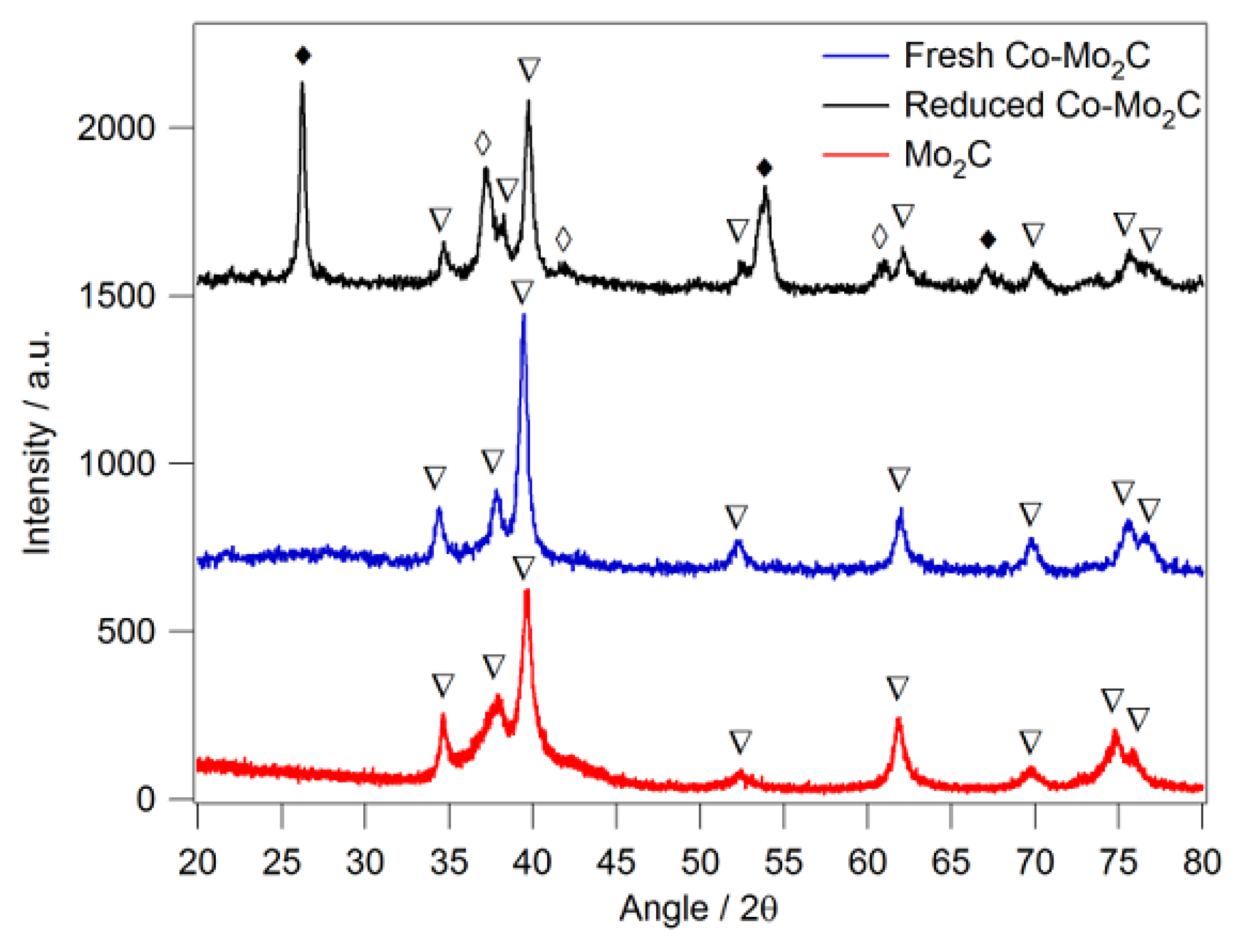
2.1. CO2 to CO
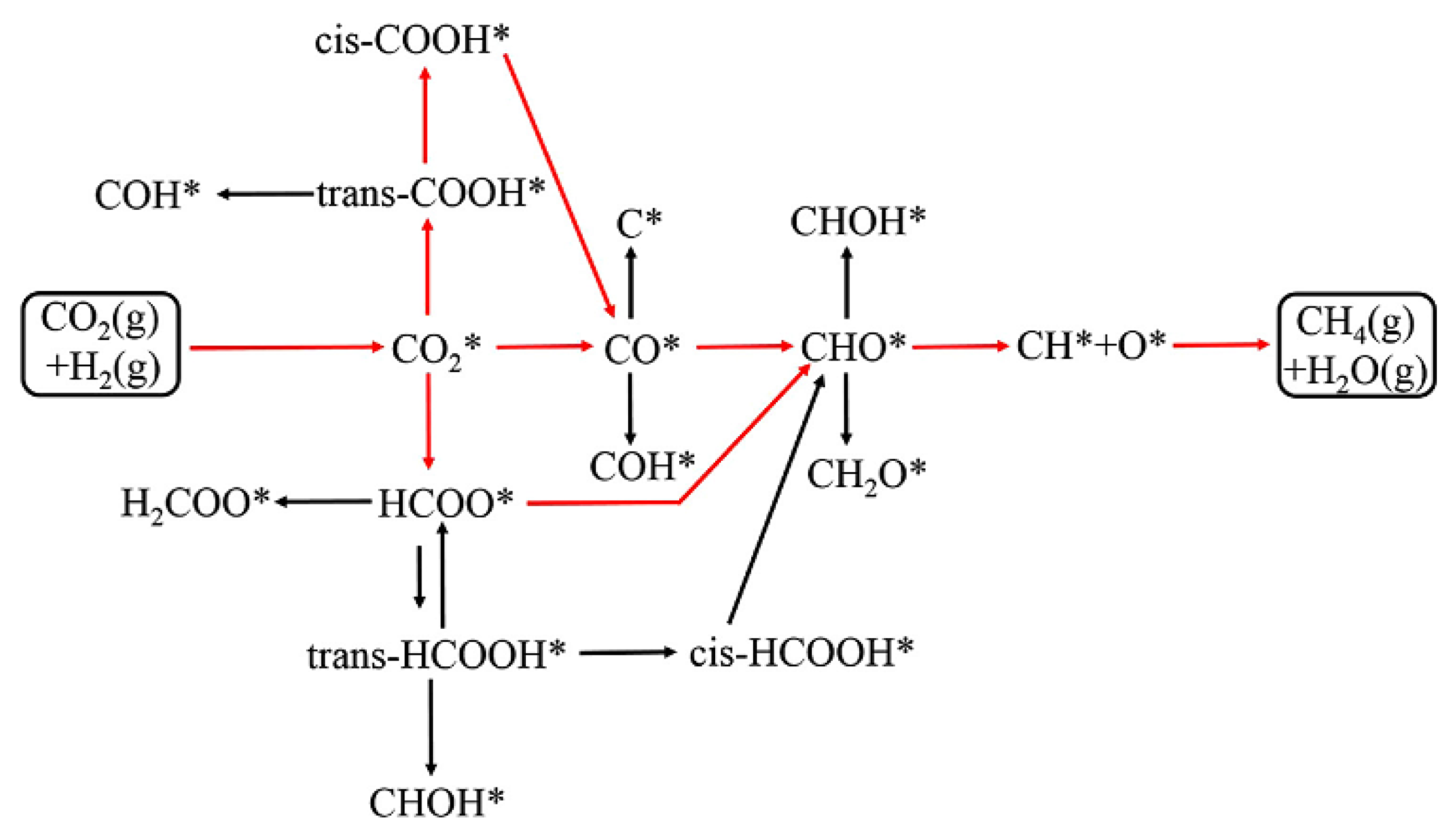
2.2. CO2 to CH4
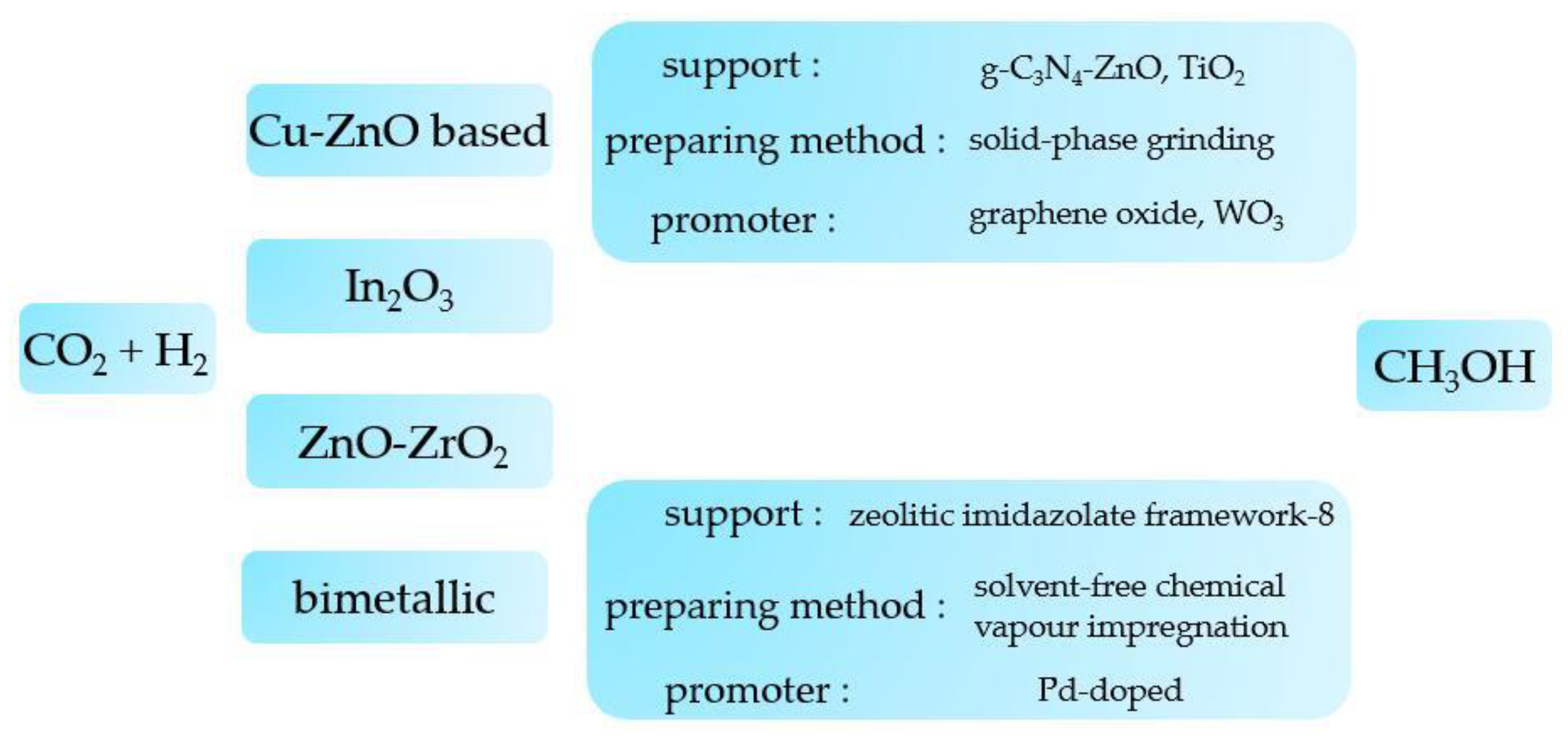
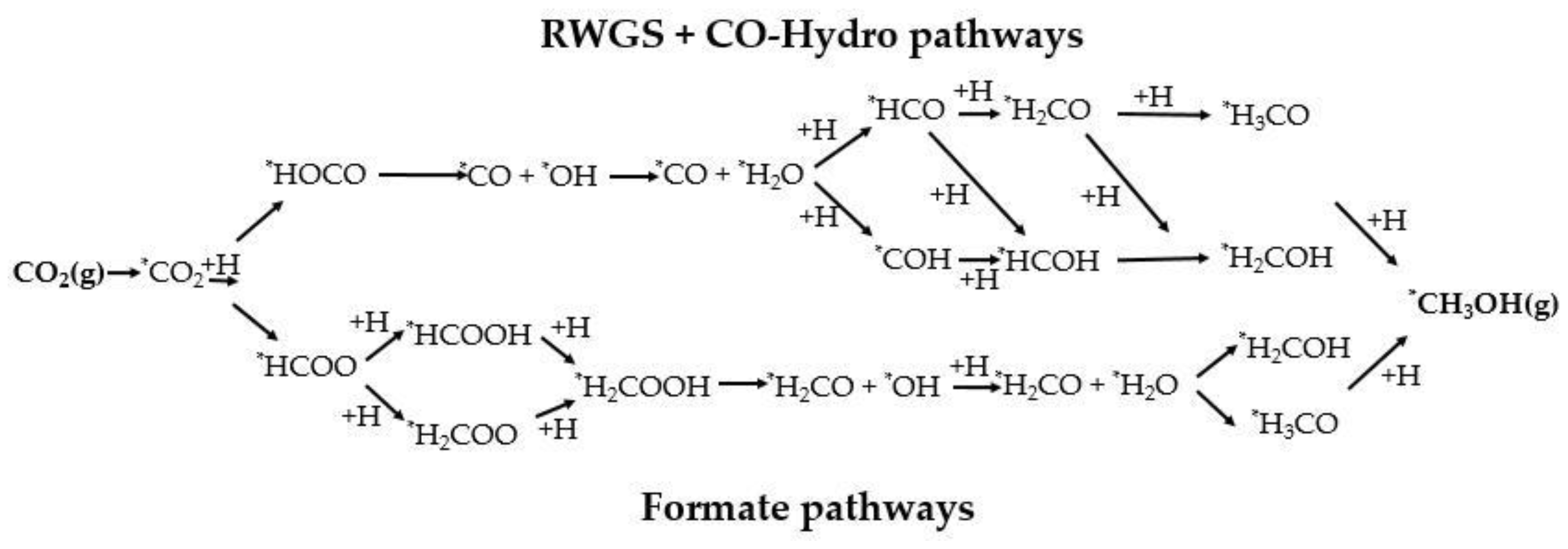
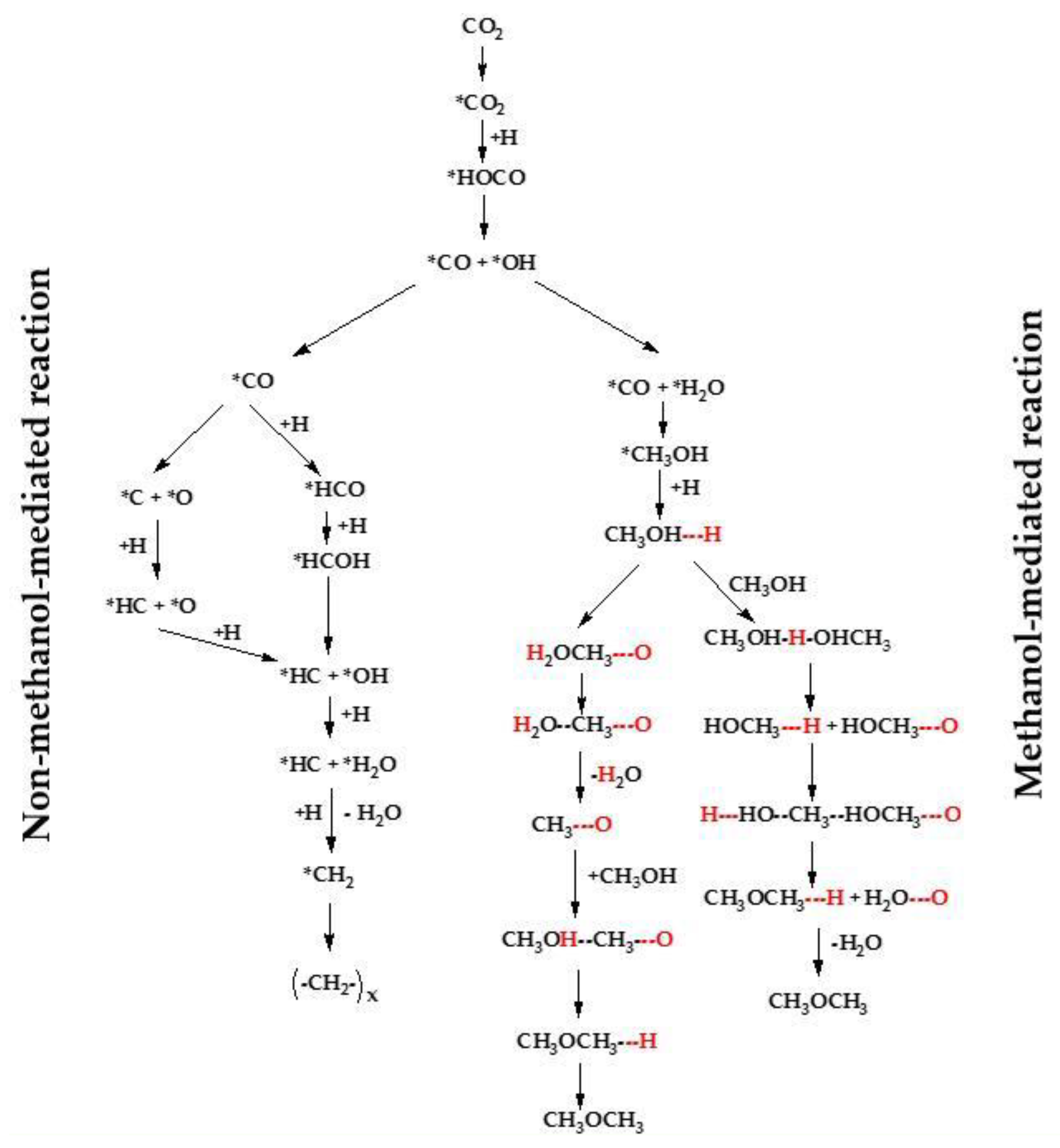
2.3. CO2 to CH3OH
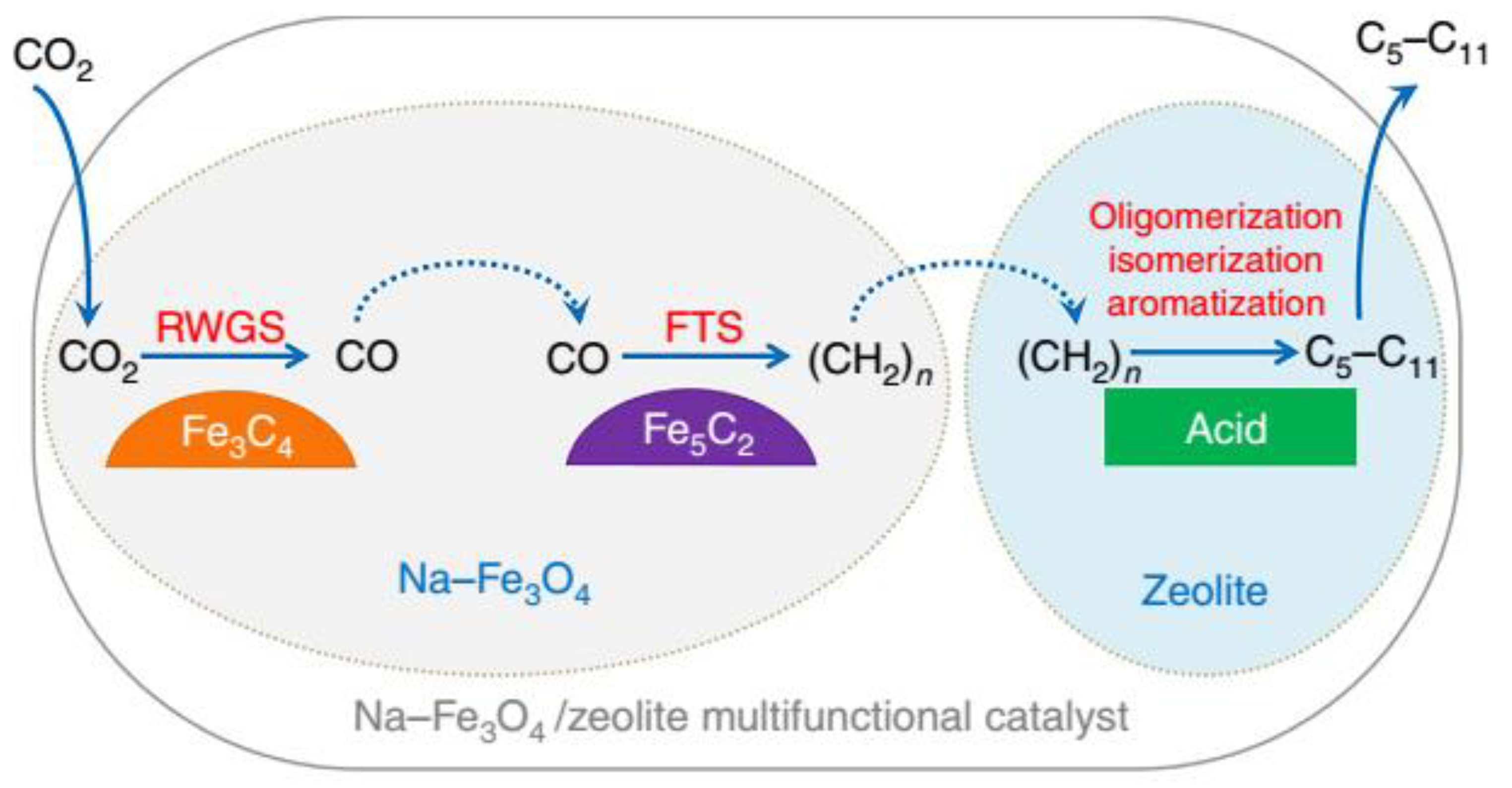
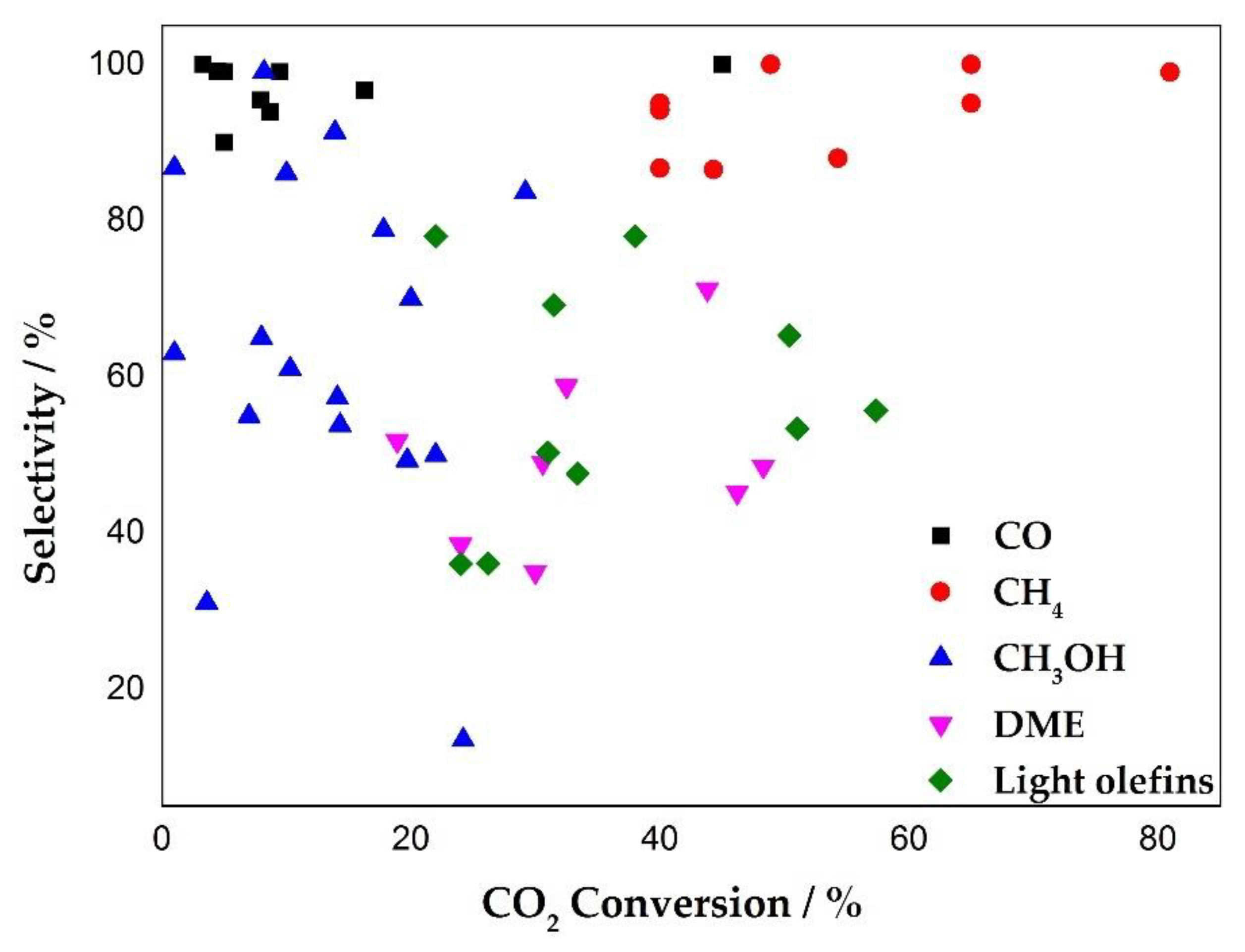
2.4. CO2 to Other Products
2.5. Opportunities of Heterogeneous Catalysis for CO2 Conversion
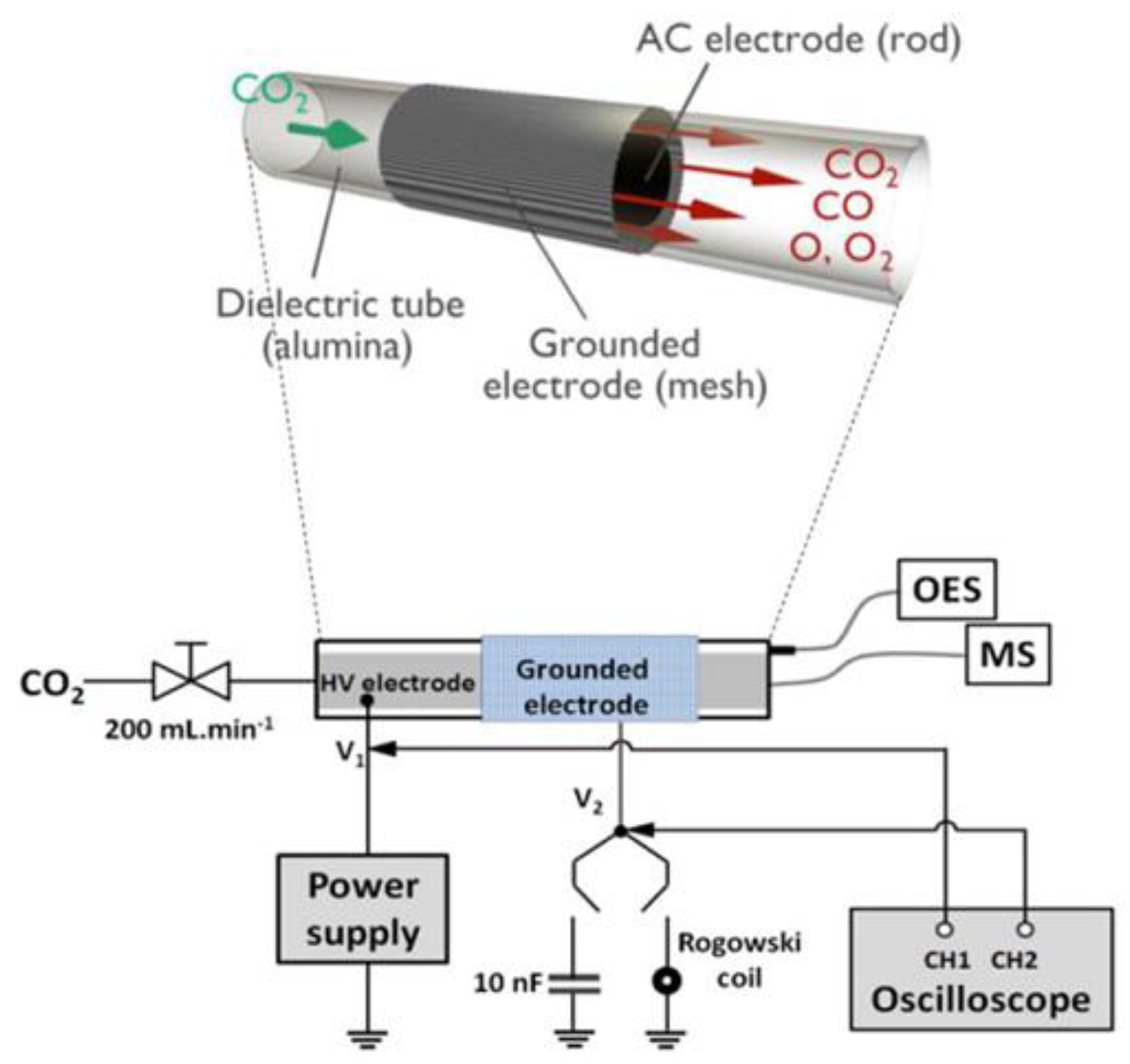
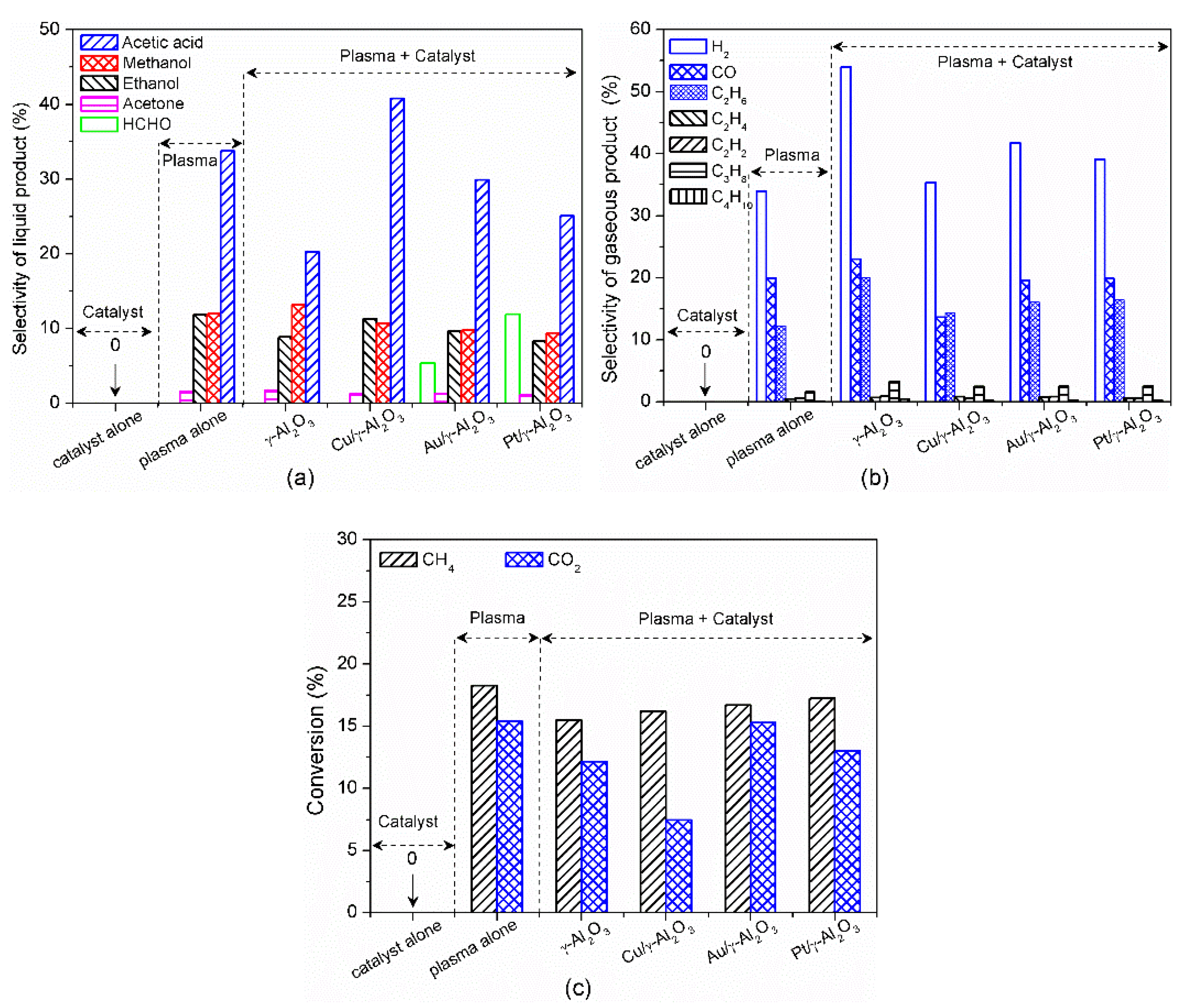
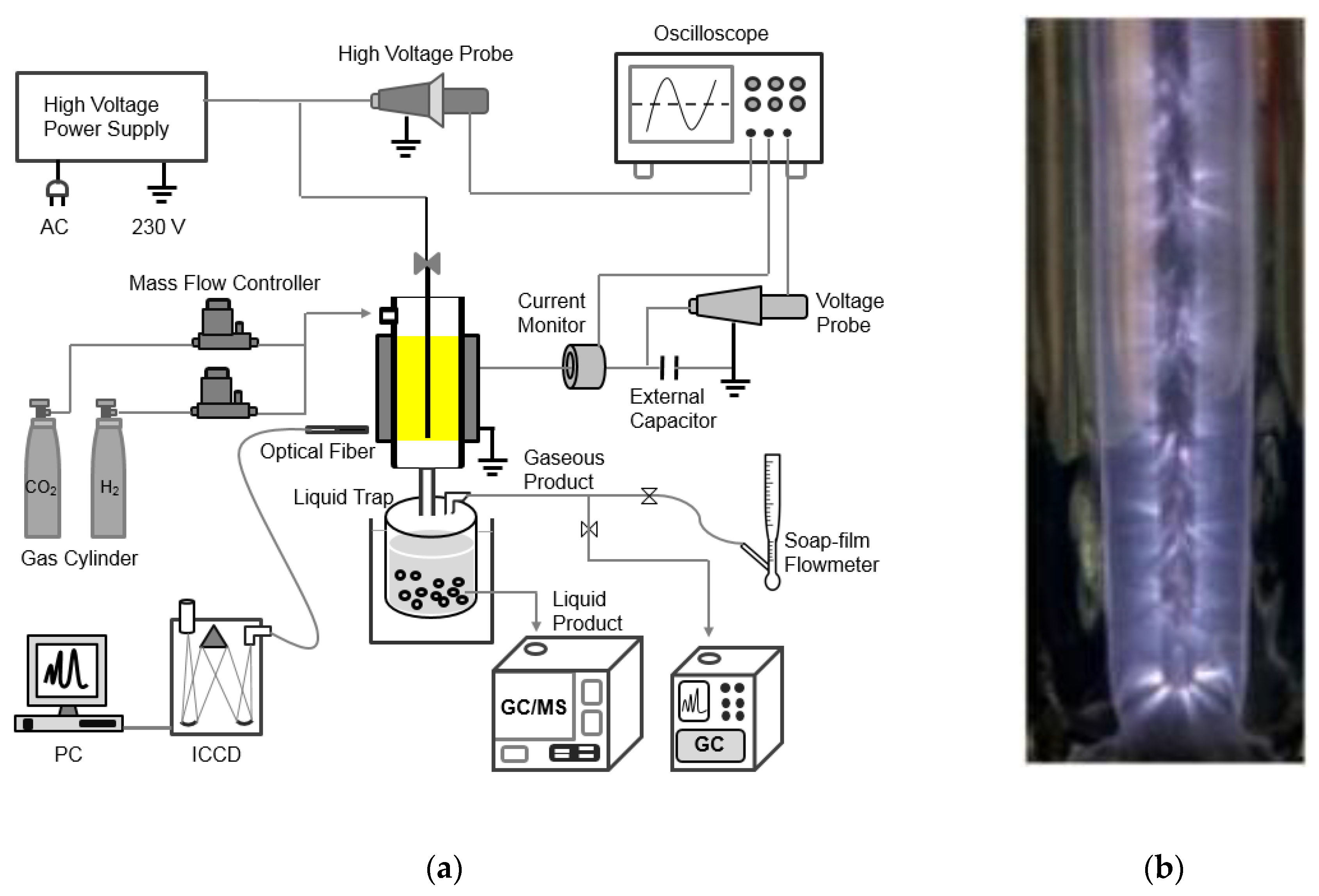
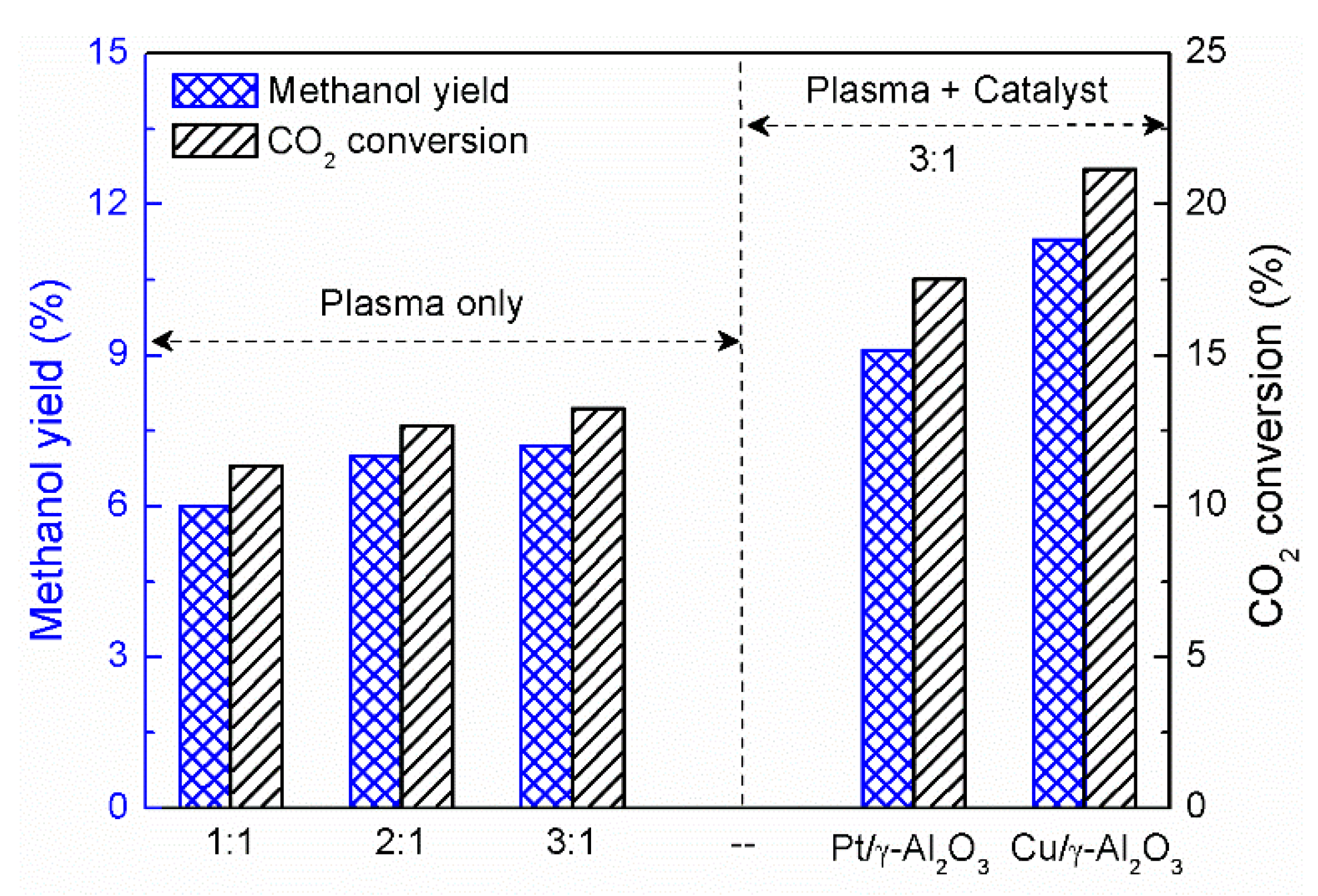
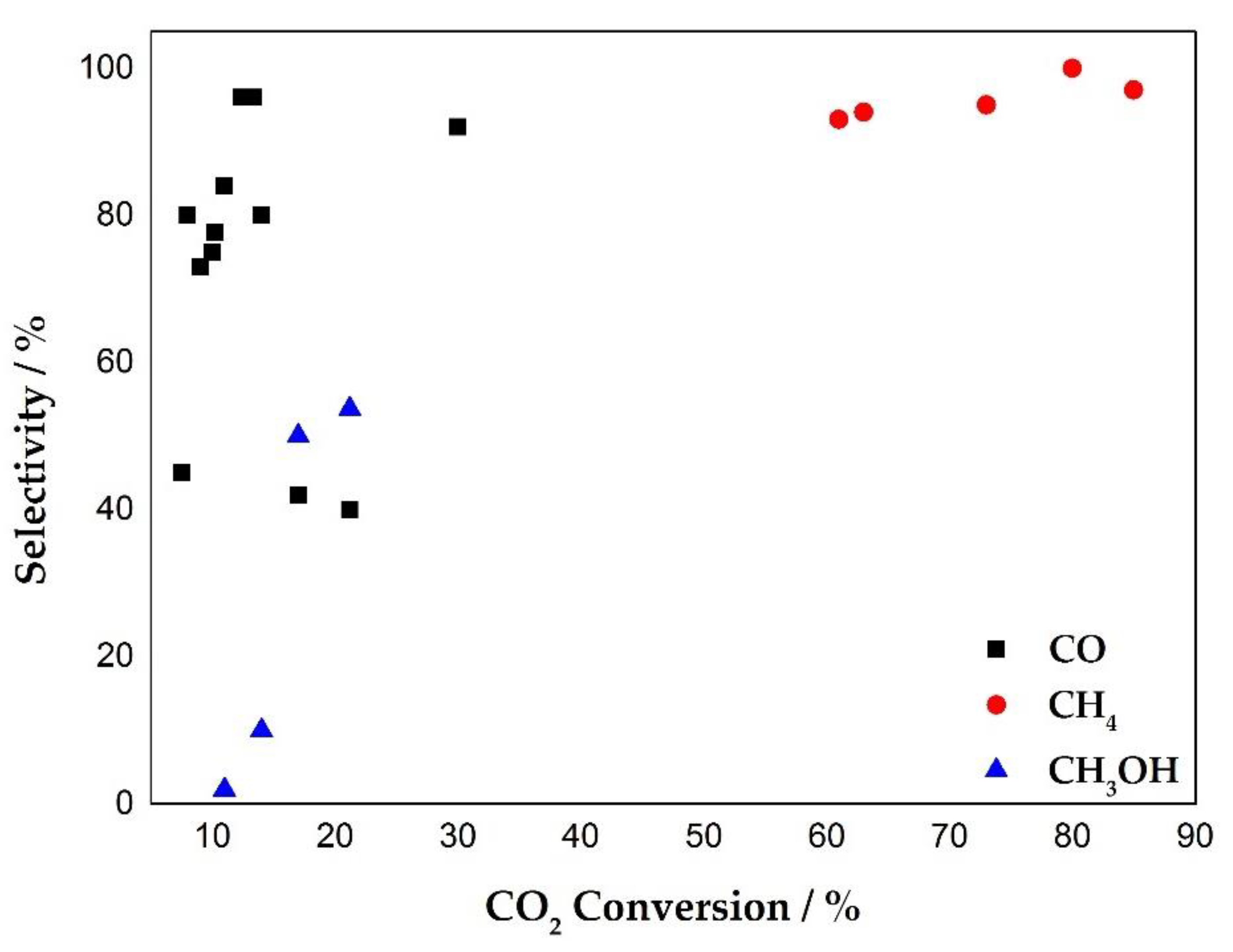
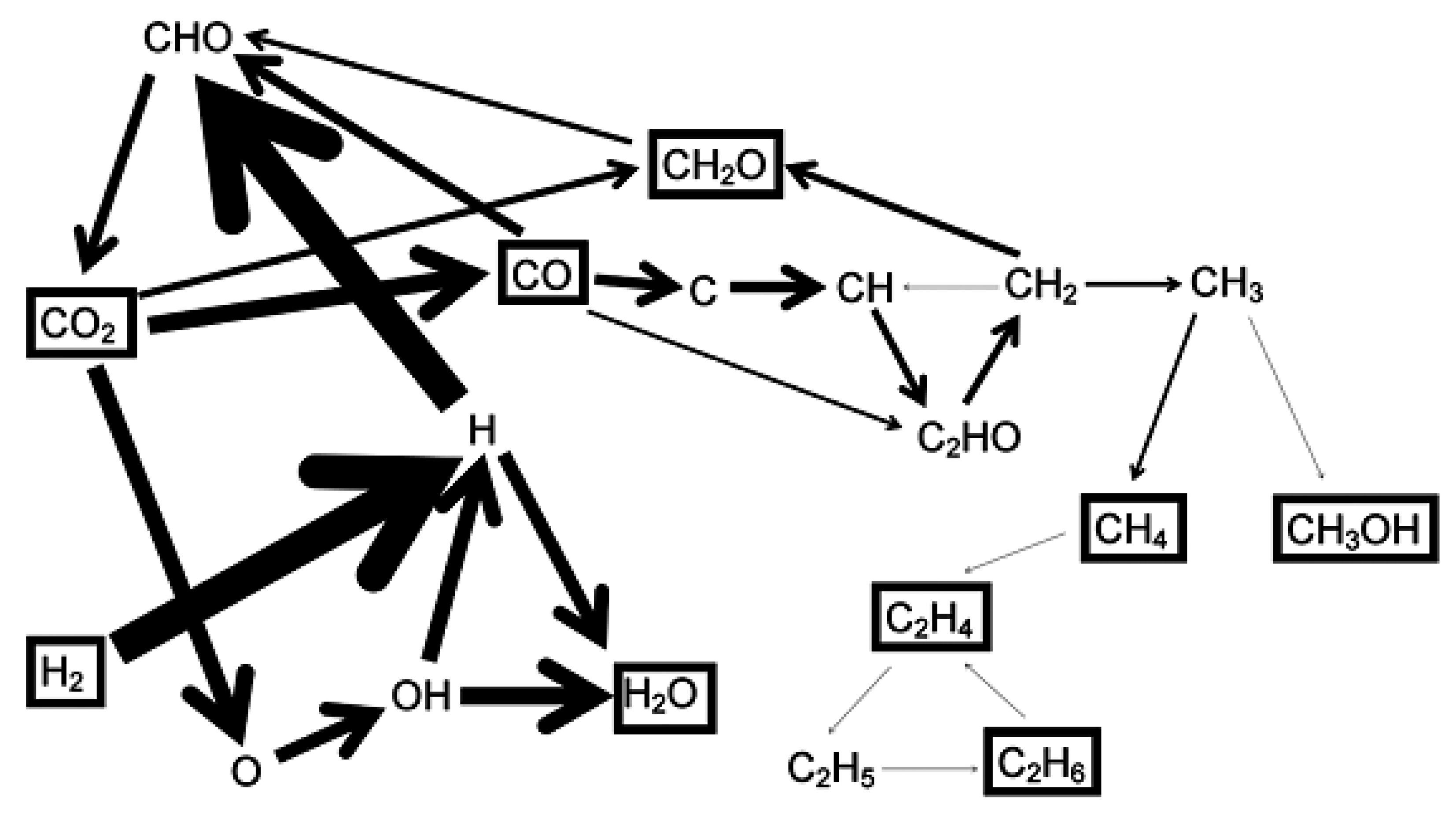
3. Plasma Catalysis
4. Outlook and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ingwersen, W.W.; Garmestani, A.S.; Gonzalez, M.A.; Templeton, J.J. A systems perspective on responses to climate change. Clean Technol. Environ. Policy 2014, 16, 719–730. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Cong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Irish, J.L.; Sleath, A.; Cialone, M.A.; Knutson, T.R.; Jensen, R.E. Simulations of Hurricane Katrina (2005) under sea level and climate conditions for 1900. Clim. Chang. 2014, 122, 635–649. [Google Scholar] [CrossRef]
- Sanz-perez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct Capture of CO2 from Ambient Air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef] [PubMed]
- Boot-Handford, M.E.; Abanades, J.C.; Anthony, E.J.; Blunt, M.J.; Brandani, S.; Dowell, N.M. Carbon capture and storage update. Energy Environ. Sci. 2014, 7, 130–189. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S. Carbon capture and storage (CCS): The way forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Bobicki, E.R.; Liu, Q.; Xu, Z.; Zeng, H. Carbon capture and storage using alkaline industrial wastes. Prog. Energy Combust. Sci. 2012, 38, 302–320. [Google Scholar] [CrossRef]
- Rahman, F.A.; Aziz, M.M.A.; Saidur, R.; Wan, W.A.; Hainin, M.R.; Putrajava, R.; Hassan, N.A. Pollution to solution: Capture and sequestration of carbon dioxide (CO2) and its utilization as a renewable energy source for a sustainable future. Renew. Sustain. Energy Rev. 2017, 71, 112–126. [Google Scholar] [CrossRef]
- Darabi, A.; Jessop, P.G.; Cunningham, M.F. CO2-responsive polymeric materials: Synthesis, self-assembly, and functional applications. Chem. Soc. Rev. 2016, 45, 4391–4436. [Google Scholar] [CrossRef]
- Dincer, I.; Acar, C. Review and evaluation of hydrogen production methods for better sustainability. Int. J. Hydrogen Energy 2015, 40, 11094–11111. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Snoeckx, R.; Bogaerts, A. Plasma technology—A novel solution for CO2 conversion? Chem. Soc. Rev. 2017, 46, 5805–5863. [Google Scholar] [CrossRef]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2018. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.G.; Jaroniec, M.; Chen, X.B. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2018. [Google Scholar] [CrossRef]
- Mota, F.M.; Kim, D.H. From CO2 methanation to ambitious long-chain hydrocarbons: Alternative fuels paving the path to sustainability. Chem. Soc. Rev. 2019, 48, 205–259. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Vaidya, P.D.; Bhanage, B.M.; Joshi, J.B. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chem. Eng. Res. Des. 2014, 92, 2557–2567. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Bando, K.K.; Soga, K.; Kunimori, K.; Ichikuni, N.; Okabe, K.; Kusama, H.; Sayama, K.; Arakawea, H. CO2 hydrogenation activity and surface structure of zeolite-supported Rh catalysts. Appl. Catal. A 1998, 173, 47–60. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO. Angew. Chem. Int. Ed. 2014, 53, 6705–6709. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, Q.; Jiang, C.; Yao, Y.; Lu, D.; Zheng, J.; Dai, Y.; Wang, H.; Yang, Y. Ru/Al2O3 catalyzed CO2 hydrogenation: Oxygen-exchange on metal-support interfaces. J. Catal. 2018, 367, 194–205. [Google Scholar] [CrossRef]
- Yan, B.; Wu, Q.; Cen, J.; Timoshenko, J.; Frenkel, A.I.; Su, D.; Chen, X.; Parise, J.B.; Stach, E.; Orlov, A.; et al. Highly active subnanometer Rh clusters derived from Rh-doped SrTiO3 for CO2 reduction. Appl. Catal. B 2018, 237, 1003–1011. [Google Scholar] [CrossRef]
- Dai, B.; Cao, S.; Xie, H.; Zhou, G.; Chen, S. Reduction of CO2 to CO via reverse water-gas shift reaction over CeO2 catalyst. Korean J. Chem. Eng. 2018, 35, 421–427. [Google Scholar] [CrossRef]
- Alayoglu, S.; Beaumont, S.K.; Zheng, F.; Pushkarev, V.V.; Zheng, H.; Iablokov, V.; Liu, Z.; Guo, J.; Kruse, N.; Somorjai, G.A. CO2 Hydrogenation Studies on Co and CoPt Bimetallic Nanoparticles Under Reaction Conditions Using TEM, XPS and NEXAFS. Top. Catal. 2011, 54, 778–785. [Google Scholar] [CrossRef]
- Kharaji, A.G.; Shariati, A.; Takassi, M.A. A Novel γ-Alumina Supported Fe-Mo Bimetallic Catalyst for Reverse Water Gas Shift Reaction. Chin. J. Chem. Eng. 2013, 21, 1007–1014. [Google Scholar] [CrossRef]
- Kharaji, A.G.; Shariati, A.; Ostadi, M. Development of Ni–Mo/Al2O3 Catalyst for Reverse Water Gas Shift (RWGS) Reaction. J. Nanosci. Nanotechnol. 2014, 14, 6841–6847. [Google Scholar] [CrossRef]
- Zhao, B.; Yan, B.; Jiang, Z.; Yao, S.; Liu, Z.; Wu, Q.; Ran, R.; Senanayake, S.D.; Weng, D.; Chen, J.G. High selectivity of CO2 hydrogenation to CO by controlling the valence state of nickel using perovskite. Chem. Commun. 2018, 54, 7354–7357. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni catalysts with La as promoter supported over Y- and BETA-zeolites for CO2 methanation. Appl. Catal. B 2018, 238, 393–403. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, T.; Xie, H.; Zheng, X. Effects of structure on the carbon dioxide methanation performance of Co-based catalysts. Int. J. Hydrogen Energy 2013, 38, 10012–10018. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, X.Y.; Jiang, Y.; Wang, Y.; Huang, Y.; Zhang, T. Crystal phase effects on the structure and performance of ruthenium nanoparticles for CO2 hydrogenation. Catal. Sci. Technol. 2014, 4, 2058–2063. [Google Scholar] [CrossRef]
- Yuan, H.; Zhu, X.; Han, J.; Wang, H.; Ge, Q. Rhenium-promoted selective CO2 methanation on Ni-based catalyst. J. CO2 Util. 2018, 26, 8–18. [Google Scholar] [CrossRef]
- Li, M.; Amari, H.; van Veen, A.C. Metal-oxide interaction enhanced CO2 activation in methanation over ceria supported nickel nanocrystallites. Appl. Catal. B 2018, 239, 27–35. [Google Scholar] [CrossRef]
- Zhan, Y.; Wang, Y.; Gu, D.; Chen, C.; Jiang, L.; Takehira, K. Ni/Al2O3-ZrO2 catalyst for CO2 methanation: The role of γ-(Al, Zr)2O3 formation. Appl. Surf. Sci. 2018, 459, 74–79. [Google Scholar] [CrossRef]
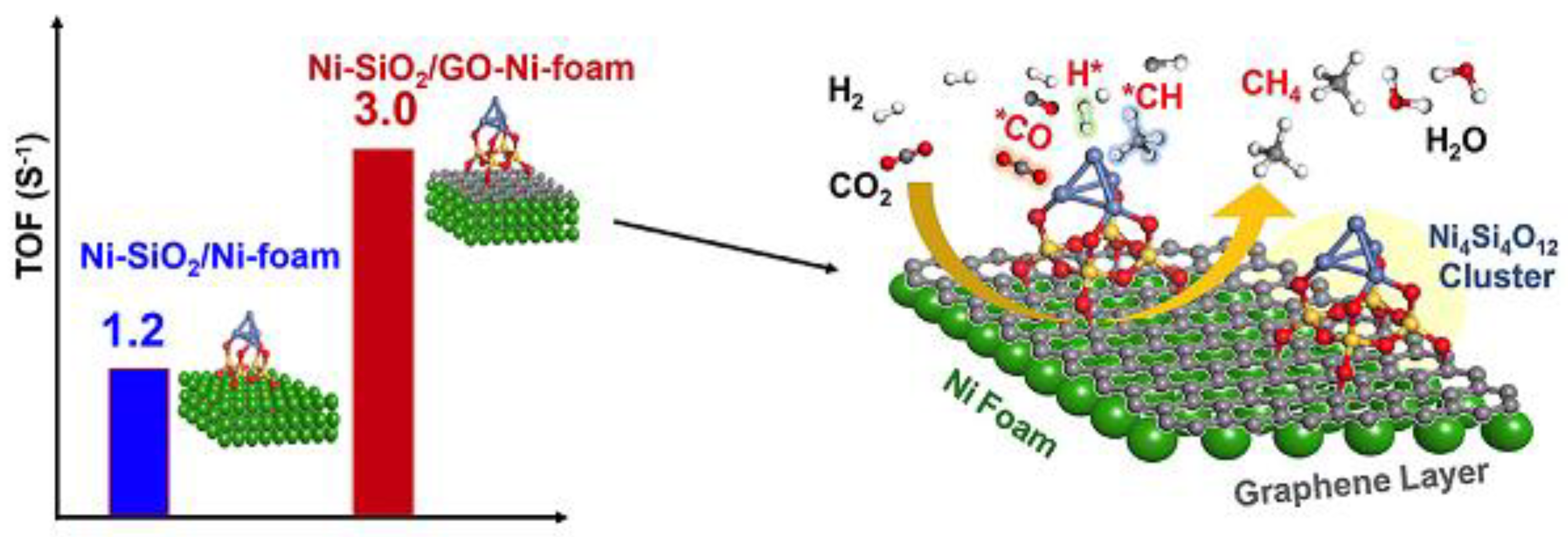
- Ma, H.; Ma, K.; Ji, J.; Tang, S.; Liu, C.; Jiang, W.; Yue, H.; Liang, B. Graphene intercalated Ni-SiO2/GO-Ni-foam catalyst with enhanced reactivity and heat-transfer for CO2 methanation. Chem. Eng. Sci. 2019, 194, 10–21. [Google Scholar] [CrossRef]
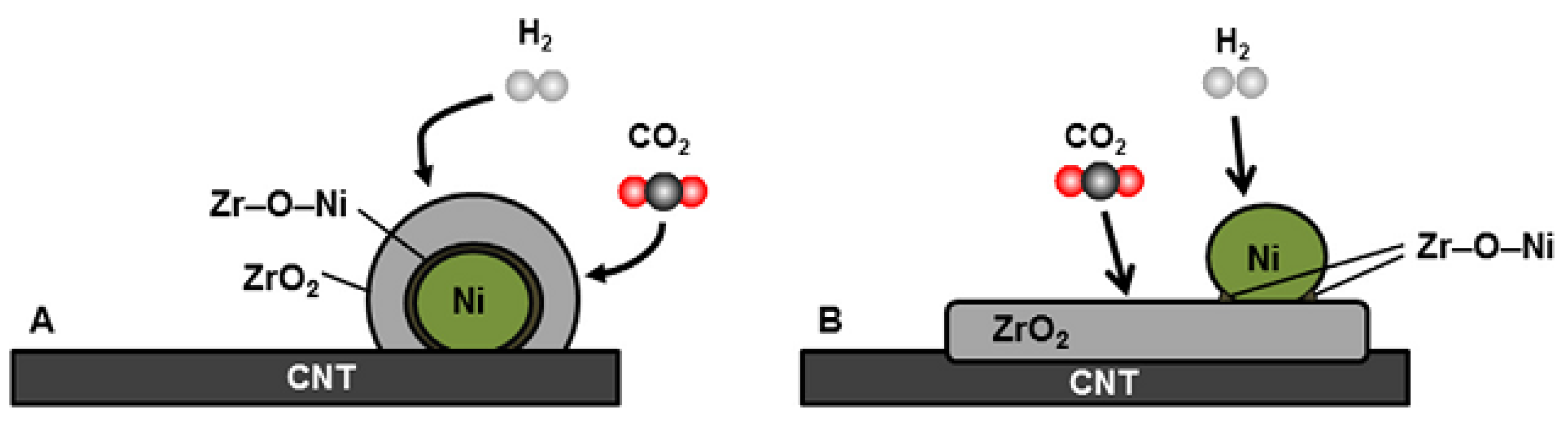
- Romero-Sáez, M.; Dongil, A.B.; Benito, N.; Espinoza-González, R.; Escalona, N.; Gracia, F. CO2 methanation over nickel-ZrO2 catalyst supported on carbon nanotubes: A comparison between two impregnation strategies. Appl. Catal. B 2018, 237, 817–825. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Lopes, J.M.; Henriques, C. Ni-Ce/Zeolites for CO2 Hydrogenation to CH4: Effect of the Metal Incorporation Order. ChemCatChem 2018, 10, 2773–2781. [Google Scholar] [CrossRef]
- Liu, H.; Xu, S.; Zhou, G.; Huang, G.; Huang, S.; Xiong, K. CO2 hydrogenation to methane over Co/KIT-6 catalyst: Effect of reduction temperature. Chem. Eng. J. 2018, 351, 65–73. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, H.; Xing, Y.; Xu, S.; Xie, H.; Xiong, K. CO2 hydrogenation to methane over mesoporous Co/SiO2 catalysts: Effect of structure. J. CO2 Util. 2018, 26, 221–229. [Google Scholar] [CrossRef]
- Gnanakumar, E.S.; Chandran, N.; Kozhevnikov, I.V.; Grau-Atienza, A.; Ramos Fernández, E.V.; Sepulveda-Escribano, A.; Shiju, N.R. Highly efficient nickel-niobia composite catalysts for hydrogenation of CO2 to methane. Chem. Eng. Sci. 2019, 194, 2–9. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Surya Prakash, G.K. Beyond oil and gas: The methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef]
- Li, W.; Lu, P.; Xu, D.; Tao, K. CO2 hydrogenation to methanol over Cu/ZnO catalysts synthesized via a facile solid-phase grinding process using oxalic acid. Korean J. Chem. Eng. 2018, 35, 110–117. [Google Scholar] [CrossRef]
- Cao, F.H.; Liu, D.H.; Hou, Q.S.; Fang, D.Y. Thermodynamic Analysis of CO2 Direct Hydrogenation Reactions. J. Nat. Gas Chem. 2001, 10, 24–33. [Google Scholar]
- Deng, K.; Hu, B.; Lu, Q.; Hong, X. Cu/g-C3N4 modified ZnO/Al2O3 catalyst: Methanol yield improvement of CO2 hydrogenation. Catal. Commun. 2017, 100, 81–84. [Google Scholar] [CrossRef]
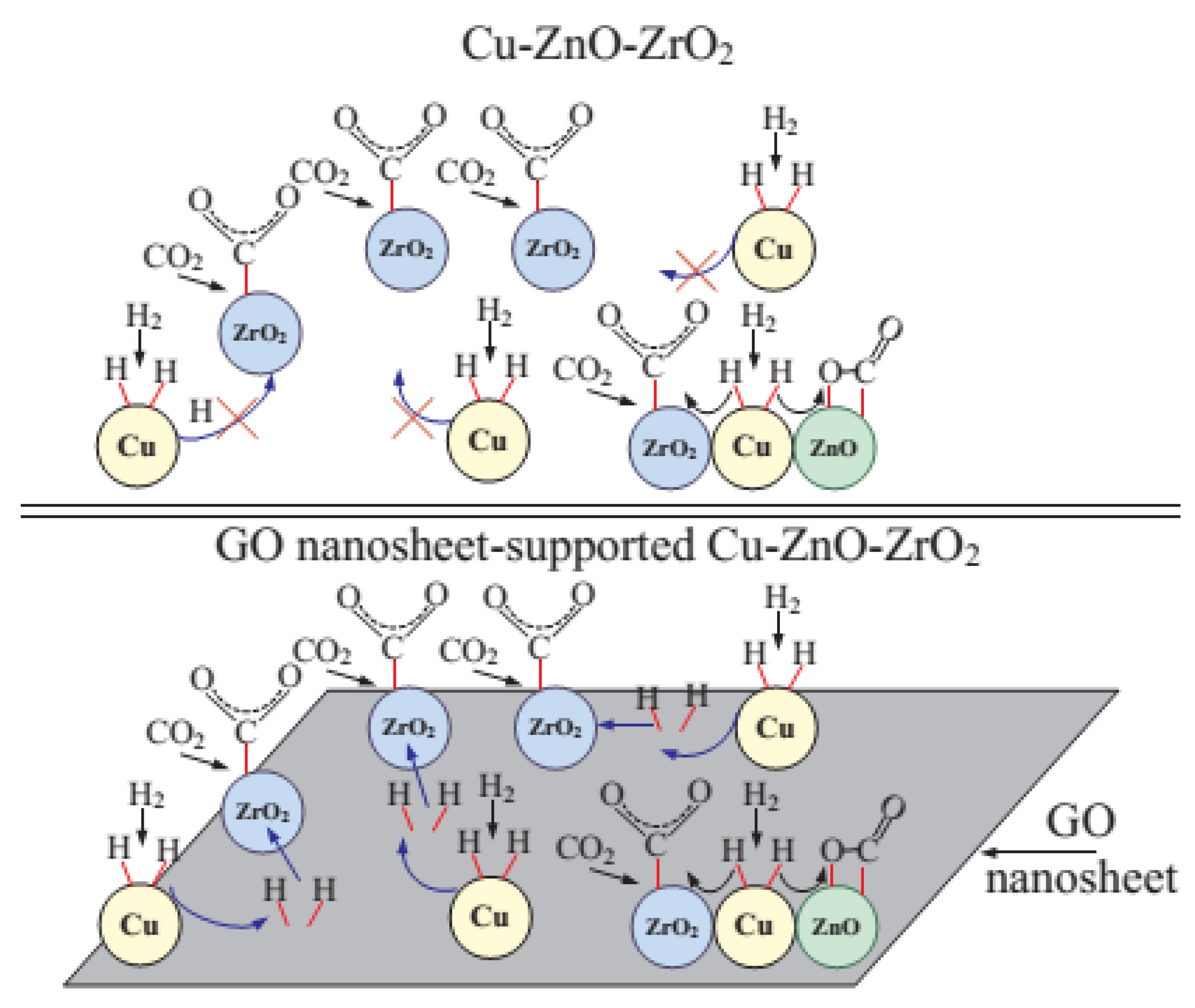
- Witoon, T.; Numpilai, T.; Phongamwong, T.; Donphai, W.; Boonyuen, C.; Warakulwit, C.; Chareonpanich, M.; Limtrakul, J. Enhanced activity, selectivity and stability of a CuO-ZnO-ZrO2 catalyst by adding graphene oxide for CO2 hydrogenation to methanol. Chem. Eng. J. 2018, 334, 1781–1791. [Google Scholar] [CrossRef]
- Wang, G.; Mao, D.; Guo, X.; Yu, J. Enhanced performance of the CuO-ZnO-ZrO2 catalyst for CO2 hydrogenation to methanol by WO3 modification. Appl. Surf. Sci. 2018, 456, 403–409. [Google Scholar] [CrossRef]
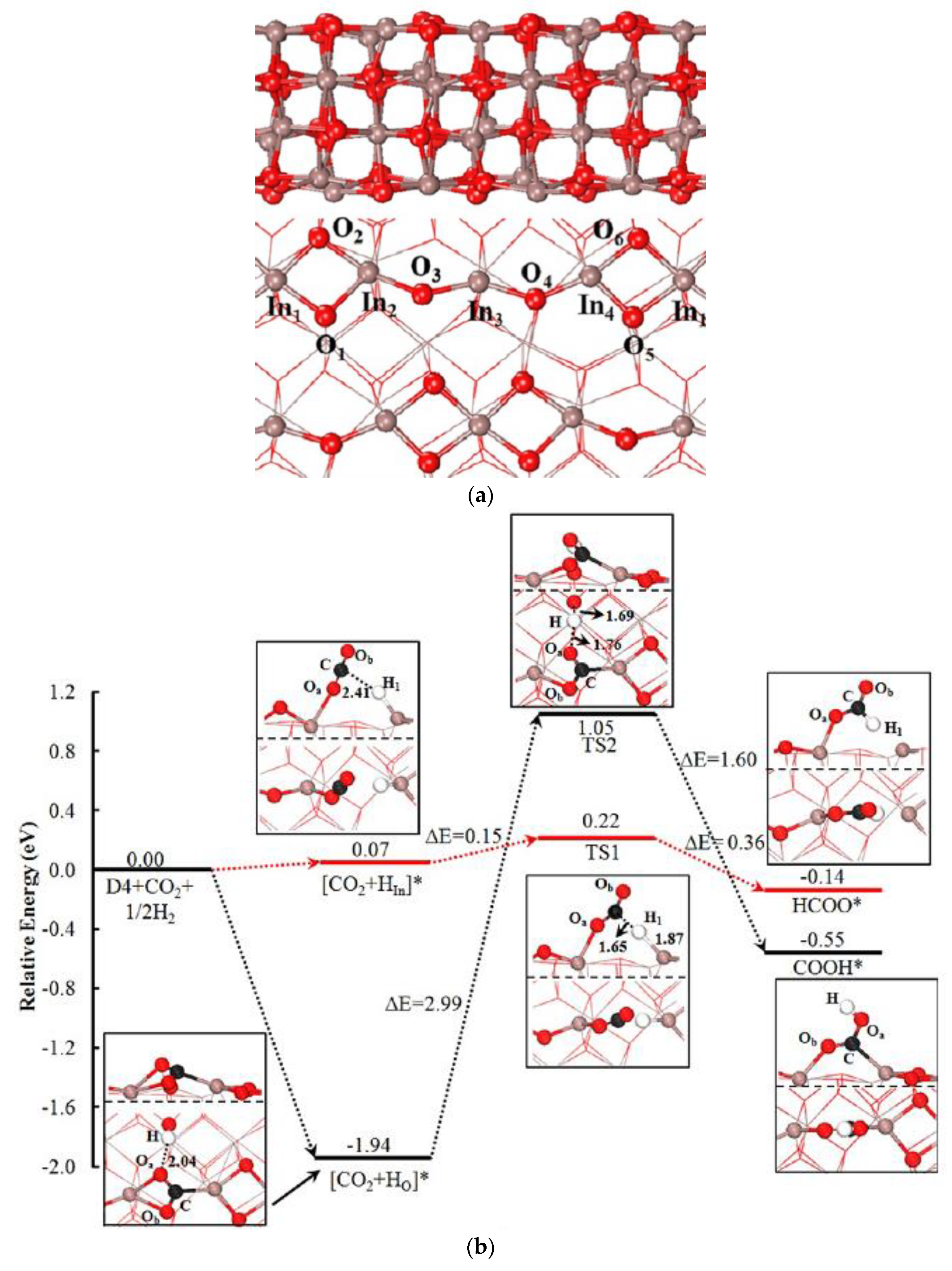
- Ye, J.; Liu, C.; Mei, D.; Ge, Q. Active Oxygen Vacancy Site for Methanol Synthesis from CO2 Hydrogenation on In2O3(110): A DFT Study. ACS Catal. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Sun, K.; Fan, Z.; Ye, J.; Yan, J.; Ge, Q.; Li, Y.; He, W.; Yang, W.; Liu, C. Hydrogenation of CO2 to methanol over In2O3 catalyst. J. CO2 Util. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Martin, O.; Martin, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferre, D.; Perez-Ramirez, J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chem. Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Li, Z.; Tang, C.; Feng, Z.; An, H.; Liu, H.; Liu, T.; Li, C. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 2017, 3, e1701290. [Google Scholar] [CrossRef]
- Jiang, X.; Jiao, Y.; Moran, C.; Nie, X.; Gong, Y.; Guo, X.; Walton, K.S.; Song, C. CO2 hydrogenation to methanol on Pd-Cu bimetallic catalysts with lower metal loadings. Catal. Commun. 2019, 118, 10–14. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X.; Nie, X.; Koizumi, N.; Guo, X.; Song, C. CO2 hydrogenation to methanol on Pd-Cu bimetallic catalysts: H2/CO2 ratio dependence and surface species. Catal. Today 2018, 316, 62–70. [Google Scholar] [CrossRef]
- Bahruji, H.; Esquius, J.R.; Bowker, M.; Hutchings, G.; Armstrong, R.D.; Jones, W. Solvent Free Synthesis of PdZn/TiO2 Catalysts for the Hydrogenation of CO2 to Methanol. Top. Catal. 2018, 61, 144–153. [Google Scholar] [CrossRef]
- Yin, Y.; Hu, B.; Li, X.; Zhou, X.; Hong, X.; Liu, G. Pd@zeolitic imidazolate framework-8 derived PdZn alloy catalysts for efficient hydrogenation of CO2 to methanol. Appl. Catal. B 2018, 234, 143–152. [Google Scholar] [CrossRef]
- Hu, B.; Yin, Y.; Liu, G.; Chen, S.; Hong, X.; Tsang, S.C.E. Hydrogen spillover enabled active Cu sites for methanol synthesis from CO2 hydrogenation over Pd doped CuZn catalysts. J. Catal. 2018, 359, 17–26. [Google Scholar] [CrossRef]
- Studt, F.; Sharafutdinov, I.; Abild-Pedersen, F.; Elkjær, C.F.; Hummelshøj, J.S.; Dahl, S.; Chorkendorff, I.; Nørskov, J.K. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 2014, 6, 320–324. [Google Scholar] [CrossRef]
- Marcos, F.C.F.; Assaf, J.M.; Assaf, E.M. CuFe and CuCo supported on pillared clay as catalysts for CO2 hydrogenation into value-added products in one-step. Mol. Catal. 2018, 458, 297–306. [Google Scholar] [CrossRef]
- Joseph, A.S.; Can, A.; Julia, S.; Wang, T.; Jens, K.N.; Frank, A.P.; Stacey, F.B. Theoretical and Experimental Studies of CoGa Catalysts for the Hydrogenation of CO2 to Methanol. Catal. Lett. 2018, 148, 3583–3591. [Google Scholar]
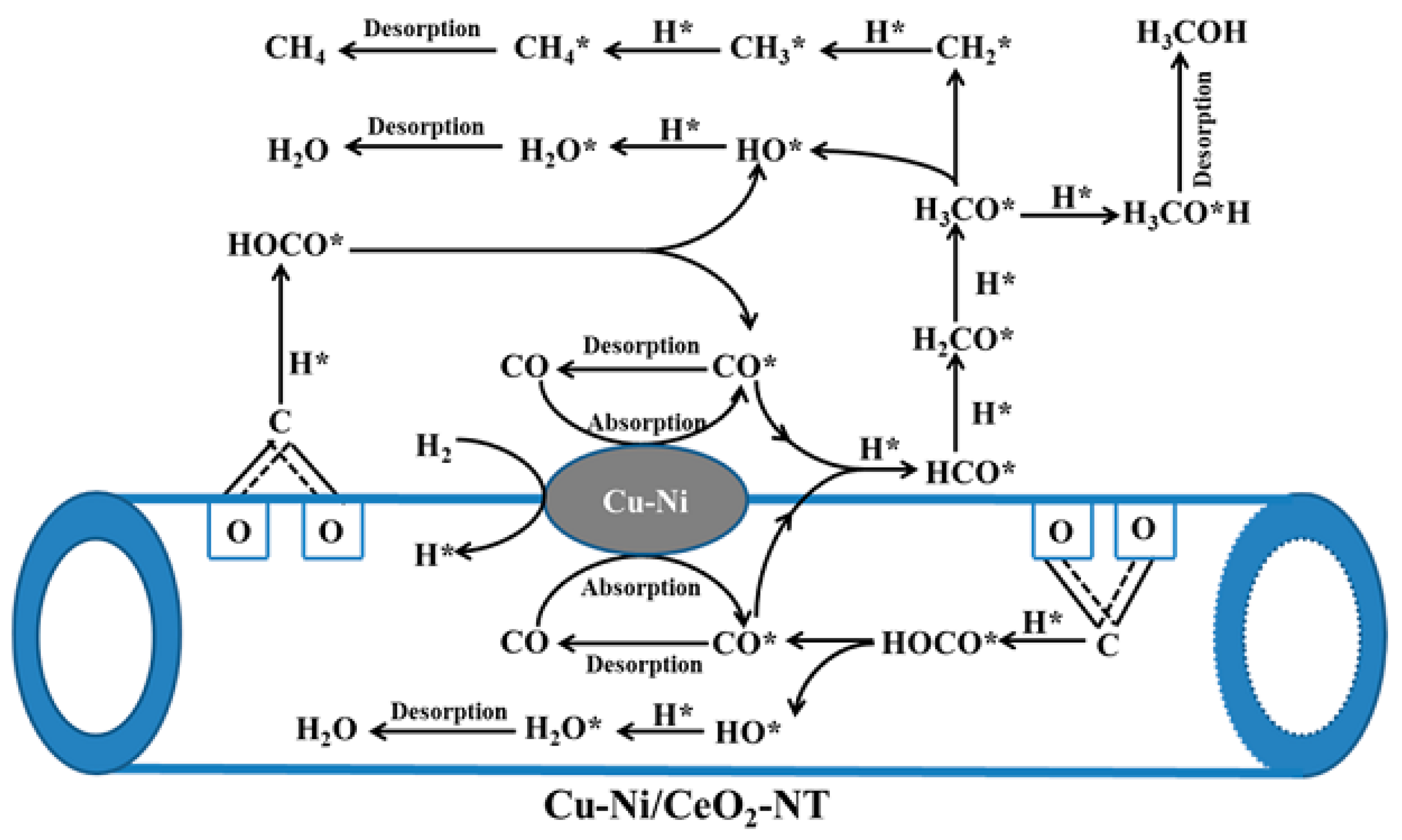
- Tan, Q.; Shi, Z.; Wu, D. CO2 Hydrogenation to Methanol over a Highly Active Cu–Ni/CeO2–Nanotube Catalyst. Ind. Eng. Chem. Res. 2018, 57, 10148–10158. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, H.; Gao, P.; Li, X.; Zhang, J.; Liu, H.; Wang, H.; Wei, W.; Sun, Y. Slurry methanol synthesis from CO2 hydrogenation over micro-spherical SiO2 support Cu/ZnO catalysts. J. CO2 Util. 2018, 26, 642–651. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, G.; Liu, Y.; Lu, Y. Monolithic Ni5Ga3/SiO2/Al2O3/Al-fiber catalyst for CO2 hydrogenation to methanol at ambient pressure. Appl. Catal. A 2018, 562, 234–240. [Google Scholar] [CrossRef]
- Lam, E.; Larmier, K.; Wolf, P.; Tada, S.; Safonova, O.V.; Copéret, C. Isolated Zr Surface Sites on Silica Promote Hydrogenation of CO2 to CH3OH in Supported Cu Catalysts. J. Am. Chem. Soc. 2018, 140, 10530–10535. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Stefancic, N.S.; Likozar, B. Correlation between synthesis pH, structure and Cu/MgO/Al2O3 heterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol. J. CO2 Util. 2018, 28, 189–199. [Google Scholar] [CrossRef]
- Shi, Z.; Tan, Q.; Wu, D. Ternary copper-cerium-zirconium mixed metal oxide catalyst for direct CO2 hydrogenation to methanol. Mater. Chem. Phys. 2018, 219, 263–272. [Google Scholar] [CrossRef]
- Bao, Y.; Huang, C.; Chen, L.; Zhang, Y.D.; Liang, L.; Wen, J.; Fu, M.; Wu, J.; Ye, D. Highly efficient Cu/anatase TiO2 {001}-nanosheets catalysts for methanol synthesis from CO2. J. Energy Chem. 2018, 27, 381–388. [Google Scholar] [CrossRef]
- Koh, M.K.; Khavarian, M.; Chai, S.P.; Mohamed, A.R. The morphological impact of siliceous porous carriers on copper-catalysts for selective direct CO2 hydrogenation to methanol. Int. J. Hydrogen Energy 2018, 43, 9334–9342. [Google Scholar] [CrossRef]
- Tasfy, S.; Zabidi, N.A.M.; Shaharum, M.S.; Subbarao, D. Methanol production via CO2 hydrogenation reaction: Effect of catalyst support. Int. J. Nanotechnol. 2017, 14, 410–421. [Google Scholar] [CrossRef]
- Li, Y.; Na, W.; Wang, H.; Gao, W. Hydrogenation of CO2 to methanol over Au–CuO/SBA-15 catalysts. J. Porous Mater. 2017, 24, 591–599. [Google Scholar] [CrossRef]
- Atakan, A.; Mäkie, P.; Söderlind, F.; Keraudy, J.; Björk, E.M.; Odén, M. Synthesis of a Cu-infiltrated Zr-doped SBA-15 catalyst for CO2 hydrogenation into methanol and dimethyl ether. Phys. Chem. Chem. Phys. 2017, 19, 19139–19149. [Google Scholar] [CrossRef]
- Rui, N.; Wang, Z.; Sun, K.; Ye, J.; Ge, Q.; Liu, C. CO2 hydrogenation to methanol over Pd/In2O3: Effects of Pd and oxygen vacancy. Appl. Catal. B 2017, 218, 488–497. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, S.; Su, X.; Fan, S.; Ma, Q.; Zhao, T. Selective formation of light olefins from CO2 hydrogenation over Fe–Zn–K catalysts. J. CO2 Util. 2015, 12, 95–100. [Google Scholar] [CrossRef]
- You, Z.; Deng, W.; Zhang, Q.; Wang, Y. Hydrogenation of carbon dioxide to light olefins over non-supported iron catalyst. Chin. J. Catal. 2013, 34, 956–963. [Google Scholar] [CrossRef]
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, C.; Yu, Y.; Su, J.; Li, Y.; Somorjai, G.A.; Yang, P. Tandem Catalysis for CO2 Hydrogenation to C2–C4 Hydrocarbons. Nano Lett. 2017, 17, 3798–3802. [Google Scholar] [CrossRef]
- Li, S.; Guo, H.; Luo, C.; Zhang, H.; Xiong, L.; Chen, X.; Ma, L. Effect of Iron Promoter on Structure and Performance of K/Cu–Zn Catalyst for Higher Alcohols Synthesis from CO2 Hydrogenation. Catal. Lett. 2013, 143, 345–355. [Google Scholar] [CrossRef]
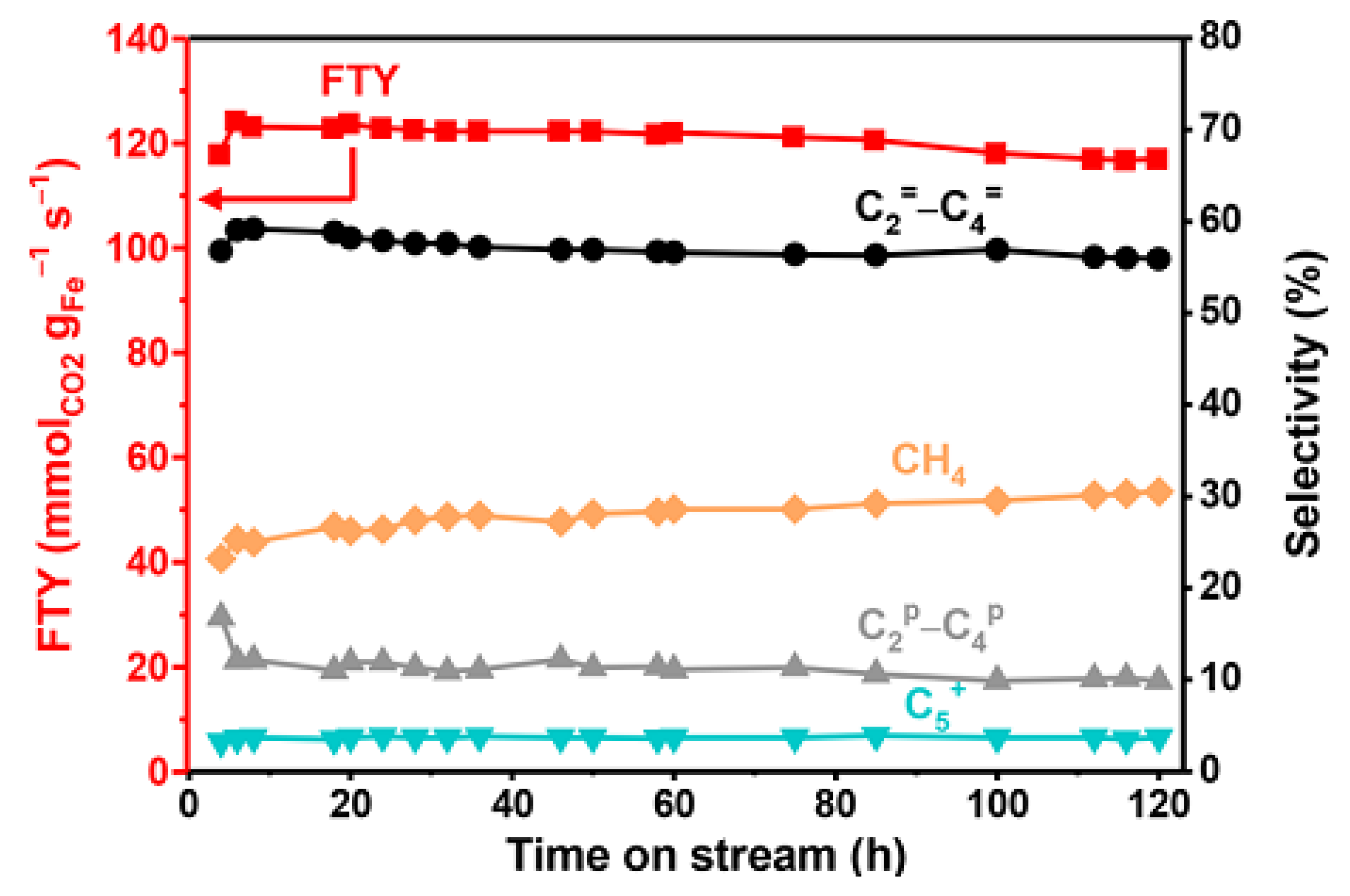
- Wei, J.; Yao, R.; Ge, Q.; Wen, Z.; Ji, X.; Fang, C.; Zhang, J.; Xu, H.; Sun, J. Catalytic Hydrogenation of CO2 to Isoparaffins over Fe-Based Multifunctional Catalysts. ACS Catal. 2018, 8, 9958–9967. [Google Scholar] [CrossRef]
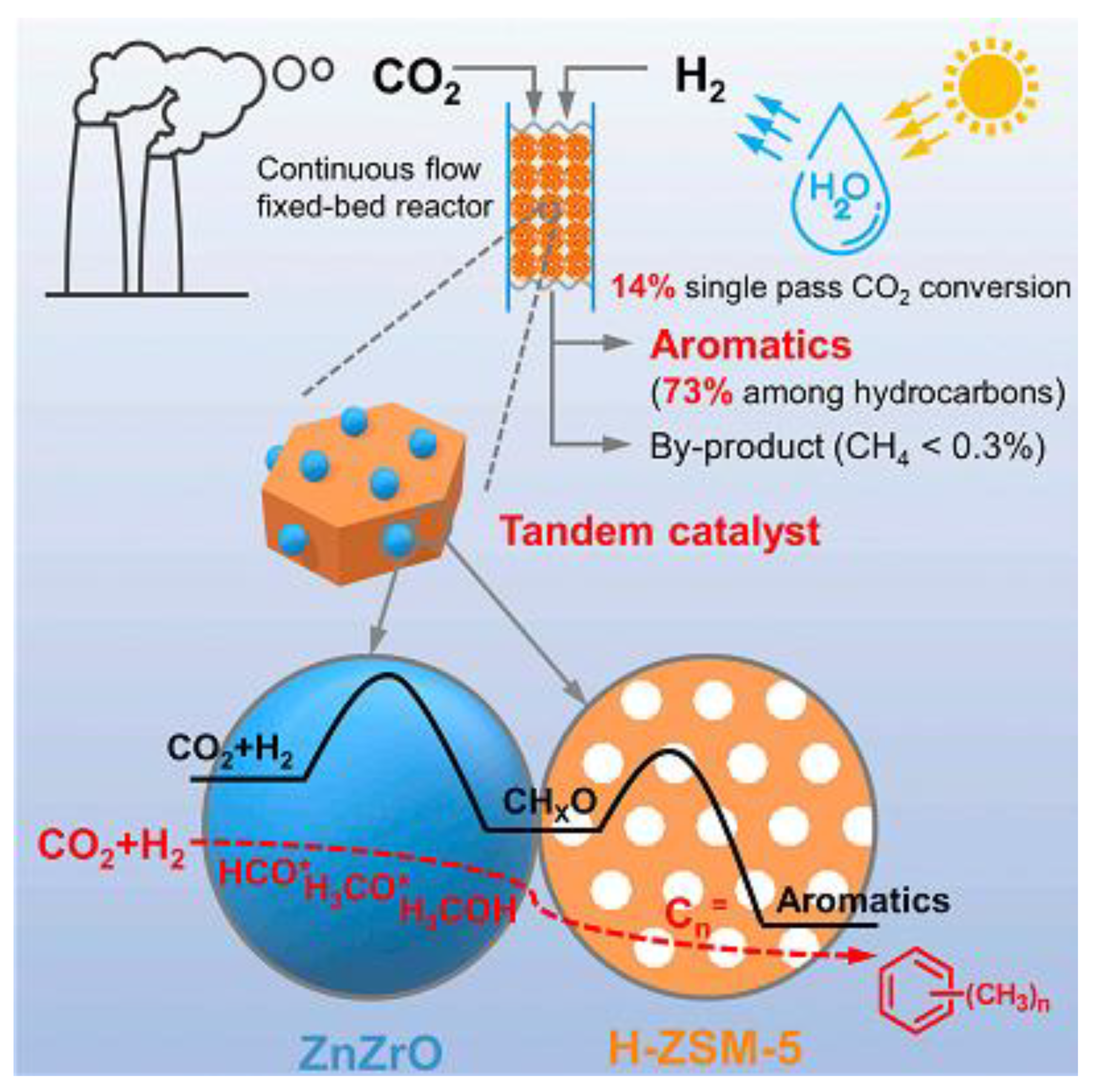
- Li, Z.; Qu, Y.; Wang, J.; Liu, H.; Li, M.; Miao, S.; Li, C. Highly Selective Conversion of Carbon Dioxide to Aromatics over Tandem Catalysts. Joule 2019, 3, 1–14. [Google Scholar] [CrossRef]
- Ni, Y.; Chen, Z.; Fu, Y.; Liu, Y.; Zhu, W.; Liu, Z. Selective conversion of CO2 and H2 into aromatics. Nat. Commun. 2018, 9, 3457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.H.; Liu, Z.M.; Lin, G.D.; Zhang, H.B. Pd/CNT-promoted CuZrO2/HZSM-5 hybrid catalysts for direct synthesis of DME from CO2/H2. Appl. Catal. A 2013, 451, 28–35. [Google Scholar] [CrossRef]
- Wu, T.; Lin, J.; Cheng, Y.; Tian, J.; Wang, S.; Xie, S.; Pei, Y.; Yan, S.; Qiao, M.; Xu, H.; et al. Porous Graphene-Confined Fe–K as Highly Efficient Catalyst for CO2 Direct Hydrogenation to Light Olefins. ACS Appl. Mater. Interfaces 2018, 10, 23439–23443. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhang, Y.; Cao, Y.; Zhang, S.; Wang, K.; Ding, F.; Wu, J. V-modified CuO–ZnO–ZrO2/HZSM-5 catalyst for efficient direct synthesis of DME from CO2 hydrogenation. Catal. Commun. 2014, 55, 49–52. [Google Scholar] [CrossRef]
- Gao, W.; Wang, H.; Wang, Y.; Guo, W.; Jia, M. Dimethyl ether synthesis from CO2 hydrogenation on La-modified CuO-ZnO-Al2O3/HZSM-5 bifunctional catalysts. J. Rare Earths 2013, 31, 470–476. [Google Scholar] [CrossRef]
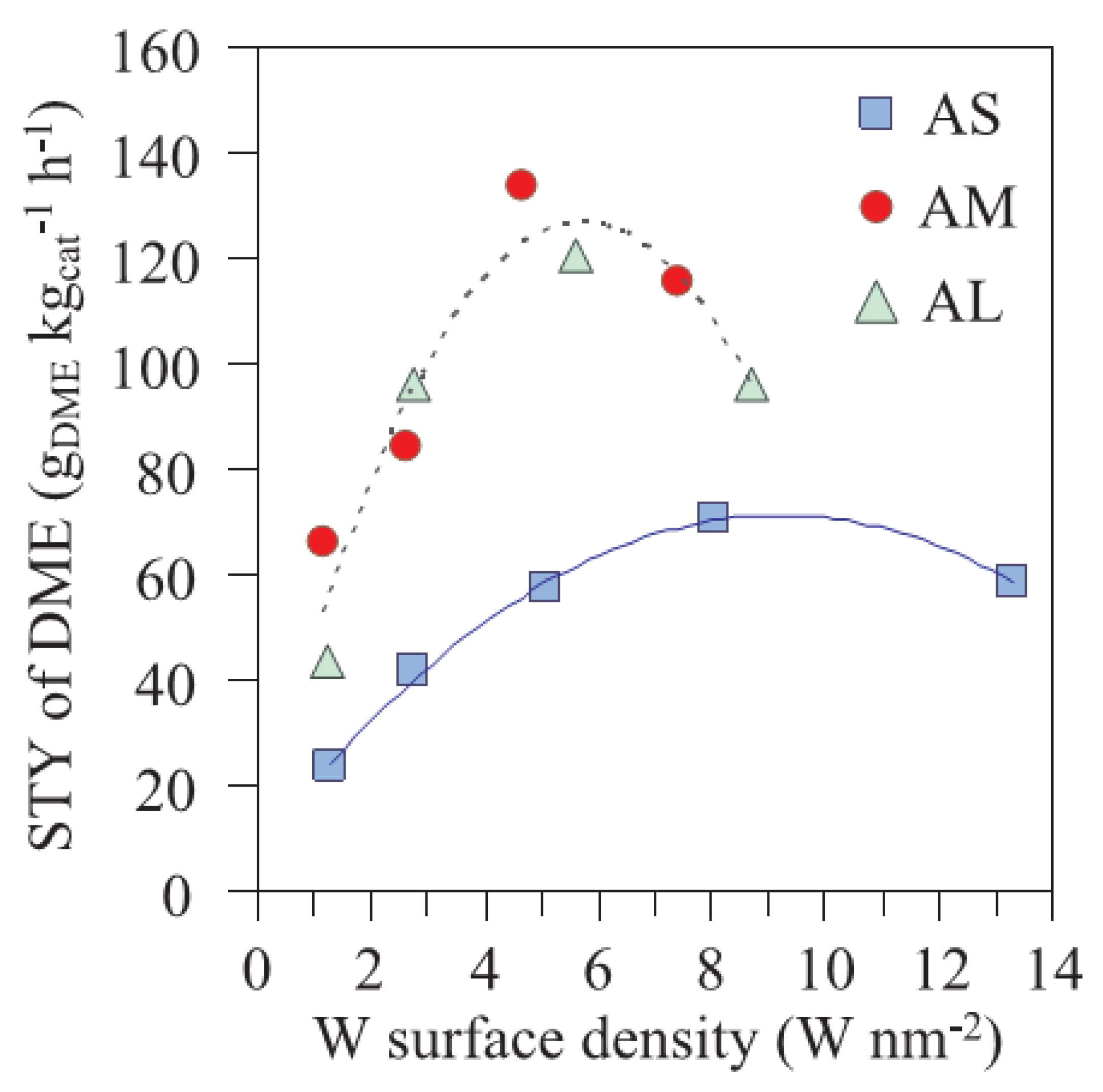
- Suwannapichat, Y.; Numpilai, T.; Chanlek, N.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J.; Witoon, T. Direct synthesis of dimethyl ether from CO2 hydrogenation over novel hybrid catalysts containing a Cu-ZnO-ZrO2 catalyst admixed with WOx/Al2O3 catalysts: Effects of pore size of Al2O3 support and W loading content. Energy Convers. Manag. 2018, 159, 20–29. [Google Scholar] [CrossRef]
- Michailos, S.; McCord, S.; Sick, V.; Stokes, G.; Styring, P. Dimethyl ether synthesis via captured CO2 hydrogenation within the power to liquids concept: A techno-economic assessment. Energy Convers. Manag. 2019, 184, 262–276. [Google Scholar] [CrossRef]
- Quadrelli, E.A.; Centi, G.; Duplan, J.L.; Perathoner, S. Carbon Dioxide Recycling: Emerging Large-Scale Technologies with Industrial Potential. ChemSusChem 2011, 4, 1194–1215. [Google Scholar] [CrossRef] [PubMed]
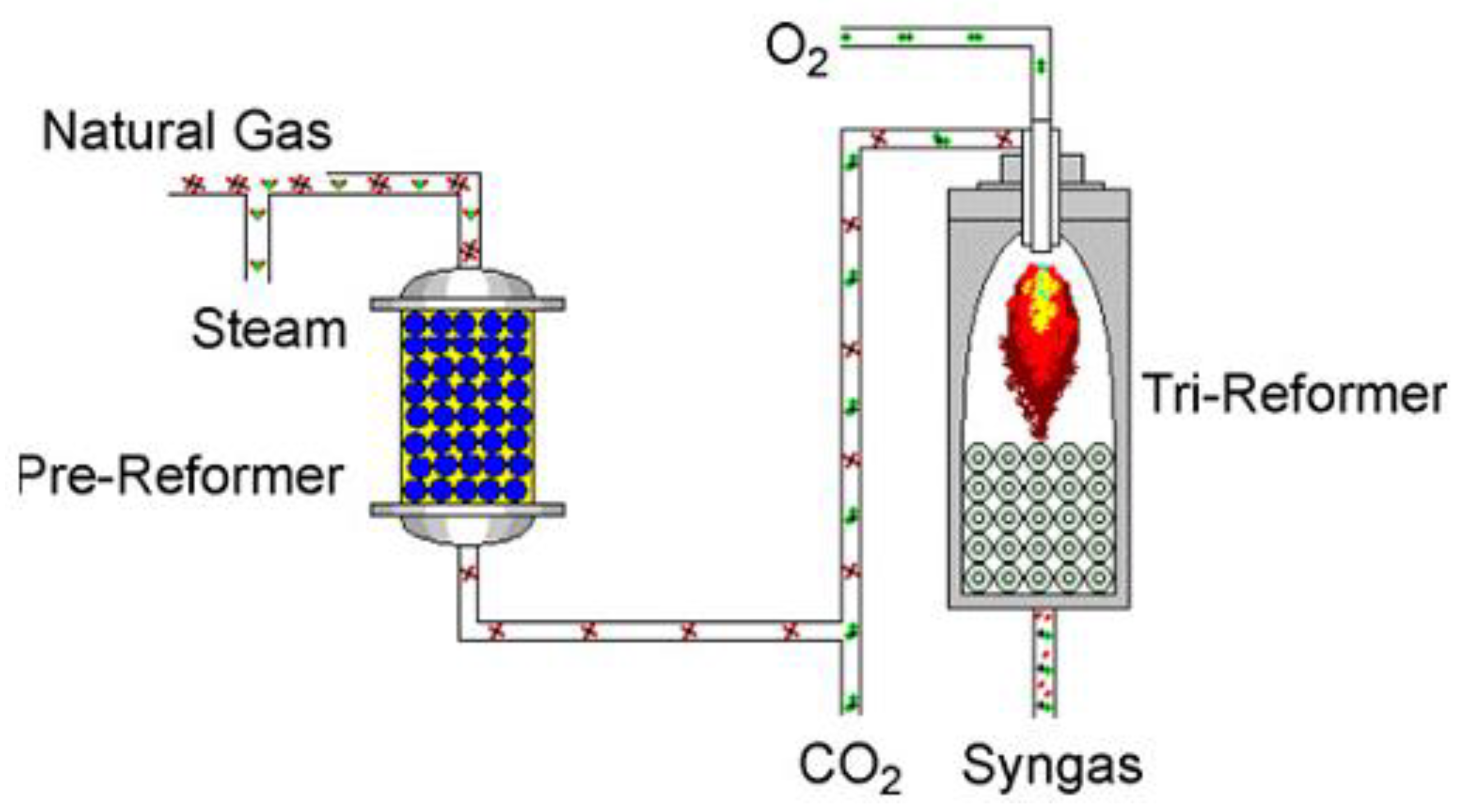
- Cho, W.; Song, T.; Mitsos, A.; McKinnon, J.T.; Ko, G.H.; Tolsma, J.E.; Denholm, D.; Park, T. Optimal design and operation of a natural gas tri-reforming reactor for DME synthesis. Catal. Today 2009, 139, 261–267. [Google Scholar] [CrossRef]
- Hirano, M.; Tatsumi, M.; Yasutake, T.; Kuroda, K. Dimethyl ether synthesis via reforming of steam/carbon dioxide and methane. J. Jpn. Pet. Inst. 2005, 48, 197–203. [Google Scholar] [CrossRef]
- Hirano, M.; Yasutake, T.; Kuroda, K. Dimethyl ether synthesis from carbon dioxide by catalytic hydrogenation (Part 3) direct synthesis using hybrid by recycling process. J. Jpn. Pet. Inst. 2007, 50, 34–43. [Google Scholar] [CrossRef]
- Bonura, G.; Migliori, M.; Frusteri, L.; Cannilla, C.; Catizzone, E.; Giordano, G.; Frusteri, F. Acidity control of zeolite functionality on activity and stability of hybrid catalysts during DME production via CO2 hydrogenation. J. CO2 Util. 2018, 24, 398–406. [Google Scholar] [CrossRef]
- Catizzone, E.; Migliori, M.; Purita, A.; Giordano, G. Ferrierite vs. γ-Al2O3: The superiority of zeolites in terms of water-resistance in vapour-phase dehydration of methanol to dimethyl ether. J. Energy Chem. 2019, 30, 162–169. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 Recycling to Dimethyl Ether: State-of-the-Art and Perspectives. Molecules 2018, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- De Falco, M.; Capocelli, M.; Centi, G. Dimethyl ether production from CO2 rich feedstocks in a one-step process: Thermodynamic evaluation and reactor simulation. Chem. Eng. J. 2016, 294, 400–409. [Google Scholar] [CrossRef]
- Fang, J.; Jin, X.F.; Huang, K. Life cycle analysis of a combined CO2 capture and conversion membrane reactor. J. Membr. Sci. 2018, 549, 142–150. [Google Scholar] [CrossRef]
- Sofia, D.; Giuliano, A.; Poletto, M.; Barletta, D. Techno-economic analysis of power and hydrogen co-production by an IGCC plant with CO2 capture based on membrane technology. Comput. Aided Process Eng. 2015, 37, 1373–1378. [Google Scholar]
- Li, Z.; Wang, J.; Qu, Y.; Liu, H.; Tang, C.; Miao, S.; Feng, Z.; An, H.; Li, C. Highly Selective Conversion of Carbon Dioxide to Lower Olefins. ACS Catal. 2017, 7, 8544–8548. [Google Scholar] [CrossRef]
- Da Silva, R.J.; Pimentel, A.F.; Monteiro, R.S.; Mota, C.J.A. Synthesis of methanol and dimethyl ether from the CO2 hydrogenation over Cu·ZnO supported on Al2O3 and Nb2O5. J. CO2 Util. 2016, 15, 83–88. [Google Scholar] [CrossRef]
- Dang, S.; Gao, P.; Liu, Z.; Chen, X.; Yang, C.; Wang, H.; Zhong, L.; Li, S.; Sun, Y. Role of zirconium in direct CO2 hydrogenation to lower olefins on oxide/zeolite bifunctional catalysts. J. Catal. 2018, 364, 382–393. [Google Scholar] [CrossRef]
- Wang, P.; Zha, F.; Yao, L.; Chang, Y. Synthesis of light olefins from CO2 hydrogenation over (CuO-ZnO)-kaolin/SAPO-34 molecular sieves. Appl. Clay Sci. 2018, 163, 249–256. [Google Scholar] [CrossRef]
- Guo, L.; Sun, J.; Ji, X.; Wei, J.; Wen, Z.; Yao, R.; Xu, H.; Ge, Q. Directly converting carbon dioxide to linear α-olefins on bio-promoted catalysts. Commun. Chem. 2018, 1, 11. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Spadaro, L.; Cannilla, C.; Arena, F.; Frusteri, F. Hybrid Cu–ZnO–ZrO2/H-ZSM5 system for the direct synthesis of DME by CO2 hydrogenation. Appl. Catal. B 2013, 140–141, 16–24. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Mezzapica, A.; Spadaro, L.; Arena, F.; Frusteri, F. Catalytic behaviour of a bifunctional system for the one step synthesis of DME by CO2 hydrogenation. Catal. Today 2014, 228, 51–57. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhang, S.; Wang, K.; Wu, J. CO2 hydrogenation to dimethyl ether over CuO–ZnO–Al2O3/HZSM-5 prepared by combustion route. RSC Adv. 2014, 4, 16391–16396. [Google Scholar] [CrossRef]
- Zha, F.; Tian, H.; Yan, J.; Chang, Y. Multi-walled carbon nanotubes as catalyst promoter for dimethyl ether synthesis from CO2 hydrogenation. Appl. Surf. Sci. 2013, 285, 945–951. [Google Scholar] [CrossRef]
- Frusteri, F.; Bonura, G.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation. Appl. Catal. B 2015, 176–177, 522–531. [Google Scholar] [CrossRef]
- Liu, R.; Tian, H.; Yang, A.; Zha, F.; Ding, J.; Chang, Y. Preparation of HZSM-5 membrane packed CuO–ZnO–Al2O3 nanoparticles for catalysing carbon dioxide hydrogenation to dimethyl ether. Appl. Surf. Sci. 2015, 345, 1–9. [Google Scholar] [CrossRef]
- Dai, C.; Zhang, A.; Liu, M.; Li, J.; Song, F.; Song, C.; Guo, X. Facile one-step synthesis of hierarchical porous carbon monoliths as superior supports of Fe-based catalysts for CO2 hydrogenation. RSC Adv. 2016, 6, 10831–10836. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 16170. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, A.; Jiang, X.; Liu, M.; Zhu, J.; Song, C.; Guo, X. Direct Transformation of Carbon Dioxide to Value-Added Hydrocarbons by Physical Mixtures of Fe5C2 and K-Modified Al2O3. Ind. Eng. Chem. Res. 2018, 57, 9120–9126. [Google Scholar] [CrossRef]
- Zhang, J.; Su, X.; Wang, X.; Ma, Q.; Fan, S.; Zhao, T.S. Promotion effects of Ce added Fe–Zr–K on CO2 hydrogenation to light olefins. React. Kinet. Mech. Catal. 2018, 124, 575–585. [Google Scholar] [CrossRef]
- Ramirez, A.; Gevers, L.; Bavykina, A.; Ould-Chikh, S.; Gascon, J. Metal Organic Framework-Derived Iron Catalysts for the Direct Hydrogenation of CO2 to Short Chain Olefins. ACS Catal. 2018, 8, 9174–9182. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, H.; Gao, P.; Chen, X.; Liu, H.; Zhong, L.; Wang, H.; Wei, W.; Sun, Y. Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons. Chin. J. Catal. 2018, 39, 1294–1302. [Google Scholar] [CrossRef]
- Bogaerts, A.; Neyts, E.; Gijbels, R.; van der Mullen, J. Gas discharge plasmas and their applications. Spectrochim. Acta Part B 2002, 57, 609–658. [Google Scholar] [CrossRef]
- Bogaerts, A.; Neyts, E.C. Plasma Technology: An Emerging Technology for Energy Storage. ACS Energy Lett. 2018, 3, 1013–1027. [Google Scholar] [CrossRef]
- Paulussen, S.; Verheyde, B.; Tu, X.; De Bie, C.; Martens, T.; Petrovic, D.; Bogaerts, A.; Sels, B. Conversion of carbon dioxide to value-added chemicals in atmospheric pressure dielectric barrier discharges. Plasma Sources Sci. Technol. 2010, 19, 034015. [Google Scholar] [CrossRef]
- Silva, T.; Britun, N.; Godfroid, T.; Snyders, R. Optical characterization of a microwave pulsed discharge used for dissociation of CO2. Plasma Sources Sci. Technol. 2014, 23, 25009. [Google Scholar] [CrossRef]
- Ozkan, A.; Dufour, T.; Silva, T.; Britun, N.; Snyders, R.; Reniers, F.; Bogaerts, A. DBD in burst mode: Solution for more efficient CO2 conversion? Plasma Sources Sci. Technol. 2016, 25, 055005. [Google Scholar] [CrossRef]
- Wang, W.; Berthelot, A.; Kolev, S.; Tu, X.; Bogaerts, A. CO2 conversion in a gliding arc plasma: 1D cylindrical discharge model. Plasma Sources Sci. Technol. 2016, 25, 065012. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Bai, M.; Tao, X.; Shang, S.; Dai, X.; Yin, Y. CO2 reforming of CH4 by atmospheric pressure glow discharge plasma: A high conversion ability. Int. J. Hydrogen Energy 2009, 34, 308–313. [Google Scholar] [CrossRef]
- Liu, J.L.; Park, H.W.; Chung, W.J.; Park, D.W. High-Efficient Conversion of CO2 in AC-Pulsed Tornado Gliding Arc Plasma. Plasma Chem. Plasma Process. 2016, 36, 437–449. [Google Scholar] [CrossRef]
- Kolb, T.; Voigt, J.H.; Gericke, K.H. Conversion of Methane and Carbon Dioxide in a DBD Reactor: Influence of Oxygen. Plasma Chem. Plasma Process. 2013, 33, 631–646. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Wu, C.; Guo, H.; Tu, X. One-Step Reforming of CO2 and CH4 into High-Value Liquid Chemicals and Fuels at Room Temperature by Plasma-Driven Catalysis. Angew. Chem. Int. Ed. 2017, 56, 13679–13683. [Google Scholar] [CrossRef]
- Zhang, K.; Eliasson, B.; Kogelschatz, U. Direct Conversion of Greenhouse Gases to Synthesis Gas and C4 Hydrocarbons over Zeolite HY Promoted by a Dielectric-Barrier Discharge. Ind. Eng. Chem. Res. 2002, 41, 1462–1468. [Google Scholar] [CrossRef]
- Eliasson, B.; Kogelschatz, U.; Xue, B.; Zhou, L.M. Hydrogenation of Carbon Dioxide to Methanol with a Discharge-Activated Catalyst. Ind. Eng. Chem. Res. 1998, 37, 3350–3357. [Google Scholar] [CrossRef]
- Zeng, Y.; Tu, X. Plasma-Catalytic CO2 Hydrogenation at Low Temperatures. IEEE Trans. Plasma Sci. 2016, 44, 405–411. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Guo, H.; Tu, X. Atmospheric Pressure and Room Temperature Synthesis of Methanol through Plasma-Catalytic Hydrogenation of CO2. ACS Catal. 2018, 8, 90–100. [Google Scholar] [CrossRef]
- De Bie, C.; van Dijk, J.; Bogaerts, A. CO2 Hydrogenation in a Dielectric Barrier Discharge Plasma Revealed. J. Phys. Chem. C 2016, 120, 25210–25224. [Google Scholar] [CrossRef]































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
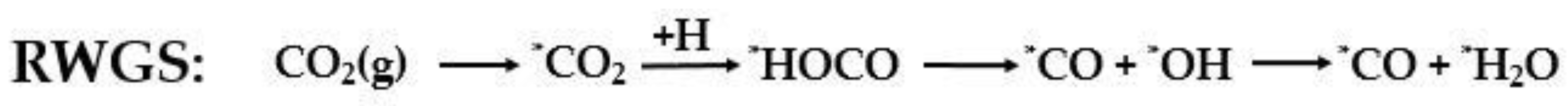{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | H2:CO2 | GHSV | Temperature (°C) | Pressure (MPa) | CO2 Conversion (%) | CO Selectivity (%) |
|---|---|---|---|---|---|---|
| Mo2C [20] | 3 | 36,000 a | 300 | 0.1 | 8.7 | 93.9 |
| Co-Mo2C [20] | 3 | 36,000 a | 300 | 0.1 | 9.5 | ~99.0 |
| Pt-TiO2 [21] | 1 | 119.7 b | 300 | 0.1 | 4.5 | 99.1 |
| Pt-SiO2 [21] | 1 | 24.7 b | 300 | 0.1 | 3.3 | 100 |
| Rh-SrTiO3 [23] | 1 | 12,000 a | 300 | N/A | 7.9 | 95.4 |
| Co-MCF-17 [25] | 3 | 60,000 b | 200-300 | 0.5 | ~5.0 | ~90.0 |
| Pt-Co-MCF-17 [25] | 3 | 60,000 b | 200-300 | 0.5 | ~5.0 | ~99.0 |
| Fe-Mo-Al2O3 [26] | 1 | 30,000 a | 600 | 1 | ~45.0 | ~100.0 |
| Mo-Al2O3 [27] | 1 | 30,000 a | 600 | 0.1 | 16 | N/A |
| Ni-Mo-Al2O3 [27] | 1 | 30,000 a | 600 | 0.1 | 34 | N/A |
| La-Fe-Ni [28] | 2 | 24,000 a | 350 | N/A | 16.3 | 96.6 |
| Catalyst | H2:CO2 | GHSV | Temperature (°C) | CO2 Conversion (%) | CH4 Selectivity (%) |
|---|---|---|---|---|---|
| Ni-La/Na-BETA [29] | 4 | 10,000 b | 350 | 65 | 100 |
| Co/meso-SiO2 [30] | 4.6 | 60,000 a | 280 | 40 | 94.1 |
| Co/KIT-6 [30] | 4.6 | 22,000 a | 280 | 48.9 | 100 |
| Ru/BF4/SiO2 [30] | 4 | 2400 b | 250 | 70.5 | N/A |
| Re-Ni(111) [32] | 4 | N/A | 250 | N/A | 100 |
| Ni/Al2O3-ZrO2 [34] | 4 | 40,000 a | 400 | ~70.0 | N/A |
| Ni-SiO2/GO [35] | 4 | 500 b | 470 | 54.3 | 88 |
| Ni-ZrO2 [36] | 4 | 75 a | 350 | ~40.0 | ~95.0 |
| Ni-Ce/USY [37] | 4 | N/A | 350 | 65 | 95 |
| Co/KIT-6 [38] | 4 | 60,000 a | 340 | 40 | 86.7 |
| Co/SiO2 [39] | 4 | 60,000 a | 360 | 44.3 | 86.5 |
| Ni-Nb2O5 [40] | 4 | 750 b | 325 | 81 | ~99.0 |
| Catalyst | H2:CO2 | GHSV | Temperature (°C) | Pressure (MPa) | CO2 Conversion (%) | CH3OH Selectivity (%) |
|---|---|---|---|---|---|---|
| Cu-ZnO [42] | 2.9 | 2160 a | 250 | 3 | 29.2 | 83.6 |
| Cu/g-C3N4-Zn/Al2O3 [44] | 3 | 6800 a | 250 | 1.2 | ~7.0 | ~55.0 |
| CuO-ZnO-ZrO2-GO [45] | 3 | 15,600 a | 240 | 2 | N/A | 75.8 |
| W-Cu-Zn-Zr [46] | 2.7 | 2400 a | 240 | 3 | 19.7 | 49.3 |
| In2O3 [48] | 3 | 15,000 a | 270 | 4 | 1.1 | 54.9 |
| In2O3 [48] | 3 | 15,000 a | 330 | 4 | 7.1 | 3 |
| In2O3 [49] | 4 | 16,000–48,000 b | 300 | 5 | N/A | 100 |
| ZnO-ZrO2 [50] | 3-4 | 24,000 a | 315-320 | 5 | >10 | 86–91 |
| Pd-Zn-TiO2 [53] | 3 | 916 b | 250 | 2 | 10.3 | 61 |
| Pd-Zn-ZIF-8 [54] | 3 | 21,600 a | 270 | 4.5 | ~22.0 | ~50.0 |
| Pd-Cu-Zn [55] | 3 | 10,800 a | 270 | 4.5 | ~8.0 | ~65.0 |
| Co5Ga3 [58] | 3 | N/A | 250 | 3 | 1 | 63 |
| CuNi2/CeO2-NT [59] | 3 | 6000 b | 260 | 3 | 17.8 | 78.8 |
| Cu-Zn-SiO2 [60] | 3 | 2000 a | 220 | 3 | 14.1 | 57.3 |
| Ni5Ga3/SiO2/Al2O3/Al [61] | 3 | 3000 a | 210 | 0.1 | ~1.0 | 86.7 |
| Cu-Zr-SiO2 [62] | 3 | N/A | 230 | 5 | N/A | 77 |
| Cu/Mg/Al [63] | 2.8 | 2000 b | 200 | 2 | 3.6 | 31 |
| Cu-Ce-Zr [64] | 3 | 7500 a | 250 | 3 | 14.3 | 53.8 |
| Cu-TiO2 [65] | 3 | 3600 a | 260 | 3 | N/A | 64.7 |
| Cu-Zn-Mn-KIT-6 [66] | 3 | 120,000 a | 180 | 4 | 8.2 | >99.0 |
| Cu-SBA-15 [67] | 3 | N/A | 210 | 2.2 | 13.9 | 91.3 |
| Au-CuO/SBA-15 [68] | 3 | 3600 b | 250 | 3 | 24.2 | 13.5 |
| Cu-Zr-SBA-15 [69] | 3 | N/A | 250 | 3.3 | 15 | N/A |
| Pd/In2O3 [70] | 4 | >21,000 a | 300 | 5 | >20.0 | >70.0 |
| Catalyst | H2:CO2 | GHSV | Temperature (°C) | Pressure (MPa) | CO2 Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|---|
| Fe-Zn-K [71] | 3 | 1000 b | 320 | 0.5 | 51.03 | olefins (53.58) |
| K-Fe [72] | 3 | 1200 a | 340 | 2 | 38 | light olefins (78) |
| In/HZSM-5 [73] | 3 | 9000 a | 340 | 3 | 13.1 | liquid fuels (78.6) |
| Ce-Pt@mSi-Co [74] | 3 | N/A | 250 | N/A | ~3.0 | C2–C4 (60) |
| K/Cu-Zn-Fe [75] | 3 | 5000 b | 300 | 6 | 42.3 | alcohol (56.43) |
| Na-Fe/HMCM-22 [76] | 2 | 4000 a | 320 | 3 | 26 | isoparaffins (74) |
| ZnZrO-HZSM-5 [77] | N/A | 1200 a | 320 | 4 | 14 | aromatic (73) |
| ZnAlOx-HZSM-5 [78] | 3 | 2000 a | 320 | 3 | 9.1 | aromatics (74) |
| Cu-Zr-Pd/HZSM-5 [79] | 3 | 25,000 a | 250 | 5 | 18.9 | DME (51.8) |
| Fe-K/HSG [80] | 3 | 26,000 a | 340 | 20 | N/A | olefins (59) |
| V-Cu-Zn-Zr/HZSM-5 [81] | 3 | 4200 b | 270 | 3 | 32.5 | DME (58.8) |
| Cu-Zn-Al-La/HZSM-5 [82] | 3 | 3000 b | 250 | 3 | 43.8 | DME (71.2) |
| ZnO-ZrO2-SAPO-34 [95] | N/A | 3600 a | 380 | 2 | 12.6 | olefins (80–90) |
| Cu-ZnO-Al2O3 [96] | 3 | 10 b | 270 | 5 | 9 | DME (31) |
| In-Zr/SAPO-34 [97] | 3 | 9000 a | 380 | 3 | 26.2 | C2+ (36.1) |
| Cu-Zn-kaolin-SAPO-34 [98] | 3 | 1800 a | 400 | 3 | 50.4 | C2–C4 (65.3) |
| Fe/C-Bio [99] | 3 | N/A | 320 | 1 | 31 | C4–18 alkenes (50.3) |
| Cu-Zn-Zr/HZSM-5 [100] | 3 | 10,000 a | 220 | 3 | 9.6 | DME (46.6) |
| Cu-Zn-Zr/HZSM-5 [101] | 3 | 9000 a | 240 | 3 | ~30 | DME (~35) |
| Cu-Zn-Al/HZSM-5 [102] | 3 | 4200 b | 270 | 3 | 30.6 | DME (49.02) |
| Cu-Zn-Al/HZSM-5 [103] | 3 | 1800 a | 262 | 3 | 46.2 | DME (45.2) |
| Cu-Zn-Zr-zeolite [104] | 3 | 10,000 b | 240 | 3 | 24 | DME (38.5) |
| Cu-Zn-Al/HZSM-5 [105] | 3 | 1800 a | 270 | 3 | 48.3 | DME (48.5) |
| Fe-K/HPCMs-1 [106] | 3 | 3600 a | 400 | 3 | 33.4 | olefins (47.6) |
| Na-Fe/HZSM-5 [107] | 3 | 4000 a | 320 | 3 | 22 | C5–C11 (78) |
| Fe5C2-/a-Al2O3 [108] | 3 | 3600 a | 400 | 3 | 31.5 | C2+ (69.2) |
| Fe-Zr-Ce-K [109] | 3 | 1000 b | 320 | 2 | 57.34 | C2–C4 (55.67) |
| Fe/C+K [110] | 3 | 24,000 a | 320 | 30 | 24 | C2–C6 (36) |
| Co-Cu/TiO2 [111] | 3 | 3000 a | 250 | 3 | 18.4 | C5+ (42.1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Yi, Y.; Wang, L.; Guo, H.; Bogaerts, A. Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2019, 9, 275. https://doi.org/10.3390/catal9030275
Liu M, Yi Y, Wang L, Guo H, Bogaerts A. Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis. Catalysts. 2019; 9(3):275. https://doi.org/10.3390/catal9030275
Chicago/Turabian StyleLiu, Miao, Yanhui Yi, Li Wang, Hongchen Guo, and Annemie Bogaerts. 2019. "Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis" Catalysts 9, no. 3: 275. https://doi.org/10.3390/catal9030275