Gold(I) Complexes with Ferrocenylphosphino Sulfonate Ligands: Synthesis and Application in the Catalytic Addition of Carboxylic Acids to Internal Alkynes in Water

 ,
,  ,
,
Abstract
:1. Introduction
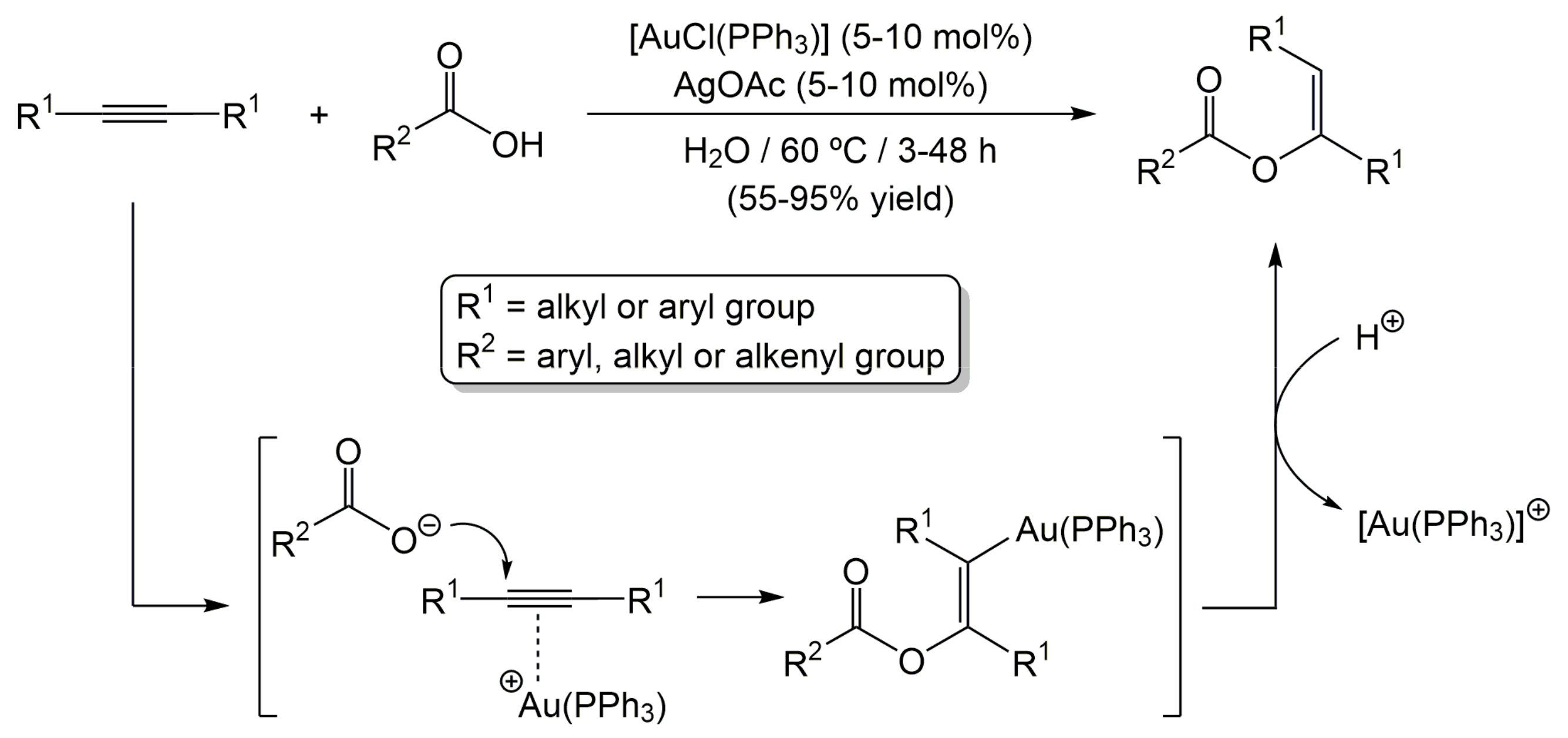
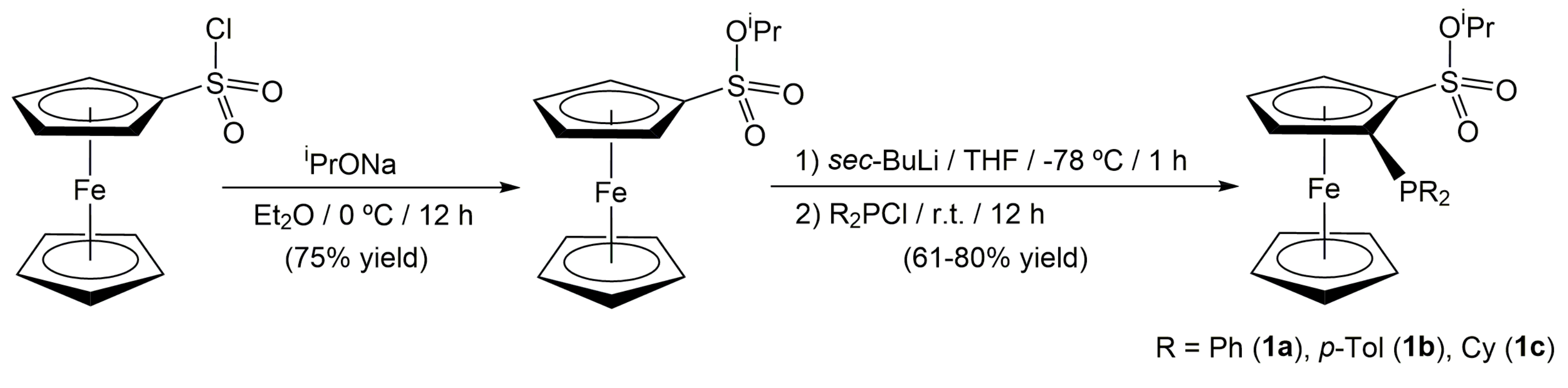
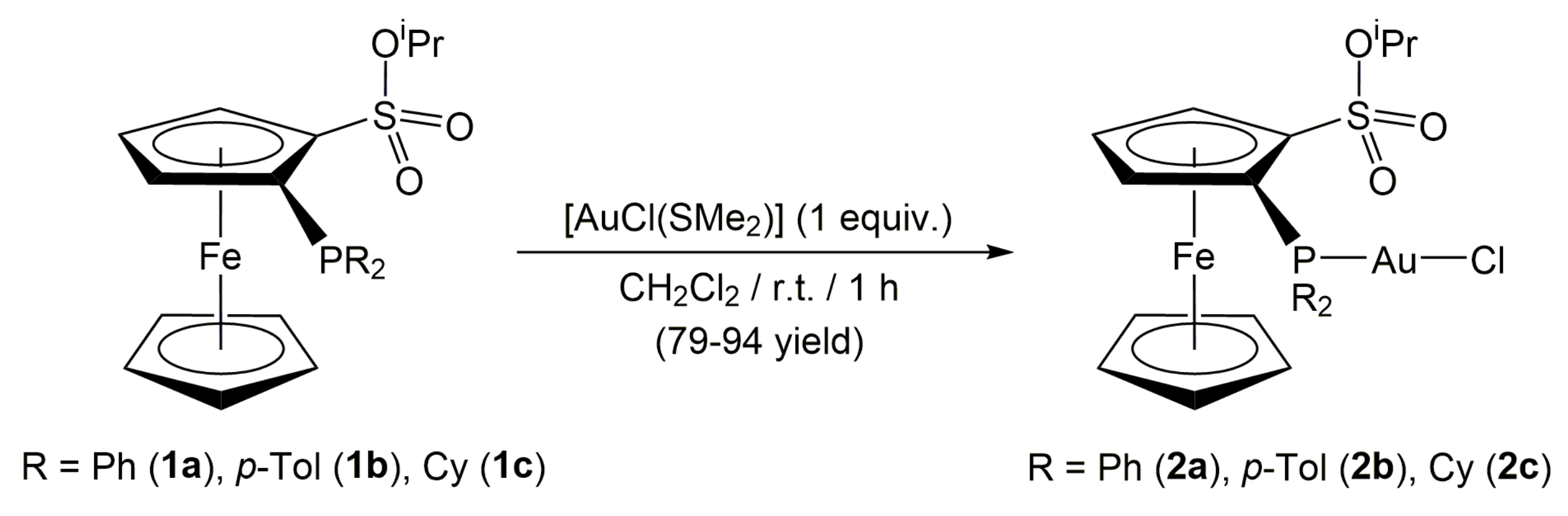
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for the Preparation of Complexes [AuCl{(η5-C5H3PR2(SO3iPr))Fe(η5-C5H5)}] (R = Ph (2a), p-Tol (2b), Cy (2c))
3.1.1. [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a)
3.1.2. [AuCl{(η5-C5H3P(p-Tol)2(SO3iPr))Fe(η5-C5H5)}] (2b)
3.1.3. [AuCl{(η5-C5H3PCy2(SO3iPr))Fe(η5-C5H5)}] (2c)
3.2. General Procedure for the Addition of Carboxylic Acids to Internal Alkynes Catalyzed by Complex 2a
3.3. Synthesis and Characterization of (Z)-Hex-3-en-3-yl 4-(((Z)-Hex-3-en-3-yl)oxy)benzoate 5af
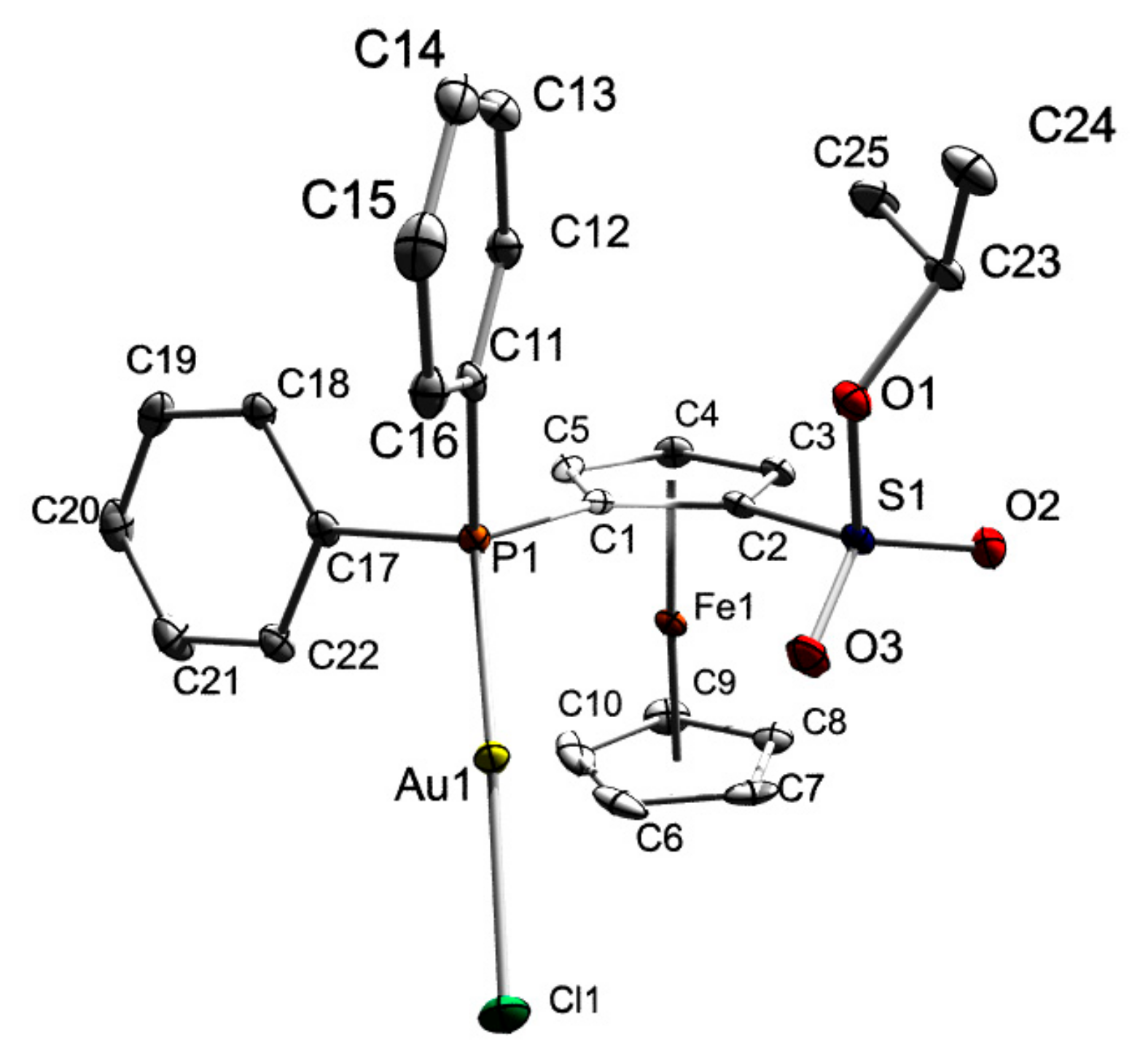
3.4. X-ray Crystal Structure Determination of Compound 2a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gooβen, L.J.; Paetzold, J. Decarbonylative Heck olefination of enol esters: Salt-free and environmentally friendly access to vinyl arenes. Angew. Chem. Int. Ed. 2004, 43, 1095–1098. [Google Scholar]
- Geibel, I.; Dierks, A.; Schmidtmann, M.; Christoffers, J. Formation of δ-lactones by cerium catalyzed, Baeyer-Villiger-type coupling of β-oxoesters, enol acetates, and dioxygen. J. Org. Chem. 2016, 81, 7790–7798. [Google Scholar] [CrossRef] [PubMed]
- Geibel, I.; Christoffers, J. Synthesis of 1,4-diketones from β-oxo esters and enol acetates by cerium-catalyzed oxidative umpolung reaction. Eur. J. Org. Chem. 2016, 2016, 918–920. [Google Scholar] [CrossRef]
- Kleman, P.; González-Liste, P.J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Highly enentioselective hydrogenation of 1-alkylvinyl benzoates: A simple, nonenzymatic access to chiral 2-alkanols. Chem. A Eur. J. 2013, 19, 16209–16212. [Google Scholar] [CrossRef] [PubMed]
- León, F.; González-Liste, P.J.; García-Garrido, S.E.; Arribas, I.; Rubio, M.; Cadierno, V.; Pizzano, A. Broad scope synthesis of ester precursors of nonfunctionalized chiral alcohols based on the asymmetric hydrogenation of α,β-dialky, α,β-diaryl, and α-alkyl-β-aryl-vinyl esters. J. Org. Chem. 2017, 82, 5852–5867. [Google Scholar] [CrossRef]
- Jia, J.; Fan, D.; Zhang, J.; Zhang, Z.; Zhang, W. An atropos biphenyl bisphosphine ligand with 2,2′-tert-butylmethylphosphino groups for the rhodium-catalyzed asymmetric hydrogenation of enol esters. Adv. Synth. Catal. 2018, 360, 3793–3800. [Google Scholar] [CrossRef]
- Corey, E.J.; Ghosh, A.K. Manganese (III)-promoted annulation of enol ethers and esters to fused spiro 2-cyclopentenones. Tetrahedron Lett. 1987, 28, 175–178. [Google Scholar] [CrossRef]
- Panda, N.; Mothkuri, R.; Pal, A.; Paital, A.R. Copper-catalyzed synthesis of α-naphthols from enol esters. Adv. Synth. Catal. 2013, 355, 2809–2814. [Google Scholar] [CrossRef]
- Panda, N.; Mishra, P.; Mattan, I. Synthesis of isocoumarins via silver (I)-mediated annulation of enol esters. J. Org. Chem. 2016, 81, 1047–1056. [Google Scholar] [CrossRef]
- Jena, R.K.; Das, U.K.; Ghorai, A.; Bhattacharjee, M. Ruthenium-catalyzed addition of carboxylic acids to propargylic alcohols: An easy route to O-dienyl esters and their tandem atom-transfer radical polymerization. Eur. J. Org. Chem. 2016, 36, 6015–6021. [Google Scholar] [CrossRef]
- Foarta, F.; Landis, C.R. Condensation oligomers with sequence control but without coupling reagents and protecting groups via asymmetric hydroformylation and hydroacyloxylation. J. Org. Chem. 2016, 81, 11250–11255. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Lee, H.-C.; Antonietti, M.; Schmidt, B.V.K.J. Free radical and RAFT polymerization of vinyl esters in metal-organic frameworks. Polym. Chem. 2017, 8, 6204–6208. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.; Yus, M. Transition-metal-catalyzed addition of heteroatom-hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Seayad, J.; Tillack, A.; Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004, 43, 3368–3398. [Google Scholar] [CrossRef]
- Hintermann, L. Recent developments in metal-catalyzed additions of oxygen nucleophiles to alkenes and alkynes. Top. Organomet. Chem. 2010, 31, 123–155. [Google Scholar]
- Patil, N.T.; Kavthe, R.D.; Shinde, V.S. Transition metal-catalyzed addition of C-, N- and O-nucleophiles to unactivated C-C multiple bonds. Tetrahedron 2012, 68, 8079–8146. [Google Scholar] [CrossRef]
- Bruneau, C. Group 8 metals-catalyzed O-H bond addition to unsaturated molecules. Top. Organomet. Chem. 2013, 43, 203–230. [Google Scholar]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. The intermolecular hydro-oxycarbonylation of internal alkynes: Current state of the art. Arkivoc 2018, ii, 17–39. [Google Scholar] [CrossRef]
- Kawatsura, M.; Namioka, J.; Kajita, K.; Yamamoto, M.; Tsuji, H.; Itoh, T. Ruthenium-catalyzed regio- and stereoselective addition of carboxylic acids to aryl and trifluoromethyl group substituted unsymmetrical internal alkynes. Org. Lett. 2011, 13, 3285–3287. [Google Scholar] [CrossRef]
- Lu, X.; Zhu, G.; Ma, S. A novel regio- and stereo-specific hydroacetoxylation reaction of 2-alkynoic acid derivatives. Tetrahedron Lett. 1992, 33, 7205–7206. [Google Scholar] [CrossRef]
- Yin, J.; Bai, Y.; Mao, M.; Zhu, G. Silver-catalyzed regio- and stereoselective addition of carboxylic acids to ynol ethers. J. Org. Chem. 2014, 79, 9179–9185. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Goundry, W.R.F.; Lam, H.W. Palladium-catalyzed hydroacyloxylation of ynamides. Chem. Commun. 2012, 48, 1505–1507. [Google Scholar] [CrossRef] [PubMed]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. Gold-catalyzed regio- and stereoselective addition of carboxylic acids to iodoalkynes: Access to (Z)-β-iodoenol esters and 1,4-disubstituted (Z)-enynyl esters. J. Org. Chem. 2017, 82, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Chary, B.C.; Kim, S. Gold (I)-catalyzed addition of carboxylic acids to alkynes. J. Org. Chem. 2010, 75, 7928–7931. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Li, Y.; Wu, G.; Cao, Z.; Zhang, L. A general ligand design for gold catalysis allowing ligand-directed anti-nucleophilic attack of alkynes. Nat. Commun. 2014, 5, 3470. [Google Scholar] [CrossRef]
- Dupuy, S.; Gasperini, D.; Nolan, S.P. Highly efficient gold (I)-catalyzed regio- and stereoselective hydrocarboxylation of internal alkynes. ACS Catal. 2015, 5, 6918–6921. [Google Scholar] [CrossRef]
- Chen, J.-F.; Li, C. Enol ester synthesis via cobalt-catalyzed regio- and stereoselective addition of carboxylic acids to alkynes. Org. Lett. 2018, 20, 6719–6724. [Google Scholar] [CrossRef]
- Dixneuf, P.H.; Cadierno, V. (Eds.) Metal-Catalyzed Reactions in Water; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Francos, J.; Cadierno, V. Metal-catalyzed intra- and intermolecular addition of carboxylic acids to alkynes in aqueous media: A review. Catalysts 2017, 7, 328. [Google Scholar] [CrossRef]
- González-Liste, P.J.; García-Garrido, S.E.; Cadierno, V. Gold (I)-catalyzed addition of carboxylic acids to internal alkynes in aqueous medium. Org. Biomol. Chem. 2017, 15, 1670–1679. [Google Scholar] [CrossRef]
- Gorin, D.J.; Sherry, B.D.; Toste, F.D. Ligands effects in homogeneous Au catalysis. Chem. Rev. 2008, 108, 3351–3378. [Google Scholar] [CrossRef]
- Sierra, D.; Contreras, C.; Francos, J.; Gómez, J.; Cadierno, V. Novel ferrocenylphosphino sulfonates: Synthesis, crystal structure and preliminary application as ligands in aqueous catalysis. J. Organomet. Chem. 2018, 854, 106–112. [Google Scholar] [CrossRef]
- Canales, F.; Gimeno, M.C.; Jones, P.G.; Laguna, A.; Sarroca, C. Substitution reaction studies on [Au2Cl2(µ-dppf)] (dppf = 1,1′-bis(diphenylphosphino)ferrocene). Synthesis of the first gold (I) complex with a µ3-2-pyridinethiolate ligand. Inorg. Chem. 1997, 36, 5206–5211. [Google Scholar] [CrossRef]
- Rampazzi, V.; Roger, J.; Amardeil, R.; Penouilh, M.-J.; Richard, P.; Fleurat-Lessard, P.; Hierson, J.-C. Gold (I) complexes of ferrocenyl polyphosphines: Aurophilic gold chloride formation and phosphine-concerted shuttling of a dinuclear [ClAu···AuCl] fragment. Inorg. Chem. 2016, 55, 10907–10921. [Google Scholar] [CrossRef] [PubMed]
- Delpont, N.; Escofet, I.; Pérez-Galán, P.; Spiegl, D.; Raducan, M.; Bour, C.; Sinisi, R.; Echavarren, A. Modular chiral gold (I) phosphite complexes. Catal. Sci. Technol. 2013, 3, 3007–3012. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, J.A.; Aponick, A. Regioselectivity in the Au-catalyzed hydration and hydroalkoxylation of alkynes. Chem. Commun. 2015, 51, 8730–8741. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Oxford, UK, 2003. [Google Scholar]
- Brandys, M.-C.; Jennings, M.C.; Puddephatt, R.J. Luminescent gold (I) macrocycles with diphosphine and 4, 4´-bipyridyl ligands. J. Chem. Soc. Dalton Trans. 2000, 4601–4606. [Google Scholar] [CrossRef]
- Tsukada, N.; Takahashi, A.; Inoue, Y. Hydrocarboxylation of unactivated internal alkynes with carboxylic acids catalyzed by dinuclear palladium complexes. Tetrahedron Lett. 2011, 52, 248–250. [Google Scholar] [CrossRef] [Green Version]
- CrysAlisPro CCD & CrysAlisPro RED; Oxford Diffraction Ltd.: Oxford, UK, 2008.
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL97: Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Wilson, A.J.C. (Ed.) International Tables for X-ray Crystallography, Volume, C; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Nardelli, M. PARST: A system of FORTRAN routines for calculating molecular structure parameters from results of crystal structure analyses. Comput. Chem. 1983, 7, 95–98. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 1999; Available online: http://www.crystalimpact.com/diamond (accessed on 1 September 2019).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
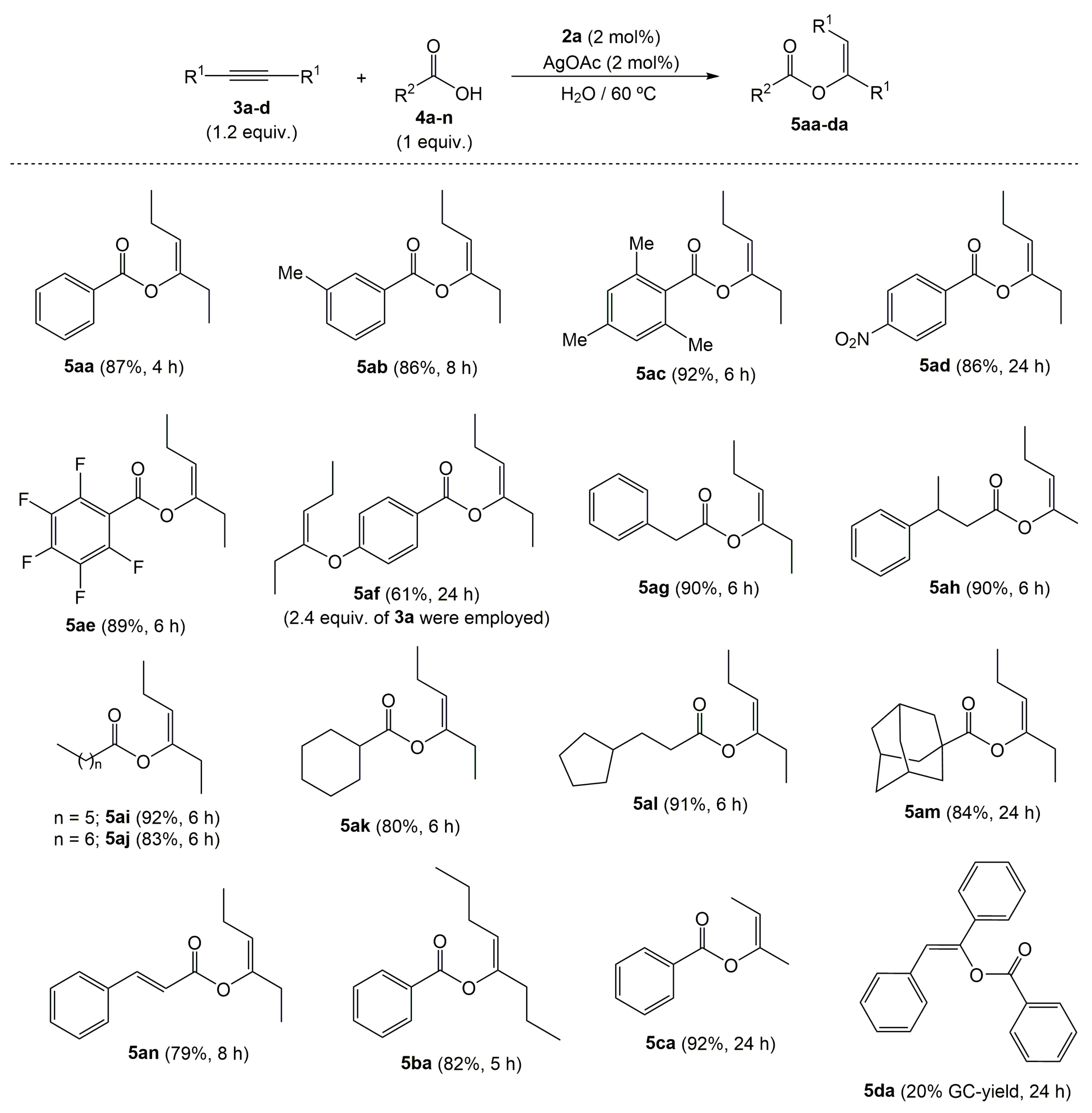{kind=link}
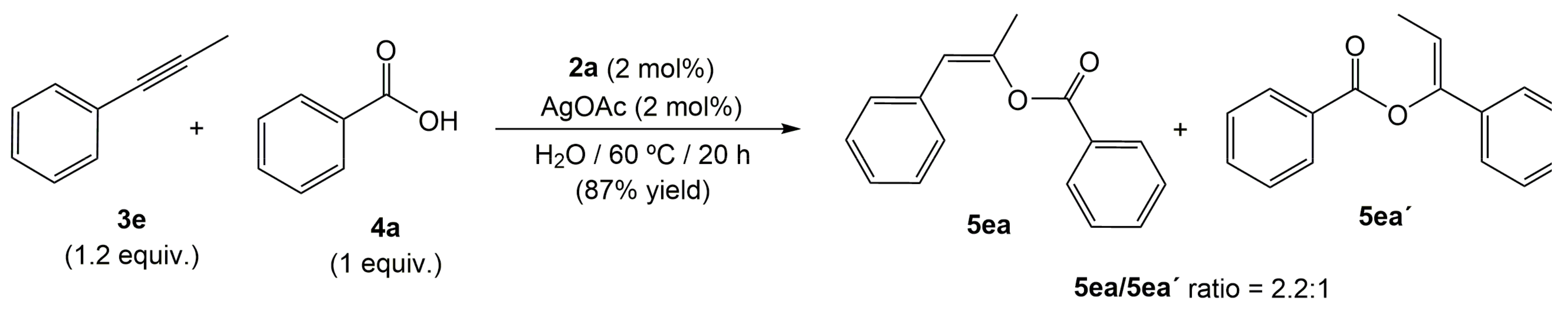{kind=link}

| Entry | Catalyst | Silver salt | Yield (%)2 |
|---|---|---|---|
| 1 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | ---- | 0 |
| 2 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | AgPF6 | 79 |
| 3 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | AgSbF6 | 49 |
| 4 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | AgNO3 | 87 |
| 5 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | AgOTs | 86 |
| 6 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | AgOTf | 82 |
| 7 | [AuCl{(η5-C5H3PPh2(SO3iPr))Fe(η5-C5H5)}] (2a) | AgOAc | 91 (87)3 |
| 8 | [AuCl{(η5-C5H3P(p-Tol)2(SO3iPr))Fe(η5-C5H5)}] (2b) | AgOAc | 89 |
| 9 | [AuCl{(η5-C5H3PCy2(SO3iPr))Fe(η5-C5H5)}] (2c) | AgOAc | 77 |
| 10 | ----------- | AgOAc | 4 |
| Chemical Formula | C25H25O3AuClFePS |
|---|---|
| fw | 724.74 |
| T (K) | 130(1) |
| cryst. syst. | monoclinic |
| space group | P 21/n |
| cryst. size mm3 | 0.24 x 0.10 x 0.07 |
| a, Å | 10.15090(10) |
| b, Å | 14.6795(2) |
| c, Å | 16.9056(2) |
| α, deg | 90 |
| β, deg | 101.7130(10) |
| γ, deg | 90 |
| Z | 4 |
| V, Å3 | 2466.65(5) |
| ρcalcd, g cm–3 | 1.952 |
| μ, mm−1 | 18.351 |
| F(000) | 1408 |
| θ range, deg | 3.0104 to 69.6789 |
| index ranges | −10 ≤ h ≤ 12; −16 ≤ k ≤ 17; −20 ≤ l ≤ 18 |
| completeness to θmax | 98% |
| no. of data collected | 12340 |
| no. of unique data | 4556 |
| no. of parameters/restrains | 332/0 |
| refinement method | full-matrix least-squares on F2 |
| goodness of fit on F2 | 1.082 |
| R1a [I > 2σ(I)] | 0.0271 |
| wR2a [I > 2σ(I)] | 0.0723 |
| R1 (all data) | 0.0292 |
| wR2 (all data) | 0.0741 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francos, J.; Moreno-Narváez, M.E.; Cadierno, V.; Sierra, D.; Ariz, K.; Gómez, J. Gold(I) Complexes with Ferrocenylphosphino Sulfonate Ligands: Synthesis and Application in the Catalytic Addition of Carboxylic Acids to Internal Alkynes in Water. Catalysts 2019, 9, 955. https://doi.org/10.3390/catal9110955
Francos J, Moreno-Narváez ME, Cadierno V, Sierra D, Ariz K, Gómez J. Gold(I) Complexes with Ferrocenylphosphino Sulfonate Ligands: Synthesis and Application in the Catalytic Addition of Carboxylic Acids to Internal Alkynes in Water. Catalysts. 2019; 9(11):955. https://doi.org/10.3390/catal9110955
Chicago/Turabian StyleFrancos, Javier, María Esther Moreno-Narváez, Victorio Cadierno, Diego Sierra, Katherine Ariz, and Johana Gómez. 2019. "Gold(I) Complexes with Ferrocenylphosphino Sulfonate Ligands: Synthesis and Application in the Catalytic Addition of Carboxylic Acids to Internal Alkynes in Water" Catalysts 9, no. 11: 955. https://doi.org/10.3390/catal9110955