Atomic Layer Deposition ZnO Over-Coated Cu/SiO2 Catalysts for Methanol Synthesis from CO2 Hydrogenation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Formation of Cu Particles with a Uniform Distribution
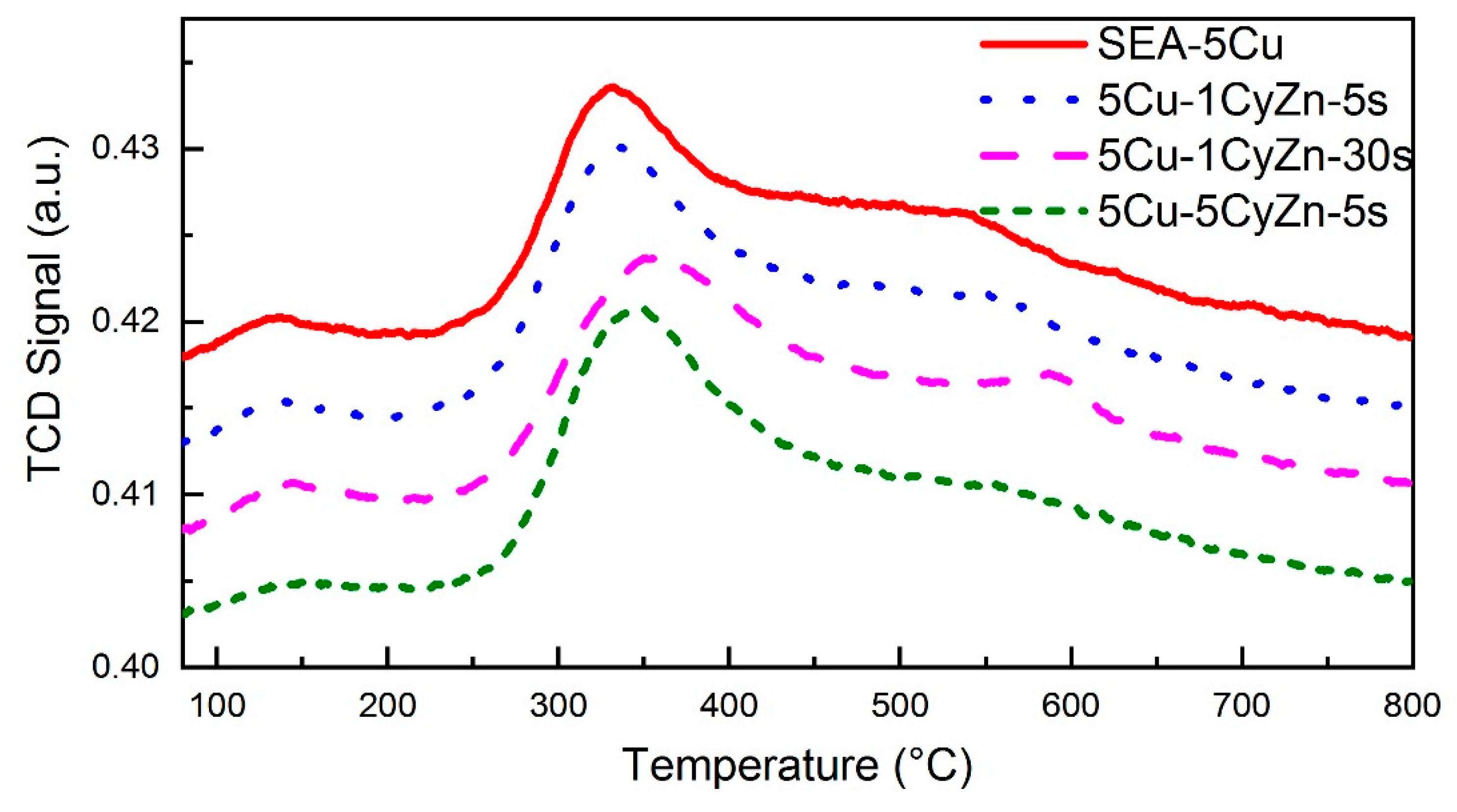
2.1.1. Reduction Properties of Samples from H2-TPR
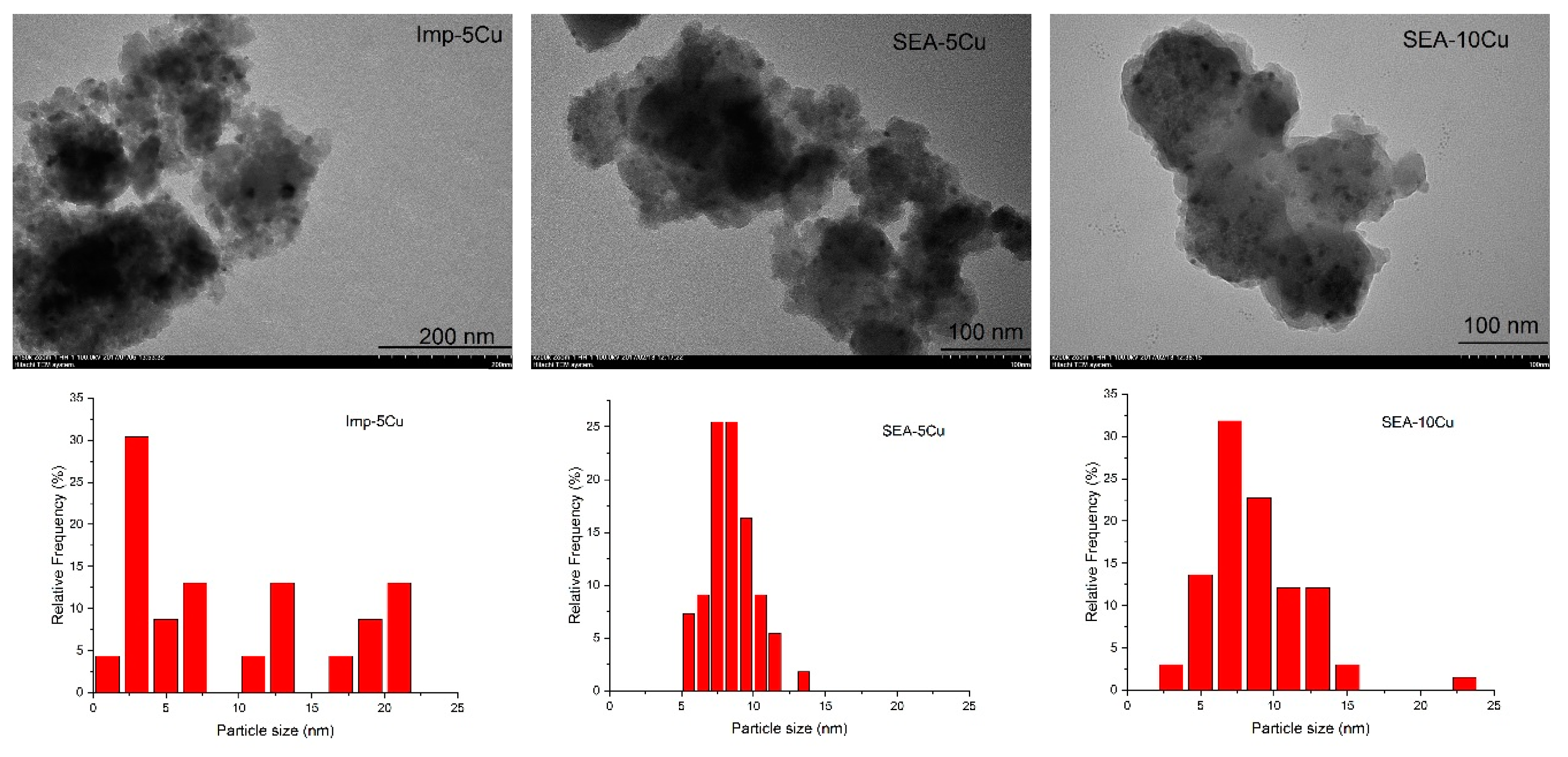
2.1.2. Samples Screened by TEM Images
2.2. Catalysts and Support Characterization
2.2.1. Results of ICP, BET, and XRD
2.2.2. Reduction Properties, Cu Particle Size, and Structure of ZnO
2.2.3. Basicity of the Catalysts from CO2-TPD
2.3. Chemical Structure from XAS Spectra
2.3.1. XANES for Cu K-Edge of SEA-5Cu and ZnO Over-Coated Cu/SiO2 Catalysts
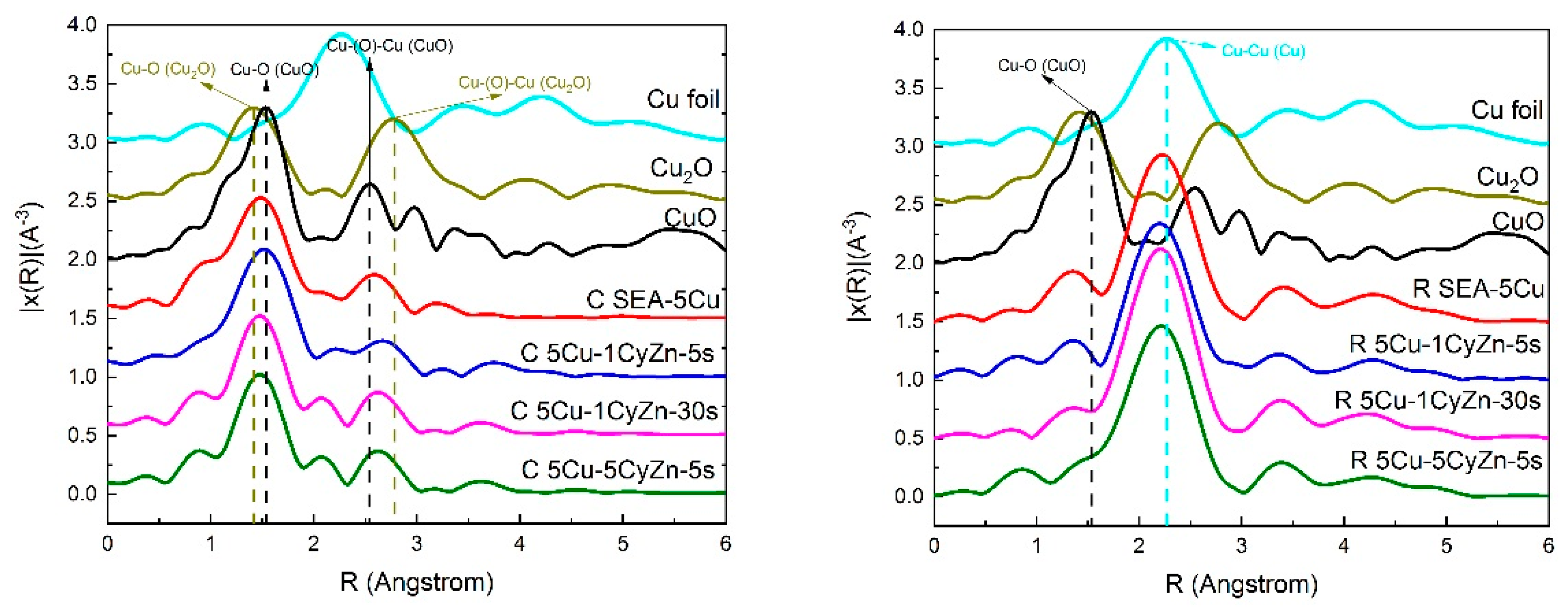
2.3.2. EXAFS for Cu K-Edge of SEA-5Cu and ZnO Over-Coated Cu/SiO2 Catalysts
2.3.3. XANES for Zn-K Edge of ZnO Over-Coated Cu/SiO2 Catalysts
2.3.4. EXAFS for Zn-K Edge of ZnO Over-Coated Cu/SiO2 Catalysts
2.4. CO2 Conversion and MeOH Synthesis
2.5. CO2 Catalytic Activation to Produce MeOH
3. Experiment
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalyst Evaluation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Le Quéré, C.; Barbero, L.; Hauck, J.; Andrew, R.M.; Canadell, J.G.; Sitch, S.; Korsbakken, J.I. Global Carbon Budget 2018. Earth Syst. Sci. Data 2018, 10, 2141–2194. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Duan, H.; Yang, C.Y.; Singh, D.R.; Chiang, B.K.; Wang, S.; Xiao, C.P.; Patel, B.J.; Danaci, C.D.; Burke, B.N.; Zhai, Y.; et al. Mesoporous Carbon-supported Cu/ZnO for Methanol Synthesis from Carbon Dioxide. Aust. J. Chem. 2014, 67, 907–914. [Google Scholar] [CrossRef]
- Deerattrakul, V.; Dittanet, P.; Sawangphruk, M.; Kongkachuichay, P. CO2 hydrogenation to methanol using Cu-Zn catalyst supported on reduced graphene oxide nanosheets. J. CO2 Util. 2016, 16, 104–113. [Google Scholar] [CrossRef]
- Dong, X.; Li, F.; Zhao, N.; Xiao, F.; Wang, J.; Tan, Y. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared by precipitation-reduction method. Appl. Catal. B Environ. 2016, 191, 8–17. [Google Scholar] [CrossRef]
- Le Valant, A.; Comminges, C.; Tisseraud, C.; Canaff, C.; Pinard, L.; Pouilloux, Y. The Cu-ZnO synergy in methanol synthesis from CO2, Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu + ZnO mechanical mixtures. J. Catal. 2015, 324, 41–49. [Google Scholar] [CrossRef]
- Liang, Z.; Gao, P.; Tang, Z.; Lv, M.; Sun, Y. Three dimensional porous Cu-Zn/Al foam monolithic catalyst for CO2 hydrogenation to methanol in microreactor. J. CO2 Util. 2017, 21, 191–199. [Google Scholar] [CrossRef]
- Karelovic, A.; Bargibant, A.; Fernández, C.; Ruiz, P. Effect of the structural and morphological properties of Cu/ZnO catalysts prepared by citrate method on their activity toward methanol synthesis from CO2 and H2 under mild reaction conditions. Catal. Today 2012, 197, 109–118. [Google Scholar] [CrossRef]
- Gesmanee, S.; Koo-Amornpattana, W. Catalytic hydrogenation of CO2 for methanol production in fixed-bed reactor using Cu-Zn supported on gamma-Al2O3. Energy Procedia 2017, 138, 739–744. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 759, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M. Meso-and nano-structuring of industrial Cu/ZnO/(Al2O3) catalysts. J. Catal. 2009, 267, 24–29. [Google Scholar] [CrossRef]
- Fujimoto, K.; Yu, Y. Spillover effect on the stabilization of Cu-Zn catalyst for CO2 hydrogenation to methanol. Stud. Surf. Sci. Catal. 1993, 77, 393–396. [Google Scholar]
- Hu, B.; Yin, Y.; Liu, G.; Chen, S.; Hong, X.; Tsang, S.C.E. Hydrogen spillover enabled active Cu sites for methanol synthesis from CO2 hydrogenation over Pd doped CuZn catalysts. J. Catal. 2018, 359, 17–26. [Google Scholar] [CrossRef]
- Waugh, K.C. Methanol Synthesis. Catal. Today 1992, 15, 51–75. [Google Scholar] [CrossRef]
- Ren, H.; Xu, C.-H.; Zhao, H.-Y.; Wang, Y.-X.; Liu, J.J.-Y.; Liu, J.J.-Y. Methanol synthesis from CO2 hydrogenation over Cu/γ-Al2O3 catalysts modified by ZnO, ZrO2 and MgO. J. Ind. Eng. Chem. 2015, 28, 261–267. [Google Scholar] [CrossRef]
- Choi, Y.; Futagami, K.; Fujitani, T.; Nakamura, J. The difference in the active sites for CO2 and CO hydrogenations on Cu / ZnO-based methanol synthesis catalysts. Catal. Lett. 2001, 73, 27–31. [Google Scholar] [CrossRef]
- Kanai, Y.; Watanabe, T.; Fujitani, T.; Saito, M.; Nakamura, J.; Uchijima, T. Evidence for the migration of ZnOx in a Cu/ZnO methanol synthesis catalyst. Catal. Lett. 1994, 27, 67–78. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Belin, T.; Ahouari, H.; Soualah, A.; Pouilloux, Y.; Le Valant, A. The Cu-ZnO synergy in methanol synthesis from CO2, Part 2: Origin of the methanol and CO selectivities explained by experimental studies and a sphere contact quantification model in randomly packed binary mixtures on Cu-ZnO coprecipitate catalysts. J. Catal. 2015, 330, 533–544. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Pronier, S.; Pouilloux, Y.; Le Valant, A. The Cu–ZnO synergy in methanol synthesis Part 3: Impact of the composition of a selective Cu@ZnOxcore–shell catalyst on methanol rate explained by experimental studies and a concentric spheres model. J. Catal. 2016, 343, 106–114. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Habrioux, A.; Pronier, S.; Pouilloux, Y.; Le Valant, A. Cu-ZnO catalysts for CO2 hydrogenation to methanol: Morphology change induced by ZnO lixiviation and its impact on the active phase formation. Mol. Catal. 2018, 446, 98–105. [Google Scholar] [CrossRef]
- Din, I.U.; Shaharun, M.S.; Naeem, A.; Tasleem, S.; Johan, M.R. Carbon nanofiber-based copper/zirconia catalyst for hydrogenation of CO2 to methanol. J. CO2 Util. 2017, 21, 145–155. [Google Scholar] [CrossRef]
- Musić, S.; Filipović-Vinceković, N.; Sekovanić, L. Precipitation of amorphous SiO2 particles and their properties. Braz. J. Chem. Eng. 2011, 28, 89–94. [Google Scholar] [CrossRef]
- Gawande, M.B.; Goswami, A.; Felpin, F.X.; Asefa, T.; Huang, X.; Silva, R.; Zou, X.; Zboril, R.; Varma, R.S. Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysis. Chem. Rev. 2016, 116, 3722–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayodele, O.B.; Tasfy, S.F.H.; Zabidi, N.A.M.; Uemura, Y. Co-synthesis of methanol and methyl formate from CO2 hydrogenation over oxalate ligand functionalized ZSM-5 supported Cu/ZnO catalyst. J. CO2 Util. 2017, 17, 273–283. [Google Scholar] [CrossRef]
- Huang, C.; Chen, S.; Fei, X.; Liu, D.; Zhang, Y. Catalytic hydrogenation of CO2 to methanol: Study of synergistic effect on adsorption properties of CO2 and H2 in CuO/ZnO/ZrO2 system. Catalysts 2015, 5, 1846–1861. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhan, H.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Fluorine-modified Cu/Zn/Al/Zr catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. Catal. Commun. 2014, 50, 78–82. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, K.; Ma, H.; Xu, X.; Wang, X. Cr, Zr-incorporated hydrotalcites and their application in the synthesis of isophorone. Catal. Commun. 2010, 11, 880–883. [Google Scholar] [CrossRef]
- Bhuiyan, M.M.R.; Lin, S.D.; Hsiao, T.C. Effect of calcination on Cu-Zn-loaded hydrotalcite catalysts for C-C bond formation derived from methanol. Catal. Today 2014, 226, 150–159. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Gopinath, C.S.; Yoshida, H.; Hattori, T. XPS, XANES and EXAFS investigations of CuO/ZnO/Al2O3/ZrO2 mixed oxide catalysts. Phys. Chem. Chem. Phys. 2002, 4, 1990–1999. [Google Scholar] [CrossRef]
- Dadlani, A.; Acharya, S.; Trejo, O.; Nordlund, D.; Peron, M.; Razavi, J.; Berto, F.; Prinz, F.B.; Torgersen, J. Revealing the Bonding Environment of Zn in ALD Zn(O,S) Buffer Layers through X-ray Absorption Spectroscopy. Appl. Mater. Interfaces 2017, 9, 39105–39109. [Google Scholar] [CrossRef] [PubMed]
- Janocha, E. Electronic Properties of ALD Zinc Oxide Interfaces and its Implication for Chalcopyrite Absorber Materials. Ph.D. Thesis, Fakultät IV—Elektrotechnik und Informatik, Technische Universität Berlin, Berlin, Germany, 2011. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Cu 1 Loading (wt%) | Zn 1 Loading (wt%) | BET Surface Area 2 (m2g−1) | Pore Volume 2 (cm3g−1) | Average Pore Size 2 (nm) | Cu0 Lattice Constant 3 (Å) |
|---|---|---|---|---|---|---|
| SiO2 | - | - | 718 | 0.41 | 3.0 | - |
| SEA-5Cu | 4.4 | - | 264 | 0.30 | 4.5 | 3.62 |
| 5Cu-1CyZn-5s | 4.4 | 0.3 | 200 | 0.21 | 4.2 | 3.62 |
| 5Cu-1CyZn-30s | 4.5 | 1.5 | 210 | 0.24 | 4.3 | 3.64 |
| 5Cu-5CyZn-5s | 4.3 | 1.3 | 238 | 0.28 | 4.6 | 3.65 |
| Catalysts | 5Cu-1CyZn-5s Element Content (wt%) | 5Cu-1CyZn-30s Element Content (wt%) | 5Cu-5CyZn-5s Elements Content (wt%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spots | Zn | Cu | Si | O | Zn | Cu | Si | O | Zn | Cu | Si | O |
| 1 | 1.9 | 87.0 | 1.8 | 9.3 | 1.1 | 10.9 | 43.7 | 44.3 | 0.1 | 5.0 | 60.7 | 34.2 |
| 2 | 0.2 | 22.6 | 42.0 | 35.2 | 1.9 | 12.3 | 48.6 | 37.2 | 4.1 | 8.0 | 43.1 | 44.8 |
| 3 | 0.3 | 17.7 | 46.8 | 35.2 | 1.1 | 8.5 | 52.8 | 37.6 | 2.4 | 9.0 | 43.2 | 45.4 |
| 4 | 0.2 | 7.8 | 45.2 | 46.8 | 1.8 | 8.3 | 50.1 | 39.8 | 1.0 | 26.5 | 34.6 | 37.9 |
| Samples | Cu Particle Size (nm) | Cu Dispersion (%) | Cu Surface Area (m2∙g−1) | The Number of Metallic Sites (μmol∙g−1) | |
|---|---|---|---|---|---|
| TEM | XRD | ||||
| SEA-5Cu | 8.4 | 7.1 | - | - | - |
| 5Cu-1CyZn-5s | 8.1 | 6.5 | 1.9 | 0.69 | 304 |
| 5Cu-1CyZn-30s | 6.7 | 5.3 | 1.7 | 0.65 | 286 |
| 5Cu-5CyZn-5s | 6.9 | 6.4 | 1.3 | 0.53 | 228 |
| Samples | Number of Basic Sites (μmol∙g−1) | Total Number of Basic Sites | ||
|---|---|---|---|---|
| Weak | Moderate | Strong | ||
| SEA-5Cu | 13 | 193 | 578 | 784 |
| 5Cu-1CyZn-5s | 19 | 301 | 838 | 1158 |
| 5Cu-1CyZn-30s | 42 | 369 | 1017 | 1428 |
| 5Cu-5CyZn-5s | 21 | 405 | 884 | 1310 |
| Samples | Configuration | ||
|---|---|---|---|
| Cu | Cu2O | CuO | |
| SEA-5Cu | 0.75 | 0.01 | 0.24 |
| 5Cu-1CyZn-5s | 0.75 | 0 | 0.25 |
| 5Cu-1CyZn-30s | 0.75 | 0 | 0.25 |
| 5Cu-5CyZn-5s | 0.79 | 0 | 0.21 |
| Catalysts 1 | CO2 Reaction Rate | MeOH Formation Rate | CO Formation Rate | MeOH Selectivity | Number of Basic Sites | Number of Metallic Sites | Ratio of Metallic to Basic Sites | TOFCO2 by ZnO | TOFH2 by Cu |
|---|---|---|---|---|---|---|---|---|---|
| (mmol∙g−1h−1) | (%) | (μmol∙g−1) | (h−1) | ||||||
| 5Cu-1CyZn-5s | 4.90 | 0.33 | 4.57 | 6.7 | 1158 | 304 | 0.26 | 4.2 | 18.3 |
| 5Cu-1CyZn-30s | 4.77 | 0.48 | 4.32 | 10.1 | 1428 | 286 | 0.19 | 3.3 | 20.0 |
| 5Cu-5CyZn-5s | 4.06 | 0.21 | 3.89 | 5.2 | 1310 | 228 | 0.17 | 3.1 | 19.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Boahene, P.E.; Hu, Y.; Dalai, A.; Wang, H. Atomic Layer Deposition ZnO Over-Coated Cu/SiO2 Catalysts for Methanol Synthesis from CO2 Hydrogenation. Catalysts 2019, 9, 922. https://doi.org/10.3390/catal9110922
Gao J, Boahene PE, Hu Y, Dalai A, Wang H. Atomic Layer Deposition ZnO Over-Coated Cu/SiO2 Catalysts for Methanol Synthesis from CO2 Hydrogenation. Catalysts. 2019; 9(11):922. https://doi.org/10.3390/catal9110922
Chicago/Turabian StyleGao, Jinglin, Philip Effah Boahene, Yongfeng Hu, Ajay Dalai, and Hui Wang. 2019. "Atomic Layer Deposition ZnO Over-Coated Cu/SiO2 Catalysts for Methanol Synthesis from CO2 Hydrogenation" Catalysts 9, no. 11: 922. https://doi.org/10.3390/catal9110922