Single and Dual Metal Oxides as Promising Supports for Carbon Monoxide Removal from an Actual Syngas: The Crucial Role of Support on the Selectivity of the Au–Cu System


Abstract
:
1. Introduction
2. Results and Discussion
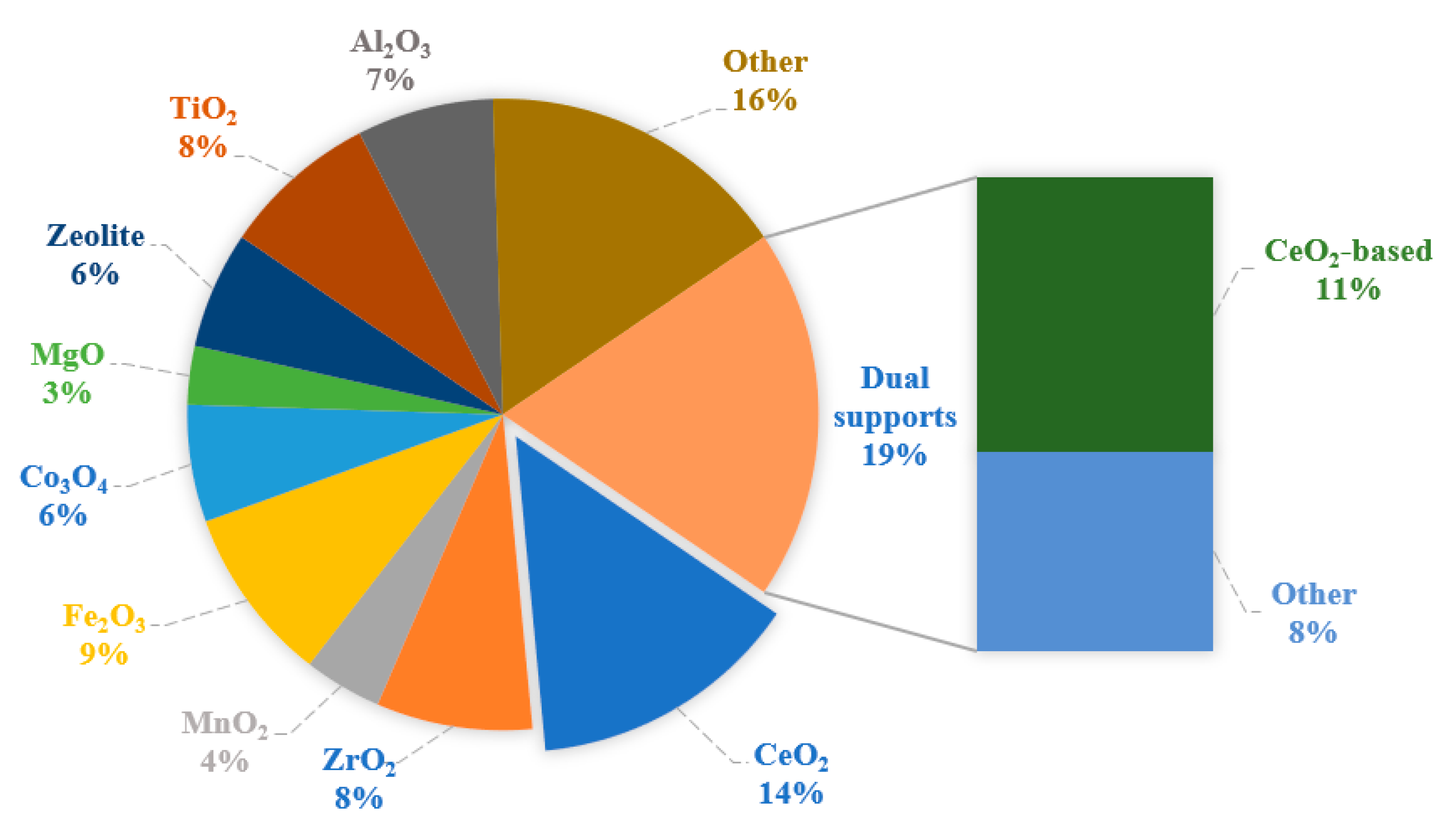
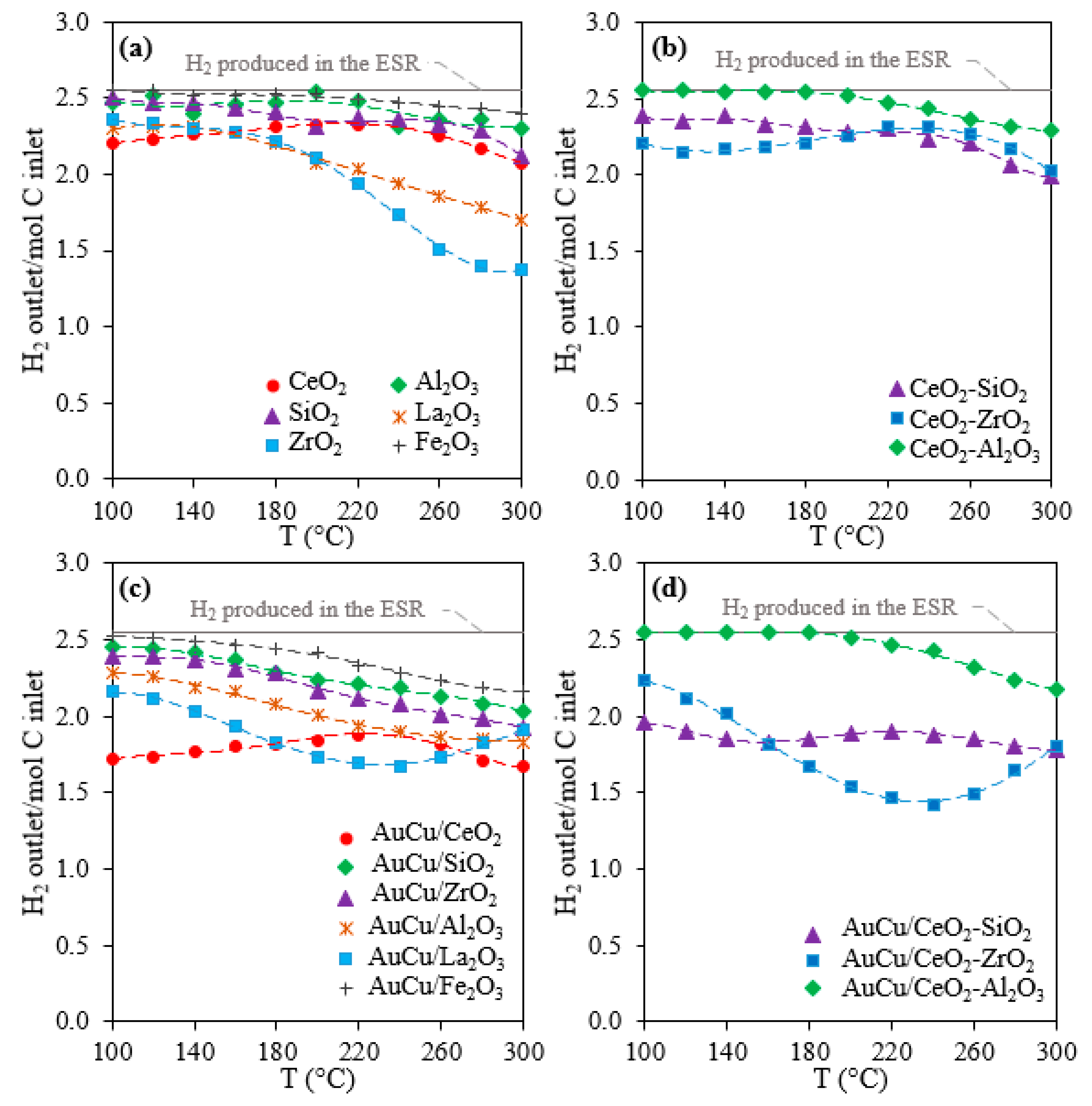
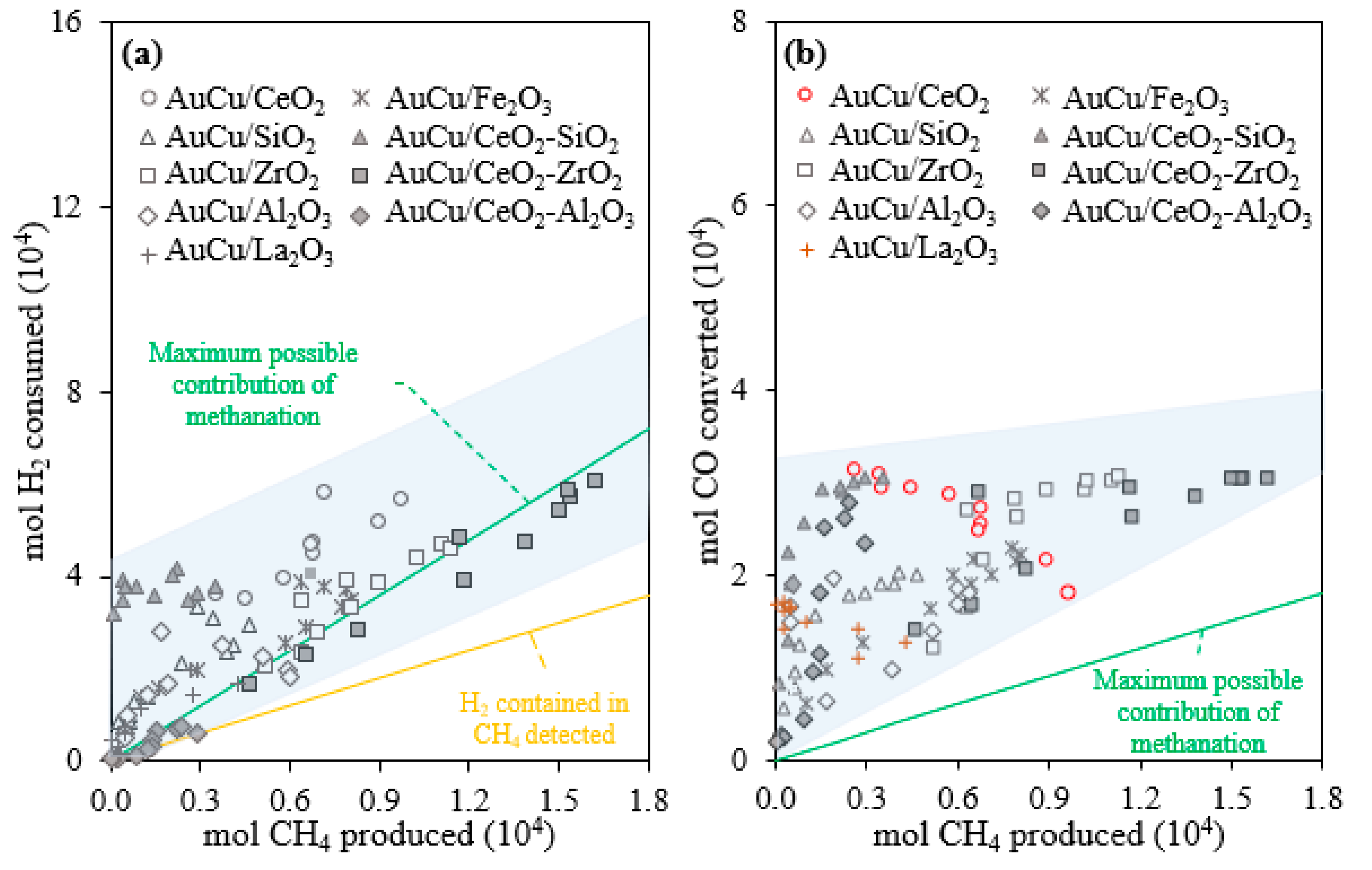
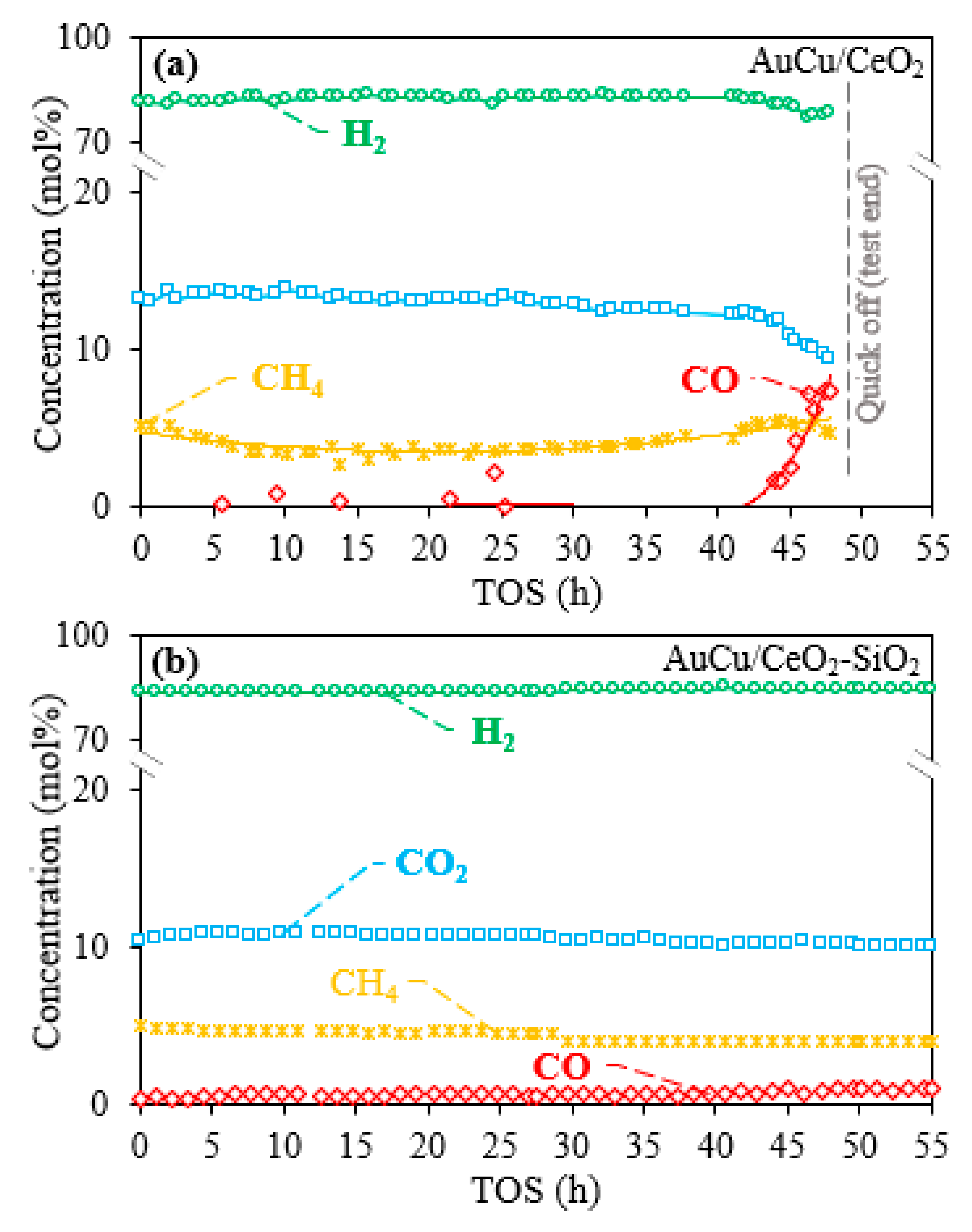
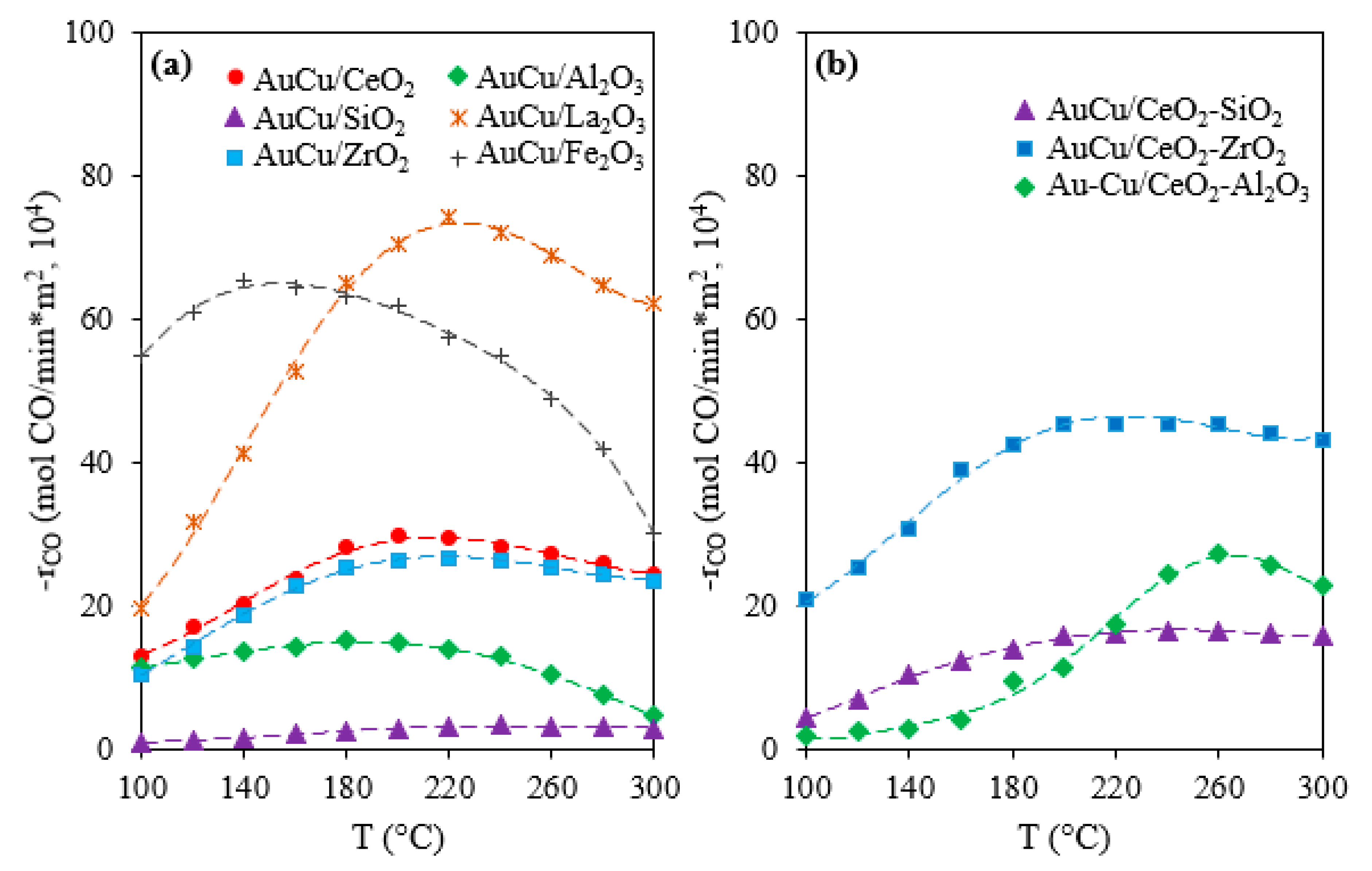
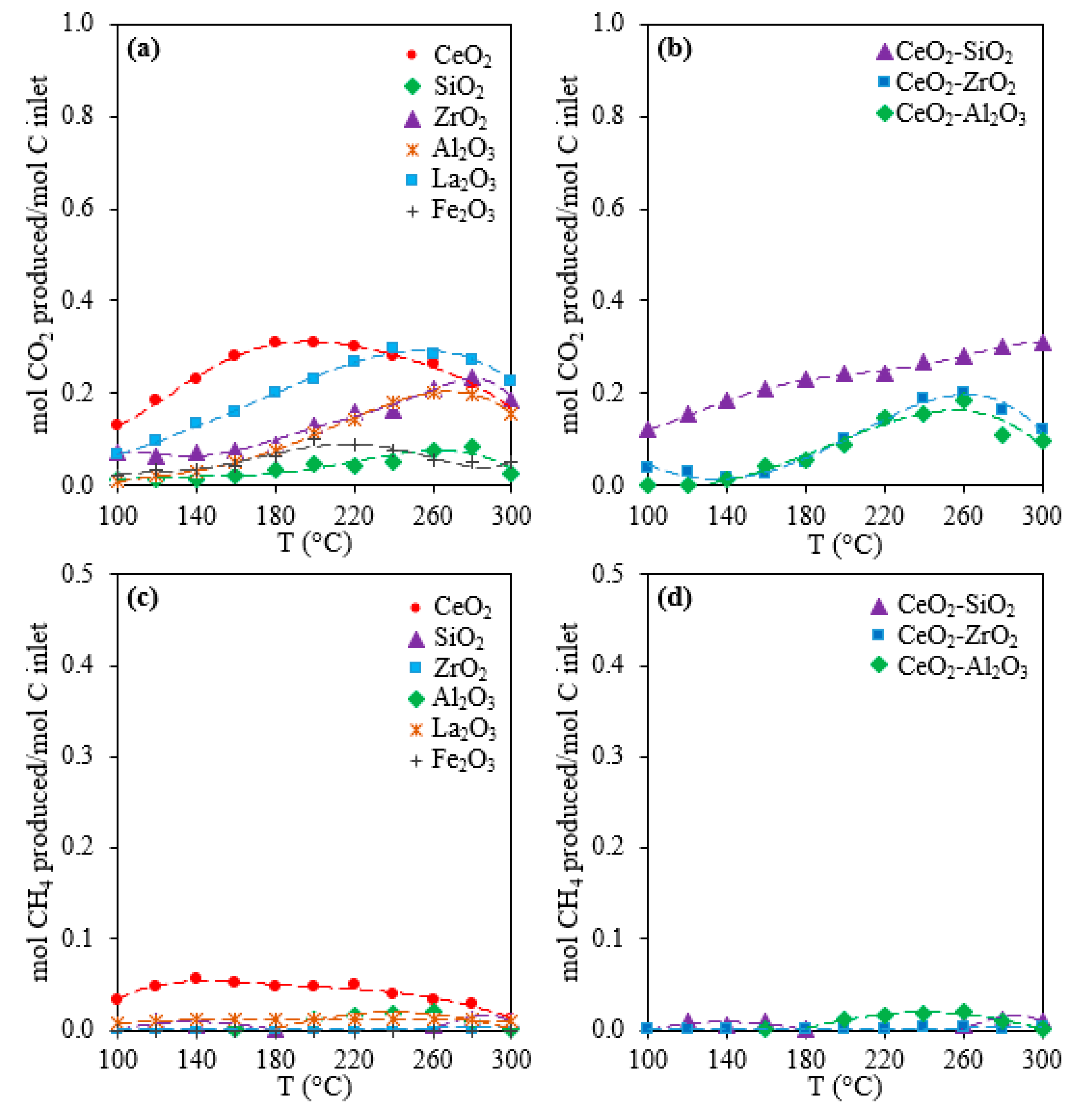
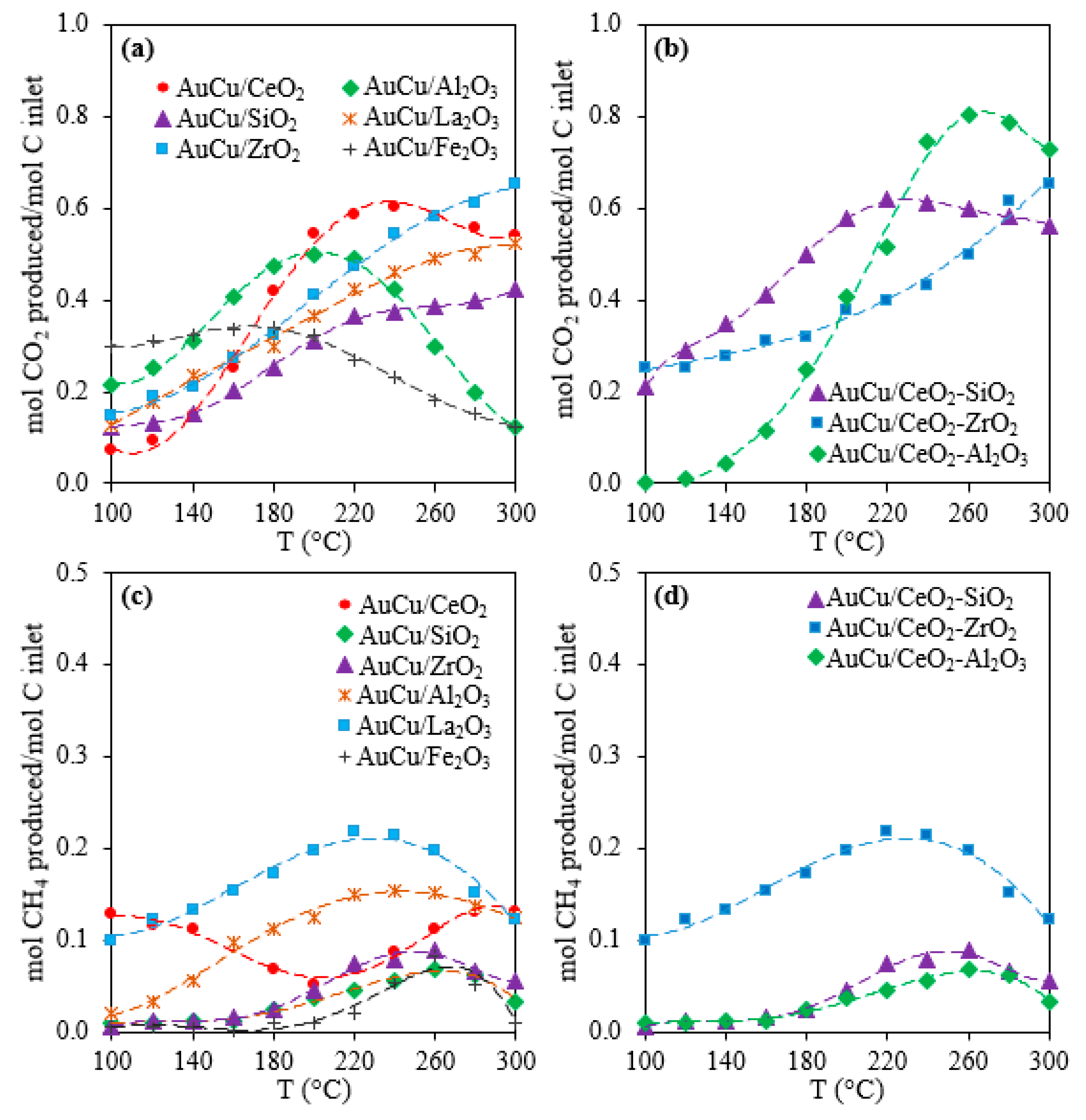
2.1. Activity, Selectivity, and Stability
2.2. Catalysts Characterization
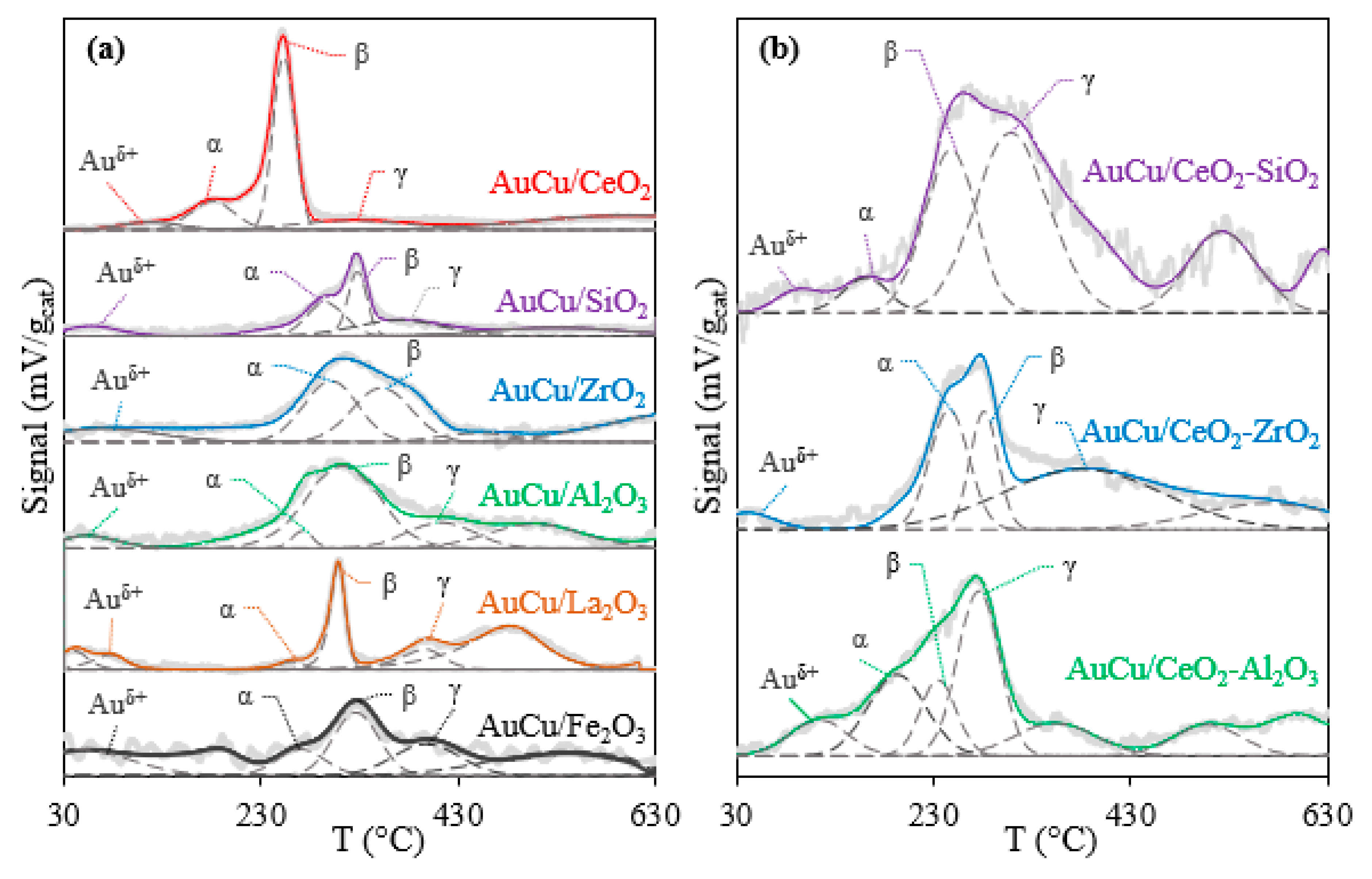
2.2.1. TPR
2.2.2. BET Area
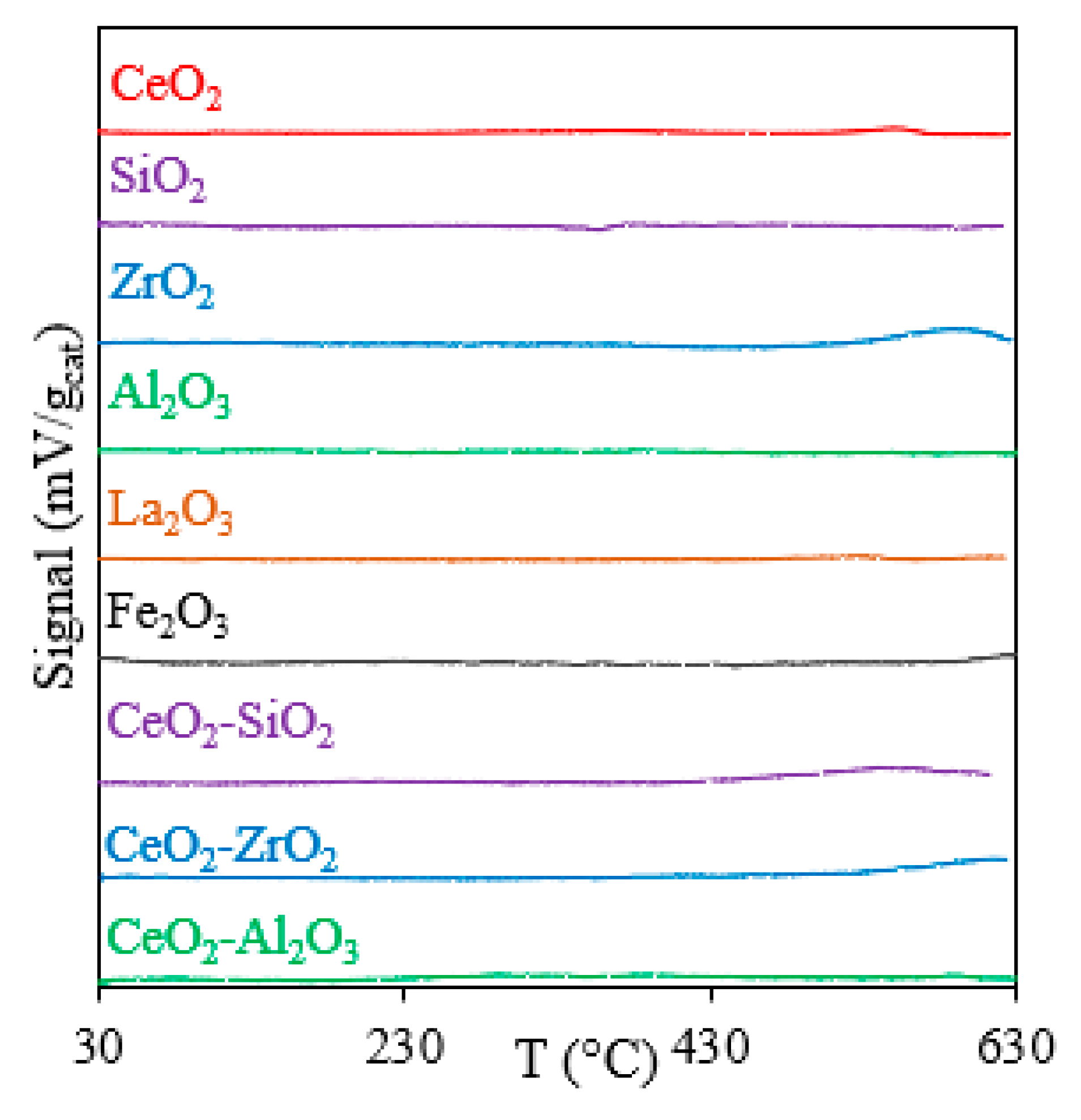
2.2.3. OSC Measurements
2.2.4. TGA
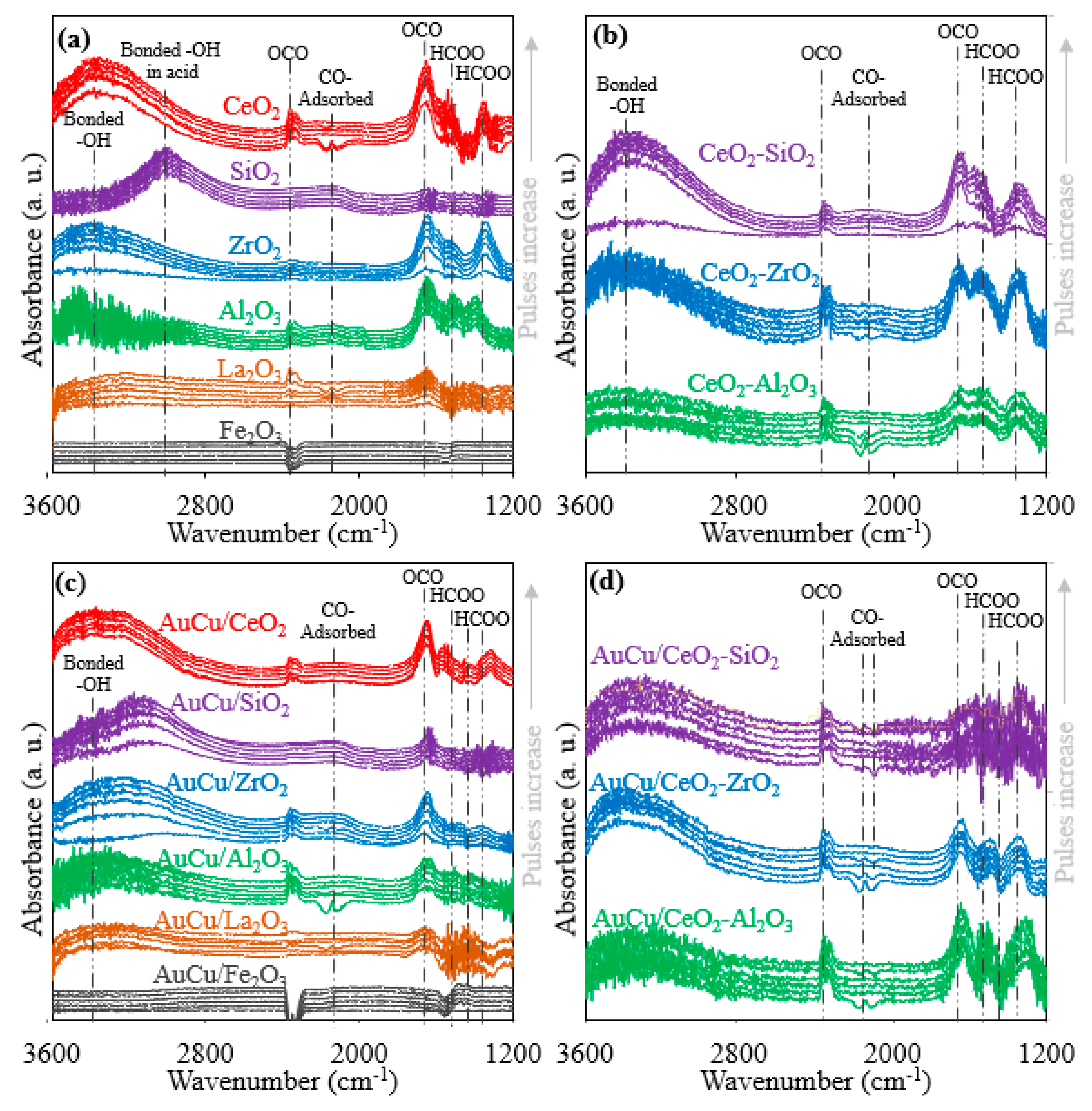
2.2.5. In Situ DRIFTS
3. Materials and Methods
3.1. Support Selection
3.2. Catalyst Synthesis
3.3. Obtaining Syngas
3.4. Catalytic Test
3.5. Characterization Tests
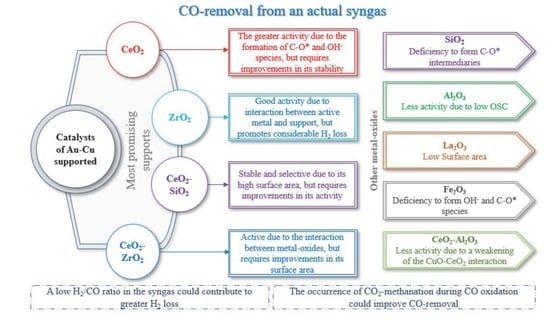
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support | BET Surface Area (m2/gcat) | OSC in Fresh Samples at 300 °C (μmol O2/gcat) | OSCC in Fresh Samples at 300 °C (μmol O2/gcat) | Weight Loss (%) | ||
|---|---|---|---|---|---|---|
| Fresh | Used | Fresh | Used | |||
| CeO2 | 52.4 | 55.4 | 61 | 135 | 1.1 | 0.6 |
| SiO2 | 466.5 | 410.6 | 41 | 49 | 1.4 | 1.1 |
| ZrO2 | 51.6 | 44.9 | 55 | 99 | 1.6 | 0.6 |
| Al2O3 | 96 | 68.6 | 36 | 55 | 1.7 | 0.2 |
| La2O3 | 14.1 | 15.3 | 21 | 68 | 5.5 | 2.1 |
| Fe2O3 | 38.1 | 36.7 | 5 | 16 | 0.7 | 0.8 |
| CeO2-SiO2 | 163.2 | 155.2 | 54 | 105 | −2.7 | 1.8 |
| CeO2-ZrO2 | 44.3 | 40.5 | 46 | 110 | 0.2 | 2.0 |
| CeO2-Al2O3 | 72.7 | 69.1 | 41 | 120 | 2.0 | 1.0 |
| Date | Active Metals | Metal Oxide I | Metal Oxide II | Journal | Digital Object Identifier (DOI) |
|---|---|---|---|---|---|
| 2012 | CuO | Fe2O3 | - | Chemical Engineering Journal | 10.1016/j.cej.2012.01.017 |
| 2012 | Pt | Other | - | Electrochimica Acta | 10.1016/j.electacta.2012.04.150 |
| 2012 | - | Fe2O3 | - | Applied Surface Science | 10.1016/j.apsusc.2011.10.092 |
| 2012 | - | NiO2 | - | Journal of Molecular Catalysis A: Chemical | 10.1016/j.molcata.2012.05.001 |
| 2013 | Ni, Co | Co3O4 | - | Journal of Alloys and Compounds | 10.1016/j.jallcom.2013.04.053 |
| 2013 | CuO | TiO2 | Al2O3 | Surface and Coatings Technology | 10.1016/j.surfcoat.2012.10.031 |
| 2013 | Co | Fe2O3 | - | Chemical Engineering Journal | 10.1016/j.ces.2013.02.002 |
| 2014 | Co | MgO | - | Process Safety and Environmental Protection | 10.1016/j.psep.2013.12.003 |
| 2014 | Pt | CeO2 | - | Chemical Engineering Journal | 10.1016/j.cej.2014.06.058 |
| 2014 | Pd | Fe2O3 | - | Journal of Catalysis | 10.1016/j.jcat.2014.06.019 |
| 2014 | Ag | Zeolite | - | Fuel | 10.1016/j.fuel.2014.07.011 |
| 2014 | Au | NiO2 | - | Applied Catalysis A: General | 10.1016/j.apcata.2014.02.003 |
| 2014 | CuO | SiO2 | CeO2 | Journal of Environmental Chemical Engineering | 10.1016/j.jece.2014.03.021 |
| 2015 | Co, Fe, Cr | CeO2 | - | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2015.03.044 |
| 2015 | - | Co3O4 | - | Applied Catalysis A: General | 10.1016/j.apcata.2014.10.024 |
| 2015 | CuO | Fe2O3 | - | Chinese Journal of Catalysis | 10.1016/S1872-2067(15)60922-6 |
| 2015 | Au | Zeolite | - | Catalysis Communications | 10.1016/j.catcom.2015.06.018 |
| 2015 | Pt | CeO2 | - | Catalysis Today | 10.1016/j.cattod.2014.12.038 |
| 2015 | Au, Cu | CeO2 | ZrO2 | Catalysis Today | 10.1016/j.cattod.2014.08.035 |
| 2015 | - | PtO2 | - | Applied Surface Science | 10.1016/j.apsusc.2015.03.108 |
| 2015 | CuO | CeO2 | ZrO2 | Journal of Industrial and Engineering Chemistry | 10.1016/j.jiec.2015.06.038 |
| 2015 | - | MnO2 | CeO2 | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2014.06.038 |
| 2016 | Pd | Fe2O3 | - | Journal of Environmental Chemical Engineering | 10.1016/j.jece.2016.10.019 |
| 2016 | - | CeO2 | ZrO2 | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2016.02.023 |
| 2016 | Au | Zn2SnO4 | - | Chinese Journal of Catalysis | 10.1016/S1872-2067(16)62468-3 |
| 2016 | - | Co3O4 | - | Catalysis Communications | 10.1016/j.catcom.2016.08.020 |
| 2016 | Au | CeO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2016.02.025 |
| 2016 | Pd | CeO2 | - | Journal of Molecular Catalysis A: Chemical | 10.1016/j.molcata.2016.08.035 |
| 2016 | - | SiO2 | Al2O3 | Journal of Molecular Graphics and Modelling | 10.1016/j.jmgm.2016.08.005 |
| 2016 | Ag | SiO2 | - | Catalysis Today | 10.1016/j.cattod.2016.05.033 |
| 2016 | - | PdO | - | Surface Science | 10.1016/j.susc.2015.08.043 |
| 2016 | - | Co3O4 | - | Applied Catalysis A: General | 10.1016/j.apcata.2016.03.027 |
| 2016 | CuO | TiO2 | - | Catalysis Communications | 10.1016/j.catcom.2016.02.001 |
| 2016 | Pt | CeO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2016.01.056 |
| 2016 | CuO | MnO2 | - | Journal of Molecular Catalysis A: Chemical | 10.1016/j.molcata.2016.08.024 |
| 2016 | CuO | Peroskita | - | Applied Clay Science | 10.1016/j.clay.2015.08.034 |
| 2016 | Pd | ZnO | - | Catalysis Today | 10.1016/j.cattod.2015.05.021 |
| 2016 | - | Fe2O3 | - | Chemical Engineering Journal | 10.1016/j.cej.2016.04.136 |
| 2016 | Au | TiO2 | - | Catalysis Today | 10.1016/j.cattod.2015.09.040 |
| 2016 | Au | Fe2O3 | CeO2 | Catalysis Today | 10.1016/j.cattod.2016.05.059 |
| 2016 | - | Co3O4 | - | Materials Letters | 10.1016/j.matlet.2016.06.108 |
| 2016 | - | Co3O4 | - | Chinese Journal of Catalysis | 10.1016/S1872-2067(15)60969-X |
| 2016 | Au | TiO2 | - | Applied Surface Science | 10.1016/j.apsusc.2016.01.285 |
| 2016 | - | Fe2O3 | - | Journal of Molecular Catalysis A: Chemical | 10.1016/j.molcata.2016.01.003 |
| 2016 | Au | Other | - | Journal of Colloid and Interface Science | 10.1016/j.jcis.2016.06.072 |
| 2016 | Au | LaPO4 | - | Journal of the Taiwan Institute of Chemical Engineers | 10.1016/j.jtice.2016.01.016 |
| 2016 | Pt | Al2O3 | - | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2016.08.170 |
| 2016 | Pt | Other | - | Surface Science | 10.1016/j.susc.2015.08.024 |
| 2017 | CuO | Nb2O5 | - | Catalysis Communications | 10.1016/j.catcom.2017.04.008 |
| 2017 | Zn, Pt | CeO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2017.04.044 |
| 2017 | Pt, Fe | Fe2O3 | Co3O4 | Chinese Journal of Catalysis | 10.1016/S1872-2067(17)62838-9 |
| 2017 | CuO | MnO2 | CeO2 | Catalysis Communications | 10.1016/j.catcom.2017.05.016 |
| 2017 | Pt | MnO2 | - | Journal of Electroanalytical Chemistry | 10.1016/j.jelechem.2016.09.031 |
| 2017 | Au | LaPO4 | - | Chinese Journal of Chemical Engineering | 10.1016/j.cjche.2017.08.008 |
| 2017 | Fe, Mn | CeO2 | - | Catalysis Today | 10.1016/j.cattod.2016.11.046 |
| 2017 | Mn | Co3O4 | - | Solid State Sciences | 10.1016/j.solidstatesciences.2017.07.006 |
| 2017 | Mn | Co3O4 | - | Fuel | 10.1016/j.fuel.2017.04.140 |
| 2017 | Au | CeO2 | - | Applied Surface Science | 10.1016/j.apsusc.2017.04.158 |
| 2017 | - | MgO | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2016.11.043 |
| 2017 | CuO | CeO2 | Zeolite | Microporous and Mesoporous Materials | 10.1016/j.micromeso.2017.02.016 |
| 2017 | - | Zeolite | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2017.06.083 |
| 2017 | Co | ZnO | - | Ceramics International | 10.1016/j.ceramint.2017.06.157 |
| 2017 | Pd | TiO2 | SnO2 | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2017.02.017 |
| 2017 | Pd | Fe2O3 | - | Fuel Processing Technology | 10.1016/j.fuproc.2017.02.037 |
| 2017 | CuO | CeO2 | - | Journal of Power Sources | 10.1016/j.jpowsour.2017.01.127 |
| 2017 | Mn | CeO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2017.03.049 |
| 2017 | Co | Co3O4 | - | Chemical Physics Letters | 10.1016/j.cplett.2017.02.085 |
| 2017 | Au | TiO2 | - | Catalysis Today | 10.1016/j.cattod.2016.05.056 |
| 2017 | CuO | CeO2 | - | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2017.02.088 |
| 2017 | CuO | CeO2 | - | Journal of Rare Earths | 10.1016/j.jre.2017.05.015 |
| 2017 | Pd | Al2O3 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2017.02.038 |
| 2017 | Pt | TiO2 | - | Molecular Catalysis | 10.1016/j.mcat.2017.01.014 |
| 2017 | - | CeO2 | Other | Catalysis Today | 10.1016/j.cattod.2017.06.017 |
| 2017 | - | Al2O3 | SnO2 | Applied Surface Science | 10.1016/j.apsusc.2017.01.058 |
| 2017 | Ag | Zeolite | - | Fuel | 10.1016/j.fuel.2016.10.037 |
| 2017 | Au | TiO2 | - | Applied Surface Science | 10.1016/j.apsusc.2016.10.076 |
| 2017 | - | Carbon | - | Molecular Catalysis | 10.1016/j.molcata.2016.12.007 |
| 2017 | Ag | SiO2 | - | Microporous and Mesoporous Materials | 10.1016/j.micromeso.2017.01.016 |
| 2017 | Pd, Rh | Al2O3 | - | Catalysis Today | 10.1016/j.cattod.2016.10.010 |
| 2017 | Au, Cu | SiO2 | - | Catalysis Today | 10.1016/j.cattod.2016.08.003 |
| 2017 | Pd | CeO2 | MnO | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2017.01.020 |
| 2017 | - | CeO2 | - | Catalysis Today | 10.1016/j.cattod.2016.04.016 |
| 2017 | Pd | Co3O4 | - | Applied Catalysis A: General | 10.1016/j.apcata.2016.12.021 |
| 2017 | Pt | CeO2 | - | Applied Catalysis A: General | 10.1016/j.apcata.2017.08.012 |
| 2017 | Mn | Co3O4 | - | Solid State Sciences | 10.1016/j.solidstatesciences.2017.07.006 |
| 2017 | Ni | ZrO2 | - | Applied Catalysis A: General | 10.1016/j.apcata.2017.02.001 |
| 2018 | - | SiO2 | Co3O4 | Microporous and Mesoporous Materials | 10.1016/j.micromeso.2017.07.016 |
| 2018 | Pt | Fe2O3 | - | Applied Catalysis A: General | 10.1016/j.apcata.2018.09.014 |
| 2018 | Pd | SiO2 | Al2O3 | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2018.06.059 |
| 2018 | Cu | CeO2 | - | Catalysis Today | 10.1016/j.cattod.2018.10.037 |
| 2018 | Cu -Ni | CeO2 | Al2O3 | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2018.12.127 |
| 2018 | Ru | TiO2 | ZrO2 | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2018.10.061 |
| 2018 | Ni | ZrO2 | - | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2018.06.173 |
| 2018 | - | ZrO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2018.03.001 |
| 2018 | Ni | ZrO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2018.06.045 |
| 2019 | Au | TiO2 | - | International Journal of Hydrogen Energy | 10.1016/j.ijhydene.2018.11.050 |
| 2019 | Cu | Co3O4 | - | Molecular Catalysis | 10.1016/j.mcat.2019.01.020 |
| 2019 | - | Other | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2018.12.022 |
| 2019 | Pt | Zeolite | - | Applied Catalysis A: General | 10.1016/j.apcata.2018.12.034 |
| 2019 | Ni | ZrO2 | - | Applied Catalysis B: Environmental | 10.1016/j.apcatb.2018.11.024 |
References
- Zhang, K.; Jiang, X. An investigation of fuel variability effect on bio-syngas combustion using uncertainty quantification. Fuel 2018, 220, 283–295. [Google Scholar] [CrossRef]
- Renzi, M.; Patuzzi, F.; Baratieri, M. Syngas feed of micro gas turbines with steam injection: Effects on performance, combustion and pollutants formation. Appl. Energy 2017, 206, 697–707. [Google Scholar] [CrossRef]
- Rosa, A. Chapter 10—Hydrogen Production. In Fundamentals of Renewable Energy Processes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 371–428. [Google Scholar]
- Mohammed, H.; Al-Othman, A.; Nancarrow, P.; Tawalbeh, M.; El Haj Assad, M. Direct hydrocarbon fuel cells: A promising technology for improving energy efficiency. Energy 2019, 172, 207–219. [Google Scholar] [CrossRef]
- Sengodan, S.; Lan, R.; Humphreys, J.; Du, D.; Xu, W.; Wang, H.; Tao, S. Advances in reforming and partial oxidation of hydrocarbons for hydrogen production and fuel cell applications. Renew. Sustain. Energy Rev. 2018, 82, 761–780. [Google Scholar] [CrossRef]
- Cifuentes, B.; Hernández, M.; Monsalve, S.; Cobo, M. Hydrogen production by steam reforming of ethanol on a RhPt/CeO2/SiO2 catalyst: Synergistic effect of the Si:Ce ratio on the catalyst performance. Appl. Catal. A Gen. 2016, 523, 283–293. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, J.; Jing, G.; Zhang, H.; Zeng, S.; Tian, X.; Zou, X.; Wen, J.; Su, H.; Zhong, C.J.; et al. Structural origin of high catalytic activity for preferential CO oxidation over CuO/CeO2 nanocatalysts with different shapes. Appl. Catal. B Environ. 2018, 239, 665–676. [Google Scholar] [CrossRef]
- Kou, T.; Si, C.; Gao, Y.; Frenzel, J.; Wang, H.; Yan, X.; Bai, Q.; Eggeler, G.; Zhang, Z. Large-scale synthesis and catalytic activity of nanoporous Cu-O system towards CO oxidation. RSC Adv. 2014, 4, 65004–65011. [Google Scholar] [CrossRef]
- Kou, T.; Si, C.; Pinto, J.; Ma, C.; Zhang, Z. Dealloying assisted high-yield growth of surfactant-free 〈110〉 highly active Cu-doped CeO2 nanowires for low-temperature CO oxidation. Nanoscale 2017, 9, 8007–8014. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Ercolino, G.; Specchia, S.; Specchia, V. Final step for CO syngas clean-up: Comparison between CO-PROX and CO-SMET processes. Int. J. Hydrogen Energy 2014, 39, 18109–18119. [Google Scholar] [CrossRef]
- Rossetti, I.; Compagnoni, M.; Torli, M. Process simulation and optimization of H2 production from ethanol steam reforming and its use in fuel cells. 2. Process analysis and optimization. Chem. Eng. J. 2015, 281, 1036–1044. [Google Scholar] [CrossRef]
- Gubán, D.; Tompos, A.; Bakos, I.; Vass, Á.; Pászti, Z.; Szabó, E.G.; Sajó, I.E.; Borbáth, I. Preparation of CO-tolerant anode electrocatalysts for polymer electrolyte membrane fuel cells. Int. J. Hydrogen Energy 2017, 42, 13741–13753. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, H.; Basu, S. Regeneration of CO poisoned Pt black anode catalyst in PEMFC using break-in procedure and KMnO4 solution. Int. J. Hydrogen Energy 2017, 42, 23814–23820. [Google Scholar] [CrossRef]
- Devrim, Y.; Albostan, A.; Devrim, H. Experimental investigation of CO tolerance in high temperature PEM fuel cells. Int. J. Hydrogen Energy 2018, 43, 18672–18681. [Google Scholar] [CrossRef]
- Kugai, J.; Fox, E.B.; Song, C. Kinetic characteristics of oxygen-enhanced water gas shift on CeO2-supported Pt–Cu and Pd–Cu bimetallic catalysts. Appl. Catal. A Gen. 2015, 497, 31–41. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, Z.G. Preferential CO oxidation on Ru/Al2O3 catalyst: An investigation by considering the simultaneously involved methanation. J. Power Sources 2006, 157, 64–77. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Laguna, O.H.; Centeno, M.A.; Odriozola, J.A. WGS and CO-PrOx reactions using gold promoted copper-ceria catalysts: “bulk CuO–CeO2 vs. CuO–CeO2/Al2O3 with low mixed oxide content”. Appl. Catal. B Environ. 2016, 197, 67–72. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, J.-C.; Li, W.-Z.; Chen, B.-H. Coke-resistant Au–Ni/MgAl2O4 catalyst for direct methanation of syngas. J. Energy Chem. 2019, 39, 198–207. [Google Scholar] [CrossRef]
- Reina, T.R.R.; Ivanova, S.; Centeno, M.A.A.; Odriozola, J.A.A. Catalytic screening of Au/CeO2-MOx/Al2O3 (M = La, Ni, Cu, Fe, Cr, Y) in the CO-PrOx reaction. Int. J. Hydrogen Energy 2015, 40, 1782–1788. [Google Scholar] [CrossRef]
- Cifuentes, B.; Bustamante, F.; Conesa, J.A.; Córdoba, L.F.; Cobo, M. Fuel-cell grade hydrogen production by coupling steam reforming of ethanol and carbon monoxide removal. Int. J. Hydrogen Energy 2018, 43, 17216–17229. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Gao, F.; Dong, L. The application of incorporation model in γ–Al2O3-supported single and dual metal oxide catalysts: A review. Cuihua Xuebao/Chin. J. Catal. 2013, 34, 1975–1985. [Google Scholar] [CrossRef]
- MacIel, C.G.; Profeti, L.P.R.; Assaf, E.M.; Assaf, J.M. Hydrogen purification for fuel cell using CuO/CeO2–Al2O3 catalyst. J. Power Sources 2011, 196, 747–753. [Google Scholar] [CrossRef]
- Águila, G.; Gracia, F.; Araya, P. CuO and CeO2 catalysts supported on Al2O3, ZrO2, and SiO2 in the oxidation of CO at low temperature. Appl. Catal. A Gen. 2008, 343, 16–24. [Google Scholar] [CrossRef]
- Zhao, Z.; Lin, X.; Jin, R.; Dai, Y.; Wang, G. High catalytic activity in CO PROX reaction of low cobalt-oxide loading catalysts supported on nano-particulate CeO2–ZrO2 oxides. Catal. Commun. 2011, 12, 1448–1451. [Google Scholar] [CrossRef]
- Moretti, E.; Rodríguez-Aguado, E.; Molina, A.I.; Rodríguez-Castellón, E.; Talon, A.; Storaro, L. Sustainable photo-assisted CO oxidation in H2-rich stream by simulated solar light response of Au nanoparticles supported on TiO2. Catal. Today 2018, 304, 135–142. [Google Scholar] [CrossRef]
- Li, S.; Tang, H.; Gong, D.; Ma, Z.; Liu, Y. Loading Ni/La2O3 on SiO2 for CO methanation from syngas. Catal. Today 2017, 297, 298–307. [Google Scholar] [CrossRef]
- Liu, S.; Wu, X.; Weng, D.; Ran, R. Ceria-based catalysts for soot oxidation: A review. J. Rare Earths 2015, 33, 567–590. [Google Scholar] [CrossRef]
- Martínez, L.M.; Laguna, O.H.; López-Cartes, C.; Centeno, M.A. Synthesis and characterization of Rh/MnO2-CeO2/Al2O3 catalysts for CO-PrOx reaction. Mol. Catal. 2017, 440, 9–18. [Google Scholar] [CrossRef]
- Gamboa-Rosales, N.K.; Ayastuy, J.L.; González-Marcos, M.P.; Gutiérrez-Ortiz, M.A. Effect of Au promoter in CuO/CeO2 catalysts for the oxygen-assisted WGS reaction. Catal. Today 2011, 176, 63–71. [Google Scholar] [CrossRef]
- Destro, P.; Marras, S.; Manna, L.; Colombo, M.; Zanchet, D. AuCu alloy nanoparticles supported on SiO2: Impact of redox pretreatments in the catalyst performance in CO oxidation. Catal. Today 2017, 282, 105–110. [Google Scholar] [CrossRef]
- Destro, P.; Kokumai, T.M.; Scarpellini, A.; Pasquale, L.; Manna, L.; Colombo, M.; Zanchet, D. The Crucial Role of the Support in the Transformations of Bimetallic Nanoparticles and Catalytic Performance. ACS Catal. 2017, 8, 1031–1037. [Google Scholar] [CrossRef]
- Ayastuy, J.L.; Gurbani, A.; Gutiérrez-Ortiz, M.A. Effect of calcination temperature on catalytic properties of Au/Fe2O3 catalysts in CO-PROX. Int. J. Hydrogen Energy 2016, 41, 19546–19555. [Google Scholar] [CrossRef]
- Lakshmanan, P.; Park, E. Preferential CO Oxidation in H2 over Au/La2O3/Al2O3 Catalysts: The Effect of the Catalyst Reduction Method. Catalysts 2018, 8, 183–193. [Google Scholar] [CrossRef]
- Córdoba, L.F.; Martínez-Hernández, A. Preferential oxidation of CO in excess of hydrogen over Au/CeO2–ZrO2 catalysts. Int. J. Hydrogen Energy 2015, 40, 16192–16201. [Google Scholar] [CrossRef]
- Arévalo, J.D.; Martinez-Hernández, Á.; Vargas, J.C.; Córdoba, L.F. Hydrogen Production and Purification by Bioethanol Steam Reforming and Preferential Oxidation of CO. Tecciencia 2018, 13, 55–64. [Google Scholar]
- Chen, Z.; Pursell, C.J.; Chandler, B.D.; Saavedra, J.; Whittaker, T.; Rioux, R.M. Controlling activity and selectivity using water in the Au–catalysed preferential oxidation of CO in H2. Nat. Chem. 2016, 8, 584–589. [Google Scholar]
- Dasireddy, V.D.B.C.; Valand, J.; Likozar, B. PROX reaction of CO in H2/H2O/CO2 Water–Gas Shift (WGS) feedstocks over Cu–Mn/Al2O3 and Cu–Ni/Al2O3 catalysts for fuel cell applications. Renew. Energy 2018, 116, 75–87. [Google Scholar] [CrossRef]
- Zhan, W.; Wang, J.; Wang, H.; Zhang, J.; Liu, X.; Zhang, P.; Chi, M.; Guo, Y.; Guo, Y.; Lu, G.; et al. Crystal Structural Effect of AuCu Alloy Nanoparticles on Catalytic CO Oxidation. J. Am. Chem. Soc. 2017, 139, 8846–8854. [Google Scholar] [CrossRef]
- Reina, T.R.; Megías-Sayago, C.; Florez, A.P.; Ivanova, S.; Centeno, M.Á.; Odriozola, J.A. H2 oxidation as criterion for PrOx catalyst selection: Examples based on Au–CoOx-supported systems. J. Catal. 2015, 326, 161–171. [Google Scholar] [CrossRef]
- Yung, M.M.; Zhao, Z.; Woods, M.P.; Ozkan, U.S. Preferential oxidation of carbon monoxide on CoOx/ZrO2. J. Mol. Catal. A Chem. 2008, 279, 1–9. [Google Scholar] [CrossRef]
- Fonseca, J.D.; Ferreira, H.S.; Bion, N.; Pirault-Roy, L.; do Carmo Rangel, M.; Duprez, D.; Epron, F. Cooperative effect between copper and gold on ceria for CO-PROX reaction. Catal. Today 2012, 180, 34–41. [Google Scholar] [CrossRef]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-Situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Jhalani, A.; Schmidt, L.D. Preferential CO oxidation in the presence of H2, H2O and CO2 at short contact-times. Catal. Lett. 2005, 104, 103–110. [Google Scholar] [CrossRef]
- Tanaka, K.I.; He, H.; Shou, M.; Shi, X. Mechanism of highly selective low temperature PROX reaction of CO in H2: Oxidation of CO via HCOO with OH. Catal. Today 2011, 175, 467–470. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, L.; Zhu, J.; Han, B.; Zhao, L.; Yu, H.; Deng, Z.; Shi, W. Study on different CeO2 structure stability during ethanol steam reforming reaction over Ir/CeO2 nanocatalysts. Appl. Catal. A Gen. 2018, 564, 226–233. [Google Scholar] [CrossRef]
- Divins, N.J.; Casanovas, A.; Xu, W.; Senanayake, S.D.; Wiater, D.; Trovarelli, A.; Llorca, J. The influence of nano-architectured CeOx supports in RhPd/CeO2 for the catalytic ethanol steam reforming reaction. Catal. Today 2015, 253, 99–105. [Google Scholar] [CrossRef]
- Li, X.; Fang, S.S.; Teo, J.; Foo, Y.L.; Borgna, A.; Lin, M.; Zhong, Z. Activation and deactivation of Au–Cu/SBA-15 catalyst for preferential oxidation of CO in H2-Rich Gas. ACS Catal. 2012, 2, 360–369. [Google Scholar] [CrossRef]
- Laguna, O.H.; Hernández, W.Y.; Arzamendi, G.; Gandía, L.M.; Centeno, M.A.; Odriozola, J.A. Gold supported on CuOx/CeO2 catalyst for the purification of hydrogen by the CO preferential oxidation reaction (PROX). Fuel 2014, 118, 176–185. [Google Scholar] [CrossRef]
- Redina, E.A.; Greish, A.A.; Mishin, I.V.; Kapustin, G.I.; Tkachenko, O.P.; Kirichenko, O.A.; Kustov, L.M. Selective oxidation of ethanol to acetaldehyde over Au–Cu catalysts prepared by a redox method. Catal. Today 2015, 241, 246–254. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, L.; Lu, J.; Chen, R.; Lei, Y.; Chen, K.; Han, C.; He, S.; Wan, G.; Luo, Y. A solvent-free method to rapidly synthesize CuO–CeO2 catalysts to enhance their CO preferential oxidation: Effects of Cu loading and calcination temperature. Mol. Catal. 2017, 443, 241–252. [Google Scholar] [CrossRef]
- Wang, X.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martínez-Arias, A.; Fernández-García, M. In Situ Studies of the Active Sites for the Water Gas Shift Reaction over Cu–CeO2 Catalysts: Complex Interaction between Metallic Copper and Oxygen Vacancies of Ceria. J. Phys. Chem. B 2006, 110, 428–434. [Google Scholar] [CrossRef]
- Wang, F.; Büchel, R.; Savitsky, A.; Zalibera, M.; Widmann, D.; Pratsinis, S.E.; Lubitz, W.; Schüth, F. In Situ EPR Study of the Redox Properties of CuO–CeO2 Catalysts for Preferential CO Oxidation (PROX). ACS Catal. 2016, 6, 3520–3530. [Google Scholar] [CrossRef]
- Mallát, T.; Szabó, S.; Petró, J.; Mendioroz, S.; Folgado, M.A. Real and apparent dispersion of carbon supported palladium-cobalt catalysts. Appl. Catal. 1989, 53, 29–40. [Google Scholar] [CrossRef]
- Barbato, P.S.; Colussi, S.; Di Benedetto, A.; Landi, G.; Lisi, L.; Llorca, J.; Trovarelli, A. Origin of High Activity and Selectivity of CuO/CeO2 Catalysts Prepared by Solution Combustion Synthesis in CO-PROX Reaction. J. Phys. Chem. C 2016, 120, 13039–13048. [Google Scholar] [CrossRef]
- Schüth, F.; Ward, M.D.; Buriak, J.M. Common Pitfalls of Catalysis Manuscripts Submitted to Chemistry of Materials. Chem. Mater. 2018, 30, 3599–3600. [Google Scholar] [CrossRef]
- Cifuentes, B.; Valero, M.; Conesa, J.; Cobo, M. Hydrogen Production by Steam Reforming of Ethanol on Rh-Pt Catalysts: Influence of CeO2, ZrO2, and La2O3 as Supports. Catalysts 2015, 5, 1872–1896. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Padmasri, A.H.; Raju, B.D.; Rao, K.S.R. Vapor phase selective hydrogenation of furfural to furfuryl alcohol over Cu-MgO coprecipitated catalysts. J. Mol. Catal. A Chem. 2007, 265, 90–97. [Google Scholar] [CrossRef]
- Li, J.; Zhan, Y.; Lin, X.; Zheng, Q. Influence of Calcination Temperature on Properties of Au/Fe2O3 Catalysts for Low Temperature Water Gas Shift Reaction. Acta Phys.-Chim. Sin. 2008, 24, 932–938. [Google Scholar] [CrossRef]
- Liu, F.; Xiao, Y.; Sun, X.; Qin, G.; Song, X.; Liu, Y. Synergistic catalysis over hollow CeO2–CaO–ZrO2 nanostructure for polycarbonate methanolysis with methanol. Chem. Eng. J. 2019, 369, 205–214. [Google Scholar] [CrossRef]
- Chen, W.; Ran, R.; Weng, D.; Wu, X.; Zhong, J.; Han, S. Influence of morphology on basicity of CeO2 and its use in 2-chloroethyl ethyl sulfide degradation. J. Rare Earths 2017, 35, 970–976. [Google Scholar] [CrossRef]
- Malik, A.S.; Zaman, S.F.; Al-Zahrani, A.A.; Daous, M.A.; Driss, H.; Petrov, L.A. Selective hydrogenation of CO2 to CH3OH and in-depth DRIFT analysis for PdZn/ZrO2 and CaPdZn/ZrO2 catalysts. Catal. Today 2019, in press. [Google Scholar] [CrossRef]
- Zhou, W.; Ma, Z.; Guo, S.; Wang, M.; Wang, J.; Xia, M.; Jia, L.; Hou, B.; Li, D.; Zhao, Y. Comparative study of CO adsorption on zirconia polymorphs with DRIFT and transmission FT-IR spectroscopy. Appl. Surf. Sci. 2018, 427, 867–873. [Google Scholar] [CrossRef]
- Agarwal, S.; Mojet, B.L.; Lefferts, L.; Datye, A.K. Ceria Nanoshapes-Structural and Catalytic Properties. In Catalysis by Materials with Well-Defined Structures; Elsevier: Amsterdam, The Netherlands, 2015; pp. 31–70. [Google Scholar]
- Leba, A.; Davran-Candan, T.; Önsan, Z.I.; Yildirim, R. DRIFTS study of selective CO oxidation over Au/γAl2O3 catalyst. Catal. Commun. 2012, 29, 6–10. [Google Scholar] [CrossRef]
- Fernández-García, S.; Collins, S.E.; Tinoco, M.; Hungría, A.B.; Calvino, J.J.; Cauqui, M.A.; Chen, X. Influence of {111} nanofaceting on the dynamics of CO adsorption and oxidation over Au supported on CeO2 nanocubes: An operando DRIFT insight. Catal. Today 2019, 336, 90–98. [Google Scholar] [CrossRef]
- Gamarra, D.; Martínez-Arias, A. Preferential oxidation of CO in rich H2 over CuO/CeO2: Operando-DRIFTS analysis of deactivating effect of CO2 and H2O. J. Catal. 2009, 263, 189–195. [Google Scholar] [CrossRef]
- Moretti, E.; Lenarda, M.; Storaro, L.; Talon, A.; Frattini, R.; Polizzi, S.; Rodríguez-Castellón, E.; Jiménez-López, A. Catalytic purification of hydrogen streams by PROX on Cu supported on an organized mesoporous ceria-modified alumina. Appl. Catal. B Environ. 2007, 72, 149–156. [Google Scholar] [CrossRef]
- Polster, C.S.; Nair, H.; Baertsch, C.D. Study of active sites and mechanism responsible for highly selective CO oxidation in H2 rich atmospheres on a mixed Cu and Ce oxide catalyst. J. Catal. 2009, 266, 308–319. [Google Scholar] [CrossRef]
- Suzuki, Y.; Hayakawa, K.; Fukuhara, C.; Watanabe, R.; Kawasaki, W. A novel nickel-based structured catalyst for CO2 methanation: A honeycomb-type Ni/CeO2 catalyst to transform greenhouse gas into useful resources. Appl. Catal. A Gen. 2016, 532, 12–18. [Google Scholar]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Kim, T.W.; Park, E.D. CO and CO2 methanation over supported Ni catalysts. Catal. Today 2017, 293–294, 89–96. [Google Scholar] [CrossRef]
- Sahoo, D.R.; Vajpai, S.; Patel, S.; Pant, K.K. Kinetic modeling of steam reforming of ethanol for the production of hydrogen over Co/Al2O3 catalyst. Chem. Eng. J. 2007, 125, 139–147. [Google Scholar] [CrossRef]
- Cifuentes, B.; Bustamante, F.; Cobo, M. Data for: Single and dual metal oxides as promising supports for carbon monoxide removal from an actual syngas. Mendeley Datasets 2019, 1–10. [Google Scholar] [CrossRef]








| Catalysta | Minimum CO Concentration in Outlet Gas (ppm)b | H/M Index | BET Surface Area (m2/gcat) | OSC in AC Samples (μmol O2/gcat) | OSCC at 300 °C (μmol O2/gcat) | |||
|---|---|---|---|---|---|---|---|---|
| AC | Spent | 100 °C | 300 °C | Fresh | Spent | |||
| AuCu/CeO2 | 75 at 210 °C | 0.9 | 60 | 58 (U) | 41 | 91 | 230 | 121 (U) |
| 50 (S) | 93 (S) | |||||||
| AuCu/SiO2 | 8320 at 240 °C | 0.7 | 364 | 277 (U) | 21 | 37 | 45 | 41 (U) |
| AuCu/ZrO2 | 507 at 225 °C | 0.8 | 58 | 47 (U) | 39 | 76 | 185 | 84 (U) |
| AuCu/Al2O3 | 745 at 180 °C | 0.8 | 90 | 65 (U) | 31 | 35 | 75 | 41 (U) |
| AuCu/La2O3 | 5365 at 225 °C | 0.4 | 19 | 18 (U) | 21 | 41 | 90 | 24 (U) |
| AuCu/Fe2O3 | 9416 at 140 °C | 0.4 | 16 | 5 (U) | NR | NR | NR | NR |
| AuCu/CeO2-SiO2 | 861 at 230 °C | 1.6 | 110 | 75 (U) | 34 | 78 | 146 | 121 (U) |
| 74 (S) | 126 (S) | |||||||
| AuCu/CeO2-ZrO2 | 941 at 210 °C | 0.9 | 42 | 30 (U) | 42 | 94 | 210 | 162 (U) |
| AuCu/CeO2-Al2O3 | 1521 at 260 °C | 1.2 | 65 | 56 (U) | 32 | 79 | 155 | 121 (U) |
| Catalyst | Syngas Type | H2/CO | T (°C) | CO Conversion (%) | H2 Loss (%) | Ref. |
|---|---|---|---|---|---|---|
| AuCu/CeO2 | Synthetic | 30 | 220 | 90 | 2 | [29] |
| AuCu/SBA-15 | Synthetic | >50 | 25 | ~100 | ~ 5a | [47] |
| Au/CuO-CeO2/Al2O3 | Synthetic | 4.5 | 350 | ~75 | NR | [17] |
| Au/CeO2-CuO2/Al2O3 | Synthetic | 50 | 110 | ~95 | ~3a | [19] |
| Au/Al2O3 | Synthetic | >50 | 80 | ~99 | ~2a | [36] |
| Au/CeO2-ZrO2 | Actual | 30 | 100 | 99 | ~2a | [35] |
| AuCu/CeO2 | Actual | 4 | 210 | 99 | 17 | This work |
| AuCu/CeO2-SiO2 | Actual | 4 | 230 | 97 | 19 | This work |
| Catalyst | Total Weight Loss (%) | Weight Loss of Spent Catalyst Samples by Temperature Intervals (mg of C/gcat*h) | |||
|---|---|---|---|---|---|
| AC | Spent | 40–250 °C | 250–600 °C | 600–1000 °C | |
| AuCu/CeO2 | 0.7 | 3.8 (U) | 17.1 (U) | 6.8 (U) | 11.8 (U) |
| 5.6 (S) | 14.5 (S) | 15.1 (U) | 18.1 (U) | ||
| AuCu/SiO2 | 0.3 | 3.7 (U) | 35.5 (U) | 3.4 (U) | 3.9 (U) |
| AuCu/ZrO2 | 0.9 | 1.6 (U) | 9.2 (U) | 2.1 (U) | 4.7 (U) |
| AuCu/Al2O3 | 0.5 | 3.7 (U) | 28.9 (U) | 2.1 (U) | 9.4 (U) |
| AuCu/La2O3 | 0.6 | 2.1 (U) | 9.2 (U) | 7.5 (U) | 2.4 (U) |
| AuCu/Fe2O3 | −0.3 | 2.5 (U) | 18.4 (U) | 8.9 (U) | NR |
| AuCu/CeO2-SiO2 | −0.9 | 0.3 (U) | 3.9 (U) | 4.8 (U) | NR |
| 1.3 (S) | 15.8 (S) | 4.1 (U) | NR | ||
| AuCu/CeO2-ZrO2 | 0.5 | 1.7 (U) | 9.2 (U) | 6.2 (U) | 0.8 (U) |
| AuCu/CeO2-Al2O3 | 0.6 | 2.6 (U) | 17.1 (U) | 2.7 (U) | 7.1 (U) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cifuentes, B.; Bustamante, F.; Cobo, M. Single and Dual Metal Oxides as Promising Supports for Carbon Monoxide Removal from an Actual Syngas: The Crucial Role of Support on the Selectivity of the Au–Cu System. Catalysts 2019, 9, 852. https://doi.org/10.3390/catal9100852
Cifuentes B, Bustamante F, Cobo M. Single and Dual Metal Oxides as Promising Supports for Carbon Monoxide Removal from an Actual Syngas: The Crucial Role of Support on the Selectivity of the Au–Cu System. Catalysts. 2019; 9(10):852. https://doi.org/10.3390/catal9100852
Chicago/Turabian StyleCifuentes, Bernay, Felipe Bustamante, and Martha Cobo. 2019. "Single and Dual Metal Oxides as Promising Supports for Carbon Monoxide Removal from an Actual Syngas: The Crucial Role of Support on the Selectivity of the Au–Cu System" Catalysts 9, no. 10: 852. https://doi.org/10.3390/catal9100852