Copper Containing Molecular Systems in Electrocatalytic Water Oxidation—Trends and Perspectives



Abstract
:
1. Introduction
1.1. On the Use of Copper in AP Systems
1.2. Methodological Approach to Cu-Based Molecular WOCs
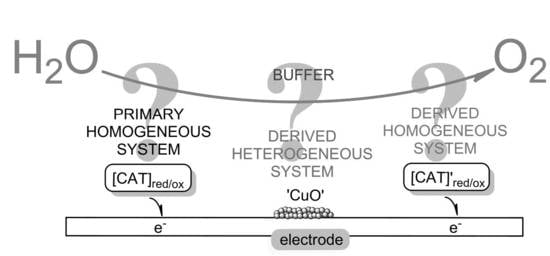
1.3. Cross-Linkages Between Homogeneous and Heterogeneous Catalysis–Molecular Systems Trespassing Borders
2. Types of Molecular Catalysts and Associated Mechanisms
2.1. Single-Site Catalysis
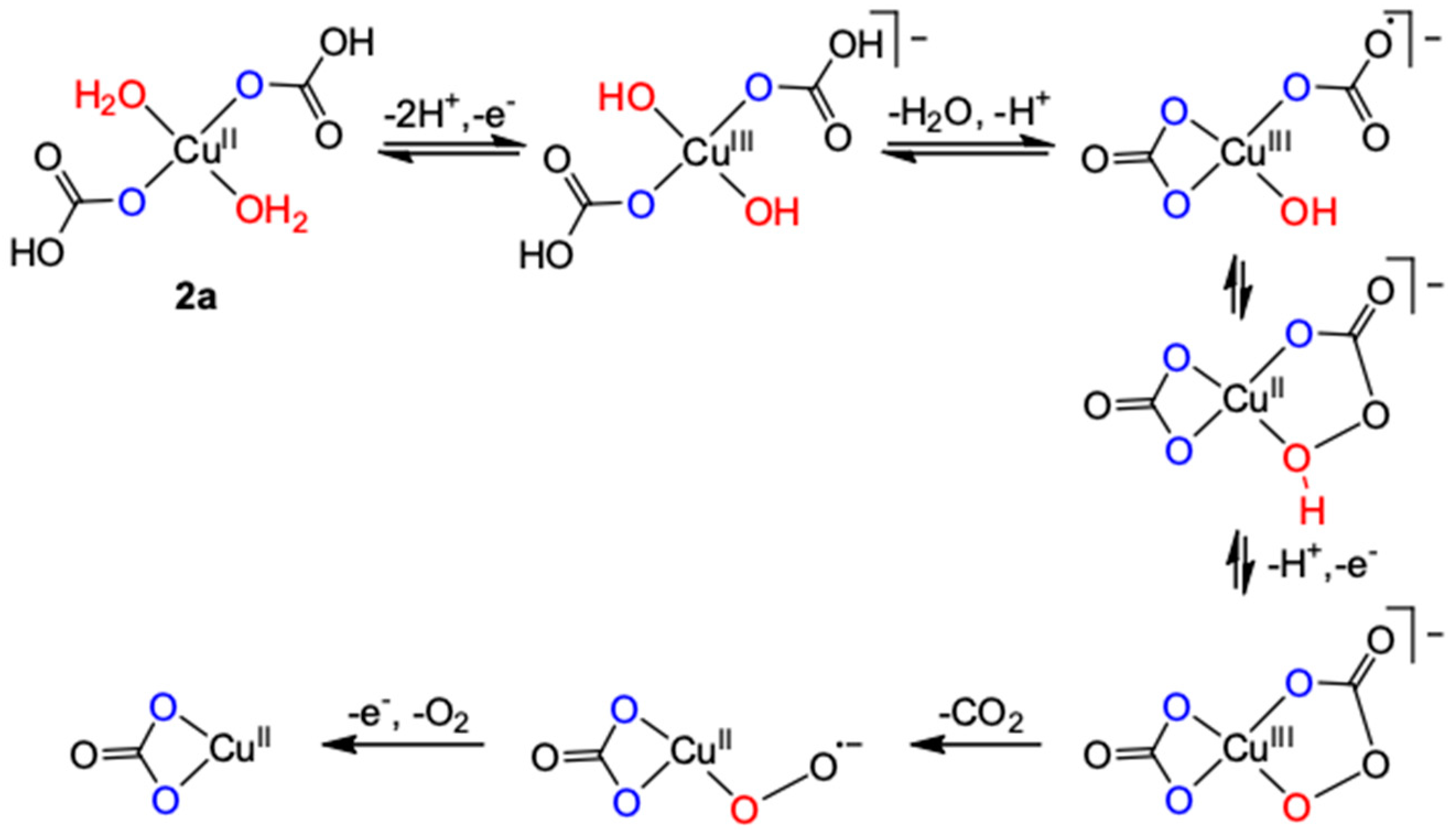
2.1.1. Inorganic Ligands
2.1.2. Organic Ligands
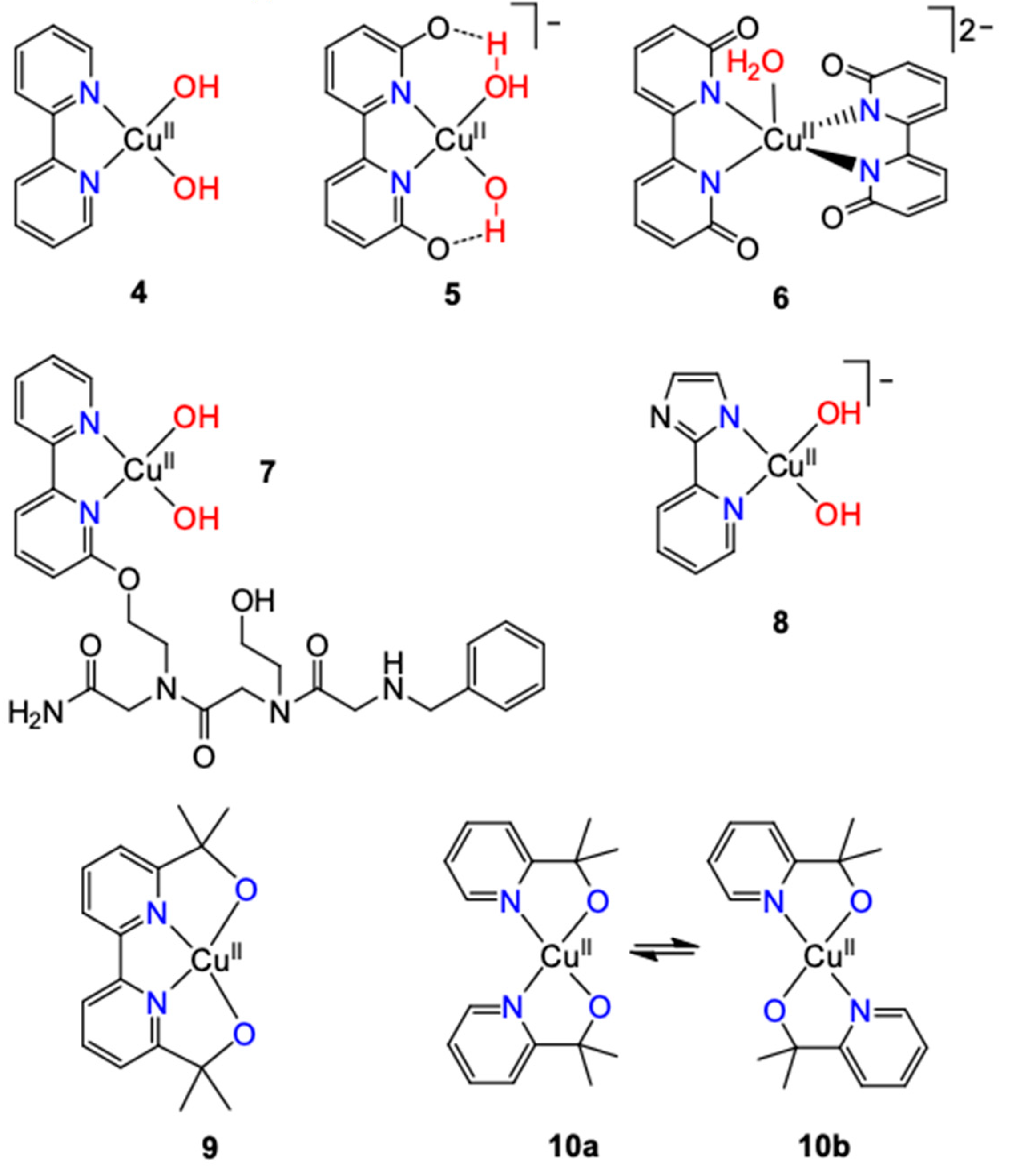
2.1.2.1. 2,2′-Bipyridine and Related Ligands
2.1.2.2. Ligands with Amine Donor Groups
2.1.2.3. Mixed Ligand Compositions
2.1.2.4. Redox Non-Innocent Ligands
2.1.2.5. Peptide Ligands
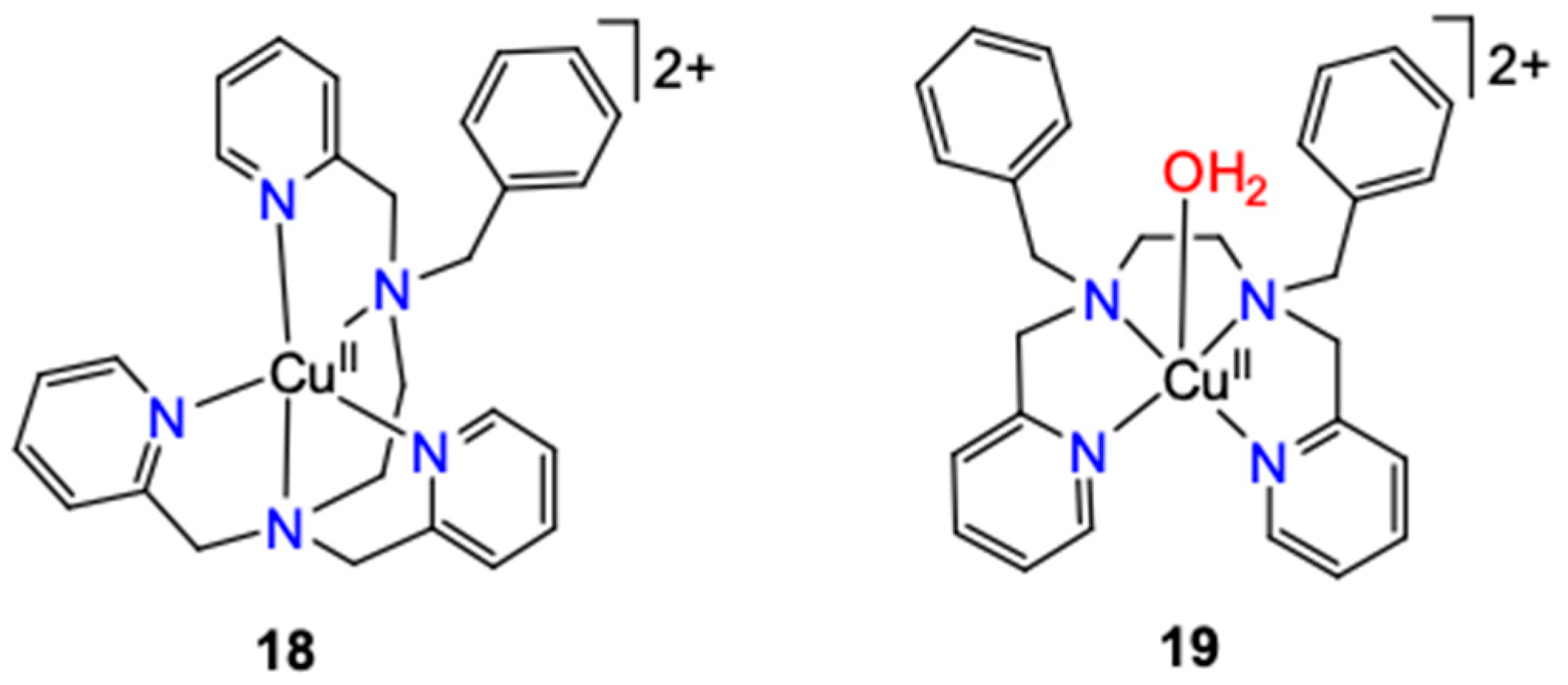
2.1.2.6. Tripodal Polypyridylmethylamine Ligands
2.1.2.7. Assisting Inorganic Co-Ligand
2.1.3. Overview of the Mechanistic Scenarios with Respect to Single-Site Catalysts
2.2. Dicopper Catalysis
2.3. Multi-Copper Catalysis
2.4. Cu Complexes as Precursors
3. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AP | Artificial photosynthesis |
| BDD | Boron doped diamond |
| CA | Chronoamperometry |
| CD | Circular dichrosim |
| CP | Chronopotentiometry |
| CPE | Controlled potential electrolysis |
| CV | Cyclic voltammetry |
| DFT | Density functional theory |
| DLS | Dynamic light scattering |
| DPV | Differential pulse voltammetry |
| η | Overpotential of OER, defined as η = (Ecat − 1.23 V + 0.059pH) |
| Ecat | Potential of the catalytic reaction |
| EC-OWLS | Electrochemical optical waveguide lightmode spectroscopy |
| EIS | Electrochemical impedance spectroscopy |
| EPR | Electron paramagnetic resonance |
| ESI-MS | Electrospray ionization mass spectrometry |
| EXAFS | Extended X-ray absorption fine structure spectroscopy |
| FTO | Fluorine doped tin oxide |
| GC | Glassy carbon |
| HER | Hydrogen Evolving Reaction |
| HYSCORE | Hyperfine sublevel correlation |
| icat, id | catalytic and diffusion controlled current, respectively |
| jcat, jd | catalytic and diffusion controlled current density, respectively |
| I2M | Interaction of two M=O units |
| ITO | Indium tin oxide |
| KIE | Kinetic isotope effect |
| kobs | pseudo-first order catalytic rate constant, s−1 (≡ TOF, kcat) |
| LSV | Linear sweep voltammetry |
| NADP(H) | Nicotinamide adenine dinucleotide phosphate |
| NHE/SHE | Normal/Standard hydrogen electrode |
| OAc− | Acetate anion |
| OEC | Oxygen evolving center |
| OER | Oxygen evolving reaction |
| OTf− | Trifluoromethyl sulfonate anion |
| PCET | Proton coupled electron transfer |
| PS II | Photosystem II |
| RHE | Reversible hydrogen electrode |
| SEM-EDS | Scanning electron microscopy, energy dispersive spectroscopy |
| SET-WNA | Single electron transfer water nucleophilic attack |
| SP-4 | Square planar coordination geometry |
| SPBY-5 | Square based bipyramidal coordination geometry |
| SWV | Square wave voltammetry |
| TPBY-5 | Trigonal bipyramidal coordination geometry |
| TOF | Turnover frequency, s−1 |
| TON | Tornover number, dimensionless |
| XANES | X-ray absorption near edge structure |
| XPS | X-Ray photoelectron spectroscopy |
| WNA | Water nucleophilic attack |
| WOC | Water oxidation (electro)catalyst |
References
- Bard, A.J.; Fox, M.A. Artificial Photosynthesis: Solar Splitting of Water to Hydrogen and Oxygen. Acc. Chem. Res. 1995, 28, 141–145. [Google Scholar] [CrossRef]
- Meyer, T.J. Chemical approaches to artificial photosynthesis. Acc. Chem. Res. 1989, 22, 163–170. [Google Scholar] [CrossRef]
- McConnell, I.; Li, G.; Brudvig, G.W. Energy Conversion in Natural and Artificial Photosynthesis. Chem. Biol. 2010, 17, 434–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, A. Energy’s Tricky Tradeoffs. Science 2010, 329, 786–787. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Karkas, M.D.; Akermark, B. Water oxidation using earth-abundant transition metal catalysts: Opportunities and challenges. Dalton Trans. 2016, 45, 14421–14461. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef]
- Hunter, B.M.; Gray, H.B.; Müller, A.M. Earth-Abundant Heterogeneous Water Oxidation Catalysts. Chem. Rev. 2016, 116, 14120–14136. [Google Scholar] [CrossRef]
- Li, J.; Güttinger, R.; Moré, R.; Song, F.; Wan, W.; Patzke, G.R. Frontiers of water oxidation: The quest for true catalysts. Chem. Soc. Rev. 2017, 46, 6124–6147. [Google Scholar] [CrossRef]
- Suen, N.-T.; Hung, S.-F.; Quan, Q.; Zhang, N.; Xu, Y.-J.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef]
- Wang, N.; Zheng, H.; Zhang, W.; Cao, R. Mononuclear first-row transition-metal complexes as molecular catalysts for water oxidation. Chin. J. Catal. 2018, 39, 228–244. [Google Scholar] [CrossRef]
- Kondo, M.; Masaoka, S. Water Oxidation Catalysts Constructed by Biorelevant First-row Metal Complexes. Chem. Lett. 2016, 45, 1220–1231. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lei, Y.-J.; Xin, Z.-J.; Lu, Y.-B.; Wang, H.-Y. Water splitting based on homogeneous copper molecular catalysts. J. Photochem. Photobiol. Chem. 2018, 355, 141–151. [Google Scholar] [CrossRef]
- Wang, J.-W.; Zhong, D.-C.; Lu, T.-B. Artificial photosynthesis: Catalytic water oxidation and CO2 reduction by dinuclear non-noble-metal molecular catalysts. Coord. Chem. Rev. 2018, 377, 225–236. [Google Scholar] [CrossRef]
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCafferty, D.G.; Meyer, T.J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef] [PubMed]
- Scholz, F.; Bond, A.M.; Compton, R.G.; Fiedler, D.A.; Inzelt, G.; Kahlert, H.; Komorsky-Lovrić, Š.; Lohse, H.; Lovrić, M.; Marken, F.; et al. (Eds.) Electroanalytical Methods; Springer: Berlin/Heidelberg, Germany, 2010; ISBN 978-3-642-02914-1. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001; ISBN 978-0-471-04372-0. [Google Scholar]
- Artero, V.; Fontecave, M. Solar fuels generation and molecular systems: Is it homogeneous or heterogeneous catalysis? Chem. Soc. Rev. 2013, 42, 2338–2356. [Google Scholar] [CrossRef]
- Barnett, S.M.; Goldberg, K.I.; Mayer, J.M. A soluble copper–bipyridine water-oxidation electrocatalyst. Nat. Chem. 2012, 4, 498–502. [Google Scholar] [CrossRef]
- Zhu, L.; Du, J.; Zuo, S.; Chen, Z. Cs(I) Cation Enhanced Cu(II) Catalysis of Water Oxidation. Inorg. Chem. 2016, 55, 7135–7140. [Google Scholar] [CrossRef]
- Chen, Z.; Meyer, T.J. Copper(II) Catalysis of Water Oxidation. Angew. Chem. Int. Ed. 2013, 52, 700–703. [Google Scholar] [CrossRef]
- Winikoff, S.G.; Cramer, C.J. Mechanistic analysis of water oxidation catalyzed by mononuclear copper in aqueous bicarbonate solutions. Catal. Sci. Technol. 2014, 4, 2484–2489. [Google Scholar] [CrossRef]
- Mizrahi, A.; Maimon, E.; Cohen, H.; Kornweitz, H.; Zilbermann, I.; Meyerstein, D. Mechanistic Studies on the Role of [CuII(CO3)n]2−2n as a Water Oxidation Catalyst: Carbonate as a Non-Innocent Ligand. Chem. Eur. J. 2018, 24, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, E.A.; Tolman, W.B. Reactivity of Dioxygen−Copper Systems. Chem. Rev. 2004, 104, 1047–1076. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-H.; Wang, J.-W.; Sahoo, P.; Zhong, D.-C.; Lu, T.-B. Electrocatalytic water oxidation by Cu(ii) ions in a neutral borate buffer solution. Chem. Commun. 2017, 53, 9324–9327. [Google Scholar] [CrossRef] [PubMed]
- Powell, K.J.; Brown, P.L.; Byrne, R.H.; Gajda, T.; Hefter, G.; Sjöberg, S.; Wanner, H. Chemical speciation of environmentally significant metals with inorganic ligands Part 2: The Cu2+-OH−, Cl−, CO32−, SO42−, and PO43− systems (IUPAC Technical Report). Pure Appl. Chem. 2007, 79, 895–950. [Google Scholar] [CrossRef]
- Li, T.-T.; Zheng, Y.-Q. Electrocatalytic water oxidation using a chair-like tetranuclear copper(ii) complex in a neutral aqueous solution. Dalton Trans. 2016, 45, 12685–12690. [Google Scholar] [CrossRef] [PubMed]
- Funes-Ardoiz, I.; Garrido-Barros, P.; Llobet, A.; Maseras, F. Single Electron Transfer Steps in Water Oxidation Catalysis. Redefining the Mechanistic Scenario. ACS Catal. 2017, 7, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Barros, P.; Funes-Ardoiz, I.; Drouet, S.; Benet-Buchholz, J.; Maseras, F.; Llobet, A. Redox Non-innocent Ligand Controls Water Oxidation Overpotential in a New Family of Mononuclear Cu-Based Efficient Catalysts. J. Am. Chem. Soc. 2015, 137, 6758–6761. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, C.; Liu, S.; Wang, J.-L.; Lin, W. A Biomimetic Copper Water Oxidation Catalyst with Low Overpotential. J. Am. Chem. Soc. 2014, 136, 273–281. [Google Scholar] [CrossRef]
- Szyrwiel, Ł.; Lukács, D.; Srankó, D.F.; Kerner, Z.; Kotynia, A.; Brasuń, J.; Setner, B.; Szewczuk, Z.; Malec, K.; Pap, J.S. Armed by Asp? C-terminal carboxylate in a Dap-branched peptide and consequences in the binding of CuII and electrocatalytic water oxidation. RSC Adv. 2017, 7, 24657–24666. [Google Scholar] [CrossRef]
- Cui, S.; Qian, M.; Liu, X.; Sun, Z.; Du, P. A Copper Porphyrin-Based Conjugated Mesoporous Polymer-Derived Bifunctional Electrocatalyst for Hydrogen and Oxygen Evolution. ChemSusChem 2016, 9, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Kuwamura, N.; Kurioka, Y.; Yoshinari, N.; Konno, T. Heterogeneous catalytic water oxidation controlled by coordination geometries of copper(II) centers with thiolato donors. Chem. Commun. 2018, 54, 10766–10769. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Ülker, E.; Karadas, F. One-Dimensional Copper(II) Coordination Polymer as an Electrocatalyst for Water Oxidation. ChemElectroChem 2017, 4, 75–80. [Google Scholar] [CrossRef]
- Gerlach, D.L.; Bhagan, S.; Cruce, A.A.; Burks, D.B.; Nieto, I.; Truong, H.T.; Kelley, S.P.; Herbst-Gervasoni, C.J.; Jernigan, K.L.; Bowman, M.K.; et al. Studies of the Pathways Open to Copper Water Oxidation Catalysts Containing Proximal Hydroxy Groups During Basic Electrocatalysis. Inorg. Chem. 2014, 53, 12689–12698. [Google Scholar] [CrossRef] [PubMed]
- Burks, D.B.; Vasiliu, M.; Dixon, D.A.; Papish, E.T. Thermodynamic Acidity Studies of 6,6′-Dihydroxy-2,2′-bipyridine: A Combined Experimental and Computational Approach. J. Phys. Chem. A 2018, 122, 2221–2231. [Google Scholar] [CrossRef]
- DePasquale, J.; Nieto, I.; Reuther, L.E.; Herbst-Gervasoni, C.J.; Paul, J.J.; Mochalin, V.; Zeller, M.; Thomas, C.M.; Addison, A.W.; Papish, E.T. Iridium Dihydroxybipyridine Complexes Show That Ligand Deprotonation Dramatically Speeds Rates of Catalytic Water Oxidation. Inorg. Chem. 2013, 52, 9175–9183. [Google Scholar] [CrossRef]
- Ghosh, T.; Fridman, N.; Kosa, M.; Maayan, G. Self-Assembled Cyclic Structures from Copper(II) Peptoids. Angew. Chem. Int. Ed. 2018, 57, 7703–7708. [Google Scholar] [CrossRef]
- Ghosh, T.; Ghosh, P.; Maayan, G. A Copper-Peptoid as a Highly Stable, Efficient and Reusable Homogeneous Water Oxidation Electrocatalyst. ACS Catal. 2018, 8, 10631–10640. [Google Scholar] [CrossRef]
- Zhang, M.-T.; Chen, Z.; Kang, P.; Meyer, T.J. Electrocatalytic Water Oxidation with a Copper(II) Polypeptide Complex. J. Am. Chem. Soc. 2013, 135, 2048–2051. [Google Scholar] [CrossRef]
- Pap, J.S.; Szyrwiel, Ł.; Srankó, D.; Kerner, Z.; Setner, B.; Szewczuk, Z.; Malinka, W. Electrocatalytic water oxidation by CuII complexes with branched peptides. Chem. Commun. 2015, 51, 6322–6324. [Google Scholar] [CrossRef]
- Farkas, E.; Srankó, D.; Kerner, Z.; Setner, B.; Szewczuk, Z.; Malinka, W.; Horvath, R.; Szyrwiel, Ł.; Pap, J.S. Self-assembled, nanostructured coatings for water oxidation by alternating deposition of Cu-branched peptide electrocatalysts and polyelectrolytes. Chem. Sci. 2016, 7, 5249–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef] [PubMed]
- Stott, L.A.; Prosser, K.E.; Berdichevsky, E.K.; Walsby, C.J.; Warren, J.J. Lowering water oxidation overpotentials using the ionisable imidazole of copper(2-(2′-pyridyl)imidazole). Chem. Commun. 2017, 53, 651–654. [Google Scholar] [CrossRef]
- Fisher, K.J.; Materna, K.L.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. Electrocatalytic Water Oxidation by a Copper(II) Complex of an Oxidation-Resistant Ligand. ACS Catal. 2017, 7, 3384–3387. [Google Scholar] [CrossRef]
- Rudshteyn, B.; Fisher, K.J.; Lant, H.M.C.; Yang, K.R.; Mercado, B.Q.; Brudvig, G.W.; Crabtree, R.H.; Batista, V.S. Water-Nucleophilic Attack Mechanism for the CuII (pyalk)2 Water-Oxidation Catalyst. ACS Catal. 2018, 8, 7952–7960. [Google Scholar] [CrossRef]
- Shopov, D.Y.; Rudshteyn, B.; Campos, J.; Batista, V.S.; Crabtree, R.H.; Brudvig, G.W. Stable Iridium(IV) Complexes of an Oxidation-Resistant Pyridine-Alkoxide Ligand: Highly Divergent Redox Properties Depending on the Isomeric Form Adopted. J. Am. Chem. Soc. 2015, 137, 7243–7250. [Google Scholar] [CrossRef] [PubMed]
- Michaelos, T.K.; Lant, H.M.C.; Sharninghausen, L.S.; Craig, S.M.; Menges, F.S.; Mercado, B.Q.; Brudvig, G.W.; Crabtree, R.H. Catalytic Oxygen Evolution from Manganese Complexes with an Oxidation-Resistant N,N,O-Donor Ligand. ChemPlusChem 2016, 81, 1129–1132. [Google Scholar] [CrossRef]
- Yu, F.; Li, F.; Hu, J.; Bai, L.; Zhu, Y.; Sun, L. Electrocatalytic water oxidation by a macrocyclic Cu(ii) complex in neutral phosphate buffer. Chem. Commun. 2016, 52, 10377–10380. [Google Scholar] [CrossRef]
- Su, X.-J.; Gao, M.; Jiao, L.; Liao, R.-Z.; Siegbahn, P.E.M.; Cheng, J.-P.; Zhang, M.-T. Electrocatalytic Water Oxidation by a Dinuclear Copper Complex in a Neutral Aqueous Solution. Angew. Chem. Int. Ed. 2015, 54, 4909–4914. [Google Scholar] [CrossRef]
- Wang, J.; Huang, H.; Lu, T. Homogeneous Electrocatalytic Water Oxidation by a Rigid Macrocyclic Copper(II) Complex. Chin. J. Chem. 2017, 35, 586–590. [Google Scholar] [CrossRef]
- Ullman, A.M.; Liu, Y.; Huynh, M.; Bediako, D.K.; Wang, H.; Anderson, B.L.; Powers, D.C.; Breen, J.J.; Abruña, H.D.; Nocera, D.G. Water Oxidation Catalysis by Co(II) Impurities in Co(III)4O4 Cubanes. J. Am. Chem. Soc. 2014, 136, 17681–17688. [Google Scholar] [CrossRef] [PubMed]
- Kafentzi, M.-C.; Papadakis, R.; Gennarini, F.; Kochem, A.; Iranzo, O.; Le Mest, Y.; Le Poul, N.; Tron, T.; Faure, B.; Simaan, A.J.; et al. Electrochemical Water Oxidation and Stereoselective Oxygen Atom Transfer Mediated by a Copper Complex. Chem. Eur. J. 2018, 24, 5213–5224. [Google Scholar] [CrossRef] [PubMed]
- Blain, I.; Bruno, P.; Giorgi, M.; Lojou, E.; Lexa, D.; Réglier, M. Copper Complexes as Functional Models for Dopamine β-Hydroxylase—Stereospecific Oxygen Atom Transfer. Eur. J. Inorg. Chem. 1998, 1998, 1297–1304. [Google Scholar] [CrossRef]
- Nestke, S.; Ronge, E.; Siewert, I. Electrochemical water oxidation using a copper complex. Dalton Trans. 2018, 47, 10737–10741. [Google Scholar] [CrossRef] [PubMed]
- Terao, R.; Nakazono, T.; Parent, A.R.; Sakai, K. Photochemical Water Oxidation Catalyzed by a Water-Soluble Copper Phthalocyanine Complex. ChemPlusChem 2016, 81, 1064–1067. [Google Scholar] [CrossRef]
- Coggins, M.K.; Zhang, M.-T.; Chen, Z.; Song, N.; Meyer, T.J. Single-Site Copper(II) Water Oxidation Electrocatalysis: Rate Enhancements with HPO42− as a Proton Acceptor at pH 8. Angew. Chem. Int. Ed. 2014, 53, 12226–12230. [Google Scholar] [CrossRef]
- Shen, J.; Wang, M.; Zhang, P.; Jiang, J.; Sun, L. Electrocatalytic water oxidation by copper(ii) complexes containing a tetra- or pentadentate amine-pyridine ligand. Chem. Commun. 2017, 53, 4374–4377. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, M.; Yang, Y.; Yao, T.; Sun, L. A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution. Angew. Chem. Int. Ed. 2014, 53, 13803–13807. [Google Scholar] [CrossRef]
- Garrido-Barros, P.; Gimbert-Suriñach, C.; Moonshiram, D.; Picón, A.; Monge, P.; Batista, V.S.; Llobet, A. Electronic π-Delocalization Boosts Catalytic Water Oxidation by Cu(II) Molecular Catalysts Heterogenized on Graphene Sheets. J. Am. Chem. Soc. 2017, 139, 12907–12910. [Google Scholar] [CrossRef] [Green Version]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef]
- Naqvi, K.R.; Melø, T.B. Reduction of tetranitromethane by electronically excited aromatics in acetonitrile: Spectra and molar absorption coefficients of radical cations of anthracene, phenanthrene and pyrene. Chem. Phys. Lett. 2006, 428, 83–87. [Google Scholar] [CrossRef]
- Fu, L.-Z.; Fang, T.; Zhou, L.-L.; Zhan, S.-Z. A mononuclear copper electrocatalyst for both water reduction and oxidation. RSC Adv. 2014, 4, 53674–53680. [Google Scholar] [CrossRef]
- Su, X.-J.; Zheng, C.; Hu, Q.-Q.; Du, H.-Y.; Liao, R.-Z.; Zhang, M.-T. Bimetallic cooperative effect on O–O bond formation: Copper polypyridyl complexes as water oxidation catalyst. Dalton Trans. 2018, 47, 8670–8675. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.-J.; Wang, H.-Y.; Xin, Z.-J.; Li, C.-B.; Lu, Y.-X.; Gao, X.-W.; Sun, H.-M.; Cao, R. A Water-Soluble Copper-Polypyridine Complex as a Homogeneous Catalyst for both Photo-Induced and Electrocatalytic O2 Evolution. Chem. Eur. J. 2016, 22, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, N.; Lei, H.; Guo, D.; Liu, H.; Zhang, Z.; Zhang, W.; Lai, W.; Cao, R. Electrocatalytic Water Oxidation by a Water-Soluble Copper(II) Complex with a Copper-Bound Carbonate Group Acting as a Potential Proton Shuttle. Inorg. Chem. 2017, 56, 13368–13375. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Sóvágó, I. Metal complexes of amino acids and peptides. In Amino Acids, Peptides and Proteins; Ryadnov, M., Hudecz, F., Eds.; Royal Society of Chemistry: Cambridge, UK, 2016; Volume 41, pp. 100–151. ISBN 978-1-78262-537-7. [Google Scholar]
- Kim, M.K.; Martell, A.E. Copper(II) Complexes of Triglycine and Tetraglycine. J. Am. Chem. Soc. 1966, 88, 914–918. [Google Scholar] [CrossRef]
- Nagy, N.V.; Szabó-Plánka, T.; Rockenbauer, A.; Peintler, G.; Nagypál, I.; Korecz, L. Great Structural Variety of Complexes in Copper(II)−Oligoglycine Systems: Microspeciation and Coordination Modes as Studied by the Two-Dimensional Simulation of Electron Paramagnetic Resonance Spectra. J. Am. Chem. Soc. 2003, 125, 5227–5235. [Google Scholar] [CrossRef] [PubMed]
- Pap, J.S.; Szyrwiel, Ł. On the Cu(III)/Cu(II) Redox Chemistry of Cu-Peptide Complexes to Assist Catalyst Design. Comments Inorg. Chem. 2017, 37, 59–77. [Google Scholar] [CrossRef]
- Szyrwiel, Ł.; Szczukowski, Ł.; Pap, J.S.; Setner, B.; Szewczuk, Z.; Malinka, W. The Cu2+ Binding Properties of Branched Peptides Based on l-2,3-Diaminopropionic Acid. Inorg. Chem. 2014, 53, 7951–7959. [Google Scholar] [CrossRef] [PubMed]
- Szyrwiel, Ł.; Pap, J.S.; Szczukowski, Ł.; Kerner, Z.; Brasuń, J.; Setner, B.; Szewczuk, Z.; Malinka, W. Branched peptide with three histidines for the promotion of CuII binding in a wide pH range – complementary potentiometric, spectroscopic and electrochemical studies. RSC Adv. 2015, 5, 56922–56931. [Google Scholar] [CrossRef]
- Jakab, N.I.; Gyurcsik, B.; Körtvélyesi, T.; Vosekalna, I.; Jensen, J.; Larsen, E. Design of histidine containing peptides for better understanding of their coordination mode toward copper(II) by CD spectroscopy. J. Inorg. Biochem. 2007, 101, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Koepke, S.J.; Light, K.M.; VanNatta, P.E.; Wiley, K.M.; Kieber-Emmons, M.T. Electrocatalytic Water Oxidation by a Homogeneous Copper Catalyst Disfavors Single-Site Mechanisms. J. Am. Chem. Soc. 2017, 139, 8586–8600. [Google Scholar] [CrossRef]
- Nagao, H.; Komeda, N.; Mukaida, M.; Suzuki, M.; Tanaka, K. Structural and Electrochemical Comparison of Copper(II) Complexes with Tripodal Ligands. Inorg. Chem. 1996, 35, 6809–6815. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, H.; Sun, Z.; Chen, H.; Xu, P.; Du, P. Nanostructured copper oxide electrodeposited from copper(II) complexes as an active catalyst for electrocatalytic oxygen evolution reaction. Electrochem. Commun. 2014, 46, 1–4. [Google Scholar] [CrossRef]
- Schröder, D.; Holthausen, M.C.; Schwarz, H. Radical-Like Activation of Alkanes by the Ligated Copper Oxide Cation (Phenanthroline)CuO+. J. Phys. Chem. B 2004, 108, 14407–14416. [Google Scholar] [CrossRef]
- Dietl, N.; van der Linde, C.; Schlangen, M.; Beyer, M.K.; Schwarz, H. Diatomic [CuO]+ and Its Role in the Spin-Selective Hydrogen- and Oxygen-Atom Transfers in the Thermal Activation of Methane. Angew. Chem. Int. Ed. 2011, 50, 4966–4969. [Google Scholar] [CrossRef] [PubMed]
- Molecular Electronic Structures of Transition Metal Complexes I; Mingos, D.M.P.; Day, P.; Dahl, J.P. (Eds.) Structure and Bonding; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2012; Volume 142, ISBN 978-3-642-27369-8. [Google Scholar]
- Hu, Q.-Q.; Su, X.-J.; Zhang, M.-T. Electrocatalytic Water Oxidation by an Unsymmetrical Di-Copper Complex. Inorg. Chem. 2018, 57, 10481–10484. [Google Scholar] [CrossRef]
- Hayashi, H.; Fujinami, S.; Nagatomo, S.; Ogo, S.; Suzuki, M.; Uehara, A.; Watanabe, Y.; Kitagawa, T. A Bis(μ-oxo)dicopper(III) Complex with Aromatic Nitrogen Donors: Structural Characterization and Reversible Conversion between Copper(I) and Bis(μ-oxo)dicopper(III) Species. J. Am. Chem. Soc. 2000, 122, 2124–2125. [Google Scholar] [CrossRef]
- Fang, T.; Fu, L.-Z.; Zhou, L.-L.; Zhan, S.-Z. A water-soluble dinuclear copper electrocatalyst, [Cu(oxpn)Cu(OH)2] for both water reduction and oxidation. Electrochim. Acta 2015, 161, 388–394. [Google Scholar] [CrossRef]
- Zhou, L.-L.; Fang, T.; Cao, J.-P.; Zhu, Z.-H.; Su, X.-T.; Zhan, S.-Z. A dinuclear copper(II) electrocatalyst both water reduction and oxidation. J. Power Sources 2015, 273, 298–304. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Kondo, M.; Kuga, R.; Kurashige, Y.; Yanai, T.; Hayami, S.; Praneeth, V.K.K.; Yoshida, M.; Yoneda, K.; Kawata, S.; et al. A pentanuclear iron catalyst designed for water oxidation. Nature 2016, 530, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Praneeth, V.K.K.; Kondo, M.; Woi, P.M.; Okamura, M.; Masaoka, S. Electrocatalytic Water Oxidation by a Tetranuclear Copper Complex. ChemPlusChem 2016, 81, 1123–1128. [Google Scholar] [CrossRef]
- Li, X.; Cheng, D.; Lin, J.; Li, Z.; Zheng, Y. Di-, Tetra-, and Hexanuclear Hydroxy-Bridged Copper(II) Cluster Compounds: Syntheses, Structures, and Properties. Cryst. Growth Des. 2008, 8, 2853–2861. [Google Scholar] [CrossRef]
- Jiang, X.; Li, J.; Yang, B.; Wei, X.-Z.; Dong, B.-W.; Kao, Y.; Huang, M.-Y.; Tung, C.-H.; Wu, L.-Z. A Bio-inspired Cu4O4 Cubane: Effective Molecular Catalysts for Electrocatalytic Water Oxidation in Aqueous Solution. Angew. Chem. Int. Ed. 2018, 57, 7850–7854. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zheng, H.; Sun, Z.; Han, A.; Du, P. Earth-Abundant Copper-Based Bifunctional Electrocatalyst for Both Catalytic Hydrogen Production and Water Oxidation. ACS Catal. 2015, 5, 1530–1538. [Google Scholar] [CrossRef]
- Yu, F.; Li, F.; Zhang, B.; Li, H.; Sun, L. Efficient Electrocatalytic Water Oxidation by a Copper Oxide Thin Film in Borate Buffer. ACS Catal. 2015, 5, 627–630. [Google Scholar] [CrossRef]
- Liu, X.; Cui, S.; Qian, M.; Sun, Z.; Du, P. In situ generated highly active copper oxide catalysts for the oxygen evolution reaction at low overpotential in alkaline solutions. Chem. Commun. 2016, 52, 5546–5549. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Du, J.; Su, X.-J.; Zhang, M.-T.; Xu, X.; Meyer, T.J.; Chen, Z. Cu(II) Aliphatic Diamine Complexes for Both Heterogeneous and Homogeneous Water Oxidation Catalysis in Basic and Neutral Solutions. ACS Catal. 2016, 6, 77–83. [Google Scholar] [CrossRef]
- Lu, C.; Wang, J.; Chen, Z. Water Oxidation by Copper-Amino Acid Catalysts at Low Overpotentials. ChemCatChem 2016, 8, 2165–2170. [Google Scholar] [CrossRef]
- Li, T.-T.; Cao, S.; Yang, C.; Chen, Y.; Lv, X.-J.; Fu, W.-F. Electrochemical Water Oxidation by In Situ -Generated Copper Oxide Film from [Cu(TEOA)(H2O)2][SO4] Complex. Inorg. Chem. 2015, 54, 3061–3067. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gao, Y.; Lu, Z.; Ye, L.; Sun, L. Copper Oxide Film In-situ Electrodeposited from Cu(II) Complex as Highly Efficient Catalyst for Water Oxidation. Electrochim. Acta 2017, 230, 501–507. [Google Scholar] [CrossRef]
- Ilhan, S.; Temel, H.; KIlIc, A. Synthesis and spectral studies of macrocyclic Cu(II) complexes by reaction of various diamines, copper(II) perchlorate and 1,4- bis (2-carboxyaldehyde phenoxy)butane. J. Coord. Chem. 2008, 61, 277–284. [Google Scholar] [CrossRef]
- Amiri, M.; Fallahi, M.; Bezaatpour, A.; Jijie, R.; Nozari-asbmarz, M.; Rouhi, M.; Boukherroub, R.; Szunerits, S. Solution Processable Cu(II)macrocycle for the Formation of Cu2O Thin Film on Indium Tin Oxide and Its Application for Water Oxidation. J. Phys. Chem. C 2018, 122, 16510–16518. [Google Scholar] [CrossRef]
- Pilkington, N.; Robson, R. Complexes of binucleating ligands. III. Novel complexes of a macrocyclic binucleating ligand. Aust. J. Chem. 1970, 23, 2225. [Google Scholar]
- Majumder, S.; Abdel Haleem, A.; Nagaraju, P.; Naruta, Y. A new preparation of a bifunctional crystalline heterogeneous copper electrocatalyst by electrodeposition using a Robson-type macrocyclic dinuclear copper complex for efficient hydrogen and oxygen evolution from water. Dalton Trans. 2017, 46, 9131–9139. [Google Scholar] [CrossRef]
- van der Ham, C.J.M.; Işık, F.; Verhoeven, T.W.G.M.; Niemantsverdriet, J.W. (Hans); Hetterscheid, D.G.H. Activation pathways taking place at molecular copper precatalysts for the oxygen evolution reaction. Catal. Today 2017, 290, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.-B.; He, Q.-Y.; Ma, X.-F.; Shi, H.-T.; Wei, X. A new copper species based on an azo-compound utilized as a homogeneous catalyst for water oxidation. Dalton Trans. 2015, 44, 351–358. [Google Scholar] [CrossRef]
- Najafpour, M.M.; Ebrahimi, F.; Safdari, R.; Ghobadi, M.Z.; Tavahodi, M.; Rafighi, P. New findings and the current controversies for water oxidation by a copper(ii)-azo complex: Homogeneous or heterogeneous? Dalton Trans. 2015, 44, 15435–15440. [Google Scholar] [CrossRef]
- Wu, Z.-S.; Chen, L.; Liu, J.; Parvez, K.; Liang, H.; Shu, J.; Sachdev, H.; Graf, R.; Feng, X.; Müllen, K. High-Performance Electrocatalysts for Oxygen Reduction Derived from Cobalt Porphyrin-Based Conjugated Mesoporous Polymers. Adv. Mater. 2014, 26, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Chen, Z.; Ye, S.; Wiley, B.J.; Meyer, T.J. Copper as a Robust and Transparent Electrocatalyst for Water Oxidation. Angew. Chem. Int. Ed. 2015, 54, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.-C.; Fu, W.-F.; Chen, Y. Self-Supported Cu-Based Nanowire Arrays as Noble-Metal-Free Electrocatalysts for Oxygen Evolution. ChemSusChem 2016, 9, 2069–2073. [Google Scholar] [CrossRef]
- Liu, X.; Cui, S.; Sun, Z.; Ren, Y.; Zhang, X.; Du, P. Self-Supported Copper Oxide Electrocatalyst for Water Oxidation at Low Overpotential and Confirmation of Its Robustness by Cu K-Edge X-ray Absorption Spectroscopy. J. Phys. Chem. C 2016, 120, 831–840. [Google Scholar] [CrossRef]
- Handoko, A.D.; Deng, S.; Deng, Y.; Cheng, A.W.F.; Chan, K.W.; Tan, H.R.; Pan, Y.; Tok, E.S.; Sow, C.H.; Yeo, B.S. Enhanced activity of H2O2 -treated copper(ii) oxide nanostructures for the electrochemical evolution of oxygen. Catal. Sci. Technol. 2016, 6, 269–274. [Google Scholar] [CrossRef]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | pH | [Ligand]/[CuII] Ratio | Electrolyte Anion | η 2 (mV) | Faraday Eff. (%) | Ref. |
|---|---|---|---|---|---|---|
| Cs(2−x)[Cu(F)x(OH2)(6−x)](2−x) (1) | 7.2 | >6000 | F−, 1 M | 445 | 94 | [20] |
| [Cu(η1-OCO2H)2(OH2)2] (2a) 3,4 | 8.2 | >250 5 | 1 M HCO3− | ~800 | 96 | [21] |
| ‘Cu-carbonate’ 6 | 10.8 | >1000 | 1 M HCO3−/CO32− | ~700 | 97 | [21] |
| {[B(OH)4]Cu(OSO3)(OH2)} (3) | 7.0 | >7250 | 0.45 M borate/1 M SO42− | 750 | ~100 | [26] |
| Catalyst | pH | Electrolyte | kobs (s−1) | η (V) | Farad. Eff. (%) | Ref. |
|---|---|---|---|---|---|---|
| [(bpy)Cu(OH)2] (4) | 12.8 | 0.1 M NaOH/NaOAc | 100 | 750 | 90 | [19] |
| [(6,6′-dhbpy)Cu(OH2)2] (5) | 12.4 | 0.1 M NaOH/NaOAc | 0.4 | 640 | 85 | [31] |
| [(6,6′-(O)2-bpy)2Cu(OH2)]2- (6) | 12.6 | 0.1 M NaOH/NaOAc | 0.356 | 477 | n.a. | [36] |
| [(BPT)Cu(OH)2] (7) | 11.5 | 0.1 M phosphate | 5.8 | ~800 | 91 | [40] |
| [Cu(pim)(OH2)(OH)] (8) | 12 | 0.1 M NaOH/NaOAc | 35 | 300 | n.a. | [45] |
| [Cu(pyalk)2] (10) | 12.5 | 0.1 M KNO3/KOH | 0.7 | 520–580 | 75 | [46,47] |
| [Cu(TMC)(OH2)]2+ (12) | 7 | 0.1 M phosphate | 30 | n.a. | 89/98 1 | [50] |
| [Cu(Lm)(OH2)]2+ (13) | 12 | 0.15 M phosphate | n.a. | ~750 | 50 | [52] |
| [Cu(IndPY2)(OH2)2]2+ (14) | 7 | 0.1 NaNO3 | 0.02 | 280 2 | 75 | [54] |
| [Cu(LOH)(OH2)]2+ (15) | 12 | 0.1 M phosphate | 0.12 | 830 | 60 | [56] |
| [Cu(PcTS)] (16) | 9.5 | 0.1 M borate | n.a. | ~570 3 | n.a. | [57] |
| [(Py3P)Cu(OH)]− (17) | 8 | 0.1/0.2 M phosphate | 20/38 | ~500 4 | 84 | [58] |
| [Cu(bztpen)]2+ (18) | 11.5 | 0.1 M phosphate | 13.1 | 440 | 91 | [59] |
| [Cu(dbzbpen)(OH2)]2+ (19) | 11.5 | 0.1 M phosphate | 18.7 | 570 | 94 | [59] |
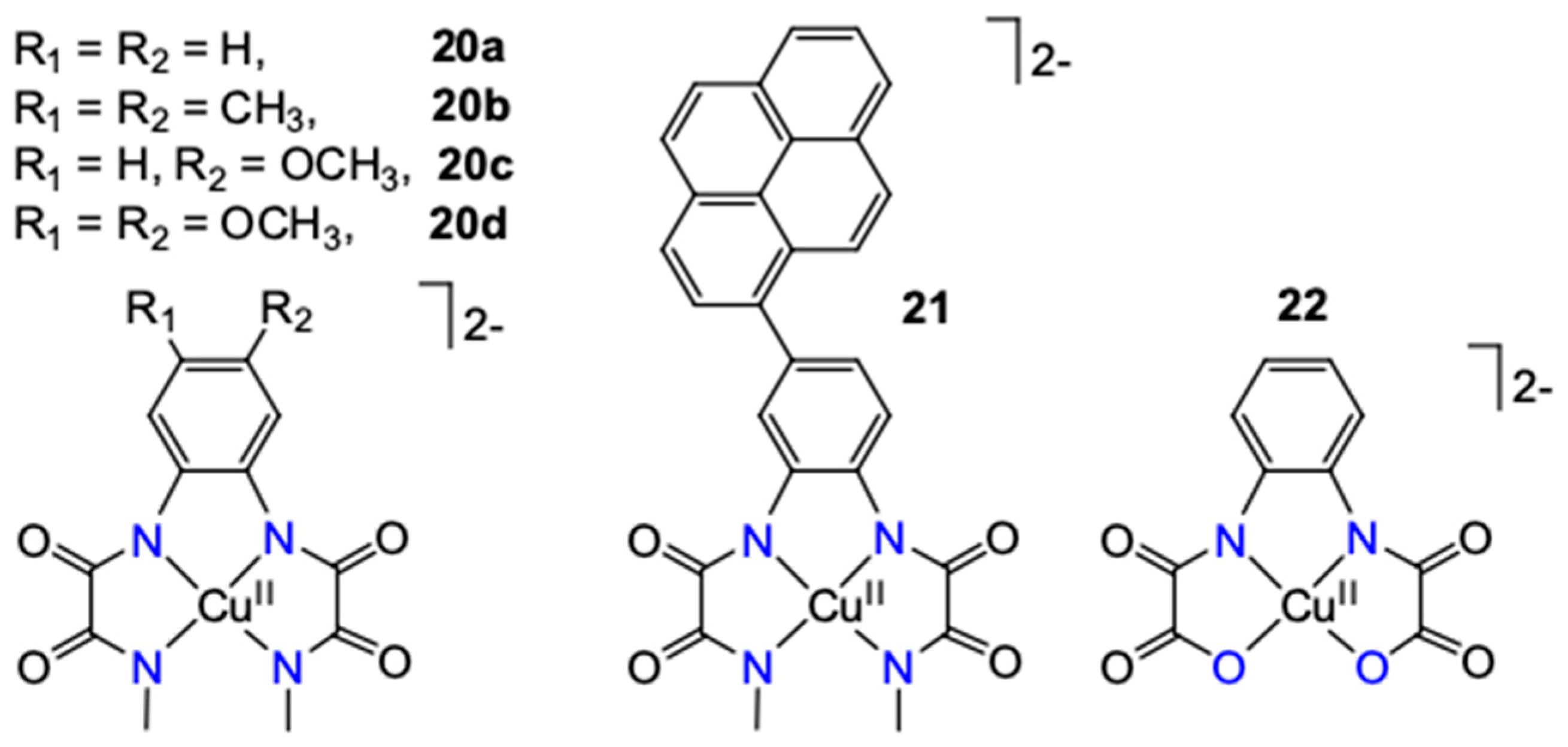
| [(L1)Cu]2− (20a) | 11.5 | phosphate, I = 0.1 M | 0.16 | 170 | ~100 | [30] |
| [(L2)Cu]2−(20b) | 11.5 | phosphate, I = 0.1 M | 0.43 | 270 | n.a. | [30] |
| [(L3)Cu]2− (20c) | 11.5 | phosphate, I = 0.1 M | 3.58 | 400 | n.a. | [30] |
| [(L4)Cu]2− (20d) | 11.5/12.5 | phosphate, I = 0.1 M | 3.56/12 | 700 | 47 | [30] |
| [(Lpy)Cu]2− (21) | 11.5 | phosphate, I = 0.1 M | 128 | 550 | ~25 5 | [61] |
| [(opba)Cu]2− (22) | 10.8 | phosphate, 0.25 M | 1.13 | 626 | 95.8 | [64] |
| [Cu(GGGG)(OH2)]2− (23) | 11 | phosphate, 0.25 M | 33 | ~650 | 99 | [41] |
| [Cu(H−23G)(OH2)] (24) | 11 | phosphate, 0.15 M | 24 | ~650 3 | 91 | [42] |
| [Cu(H−22GH)(OH2)] (25) | 11 | phosphate, 0.15 M | 53 | ~620 3 | 95 | [42] |
| [Cu(H−2GGD)(OH2)]− (26) | 11 | phosphate, 0.20 M | 10 | ~670 3 | n.a. | [32] |
| [Cu(H−22GD)(OH2)]− (27) | 11 | phosphate, 0.20 M | 16 | ~670 3 | 90 | [32] |
| [Cu(TPA)(OH2)]2+ (28) | 7.0 | phosphate, 0.1 M | 0.10 | 970 | >90 | [65] |
| [Cu(F3TPA)(OH2)]2+ (28f) | 8.5 | borate, 0.1 M | 0.38 | 610 | 93 | [66] |
| [Cu(Lp)(OCO2H)]− (29) | 10 | carbonate, 0.1 M | 20.1 | 650 | 95 | [67] |
| Catalyst | pH | Electrolyte | kobs (s−1) | η (mV) | Faraday Eff. (%) | Ref. |
|---|---|---|---|---|---|---|
| [(BPMAN)Cu2(μ-OH)]3+ (30) | 7.0 | phosphate, 0.1 M | 0.6 | 1050 | 98 | [51] |
| [(TPMAN)Cu2(μ-OH)]3+ (31) | 7.0 | phosphate, 0.1 M | 0.78 | 780 | n.a. | [81] |
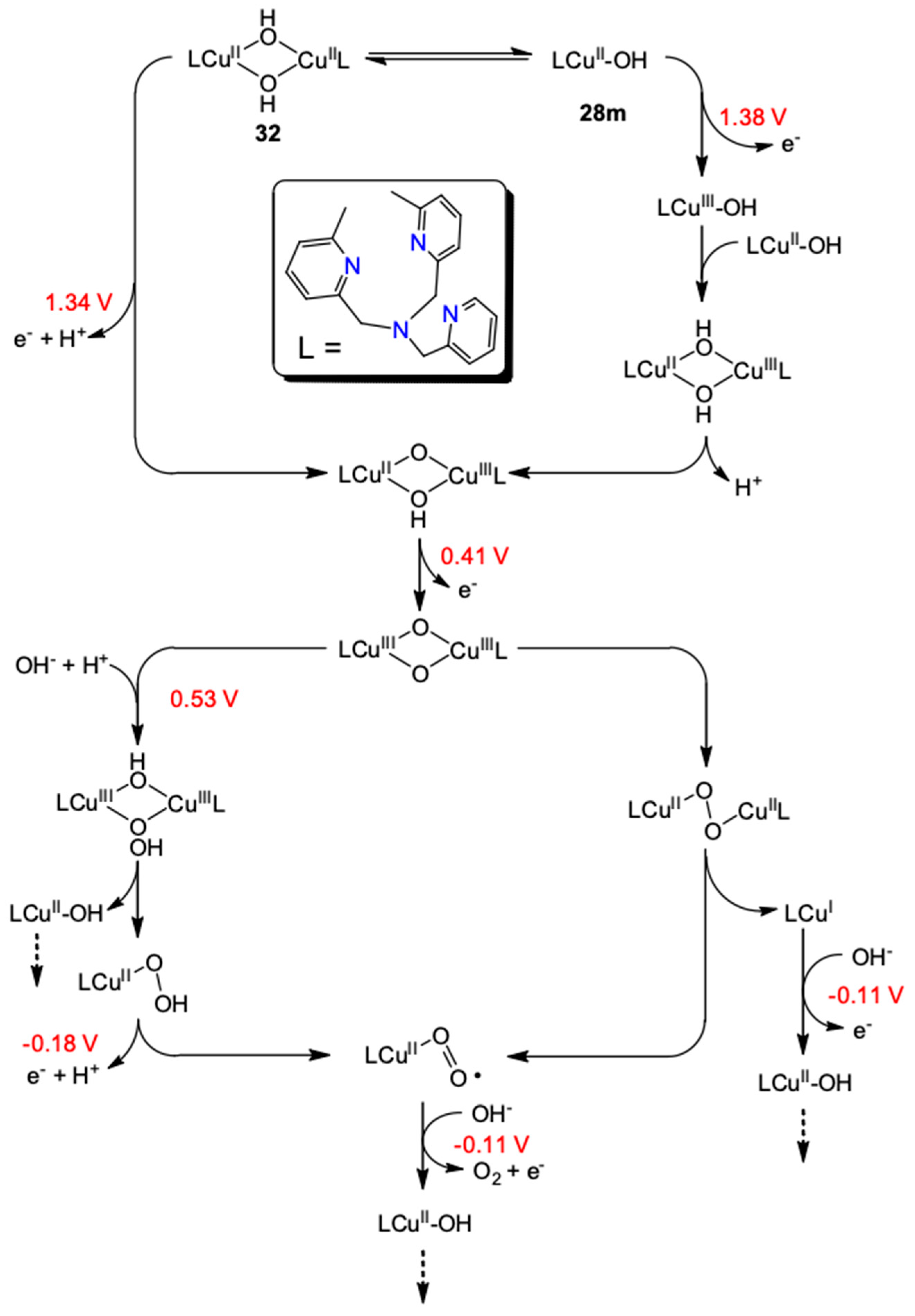
| {[(Me2TMPA)Cu]2-(μ-OH)2}2+ (32) | 12.5 | NaOH/NaOTf | 33 | 1007 | >90 | [75] |
| [Cu(Me2oxpn)Cu(OH)2] (34) 1 | 10.4 | phosphate, 0.25 M | ~2.14 | 636 | 90 | [84] |
| Catalyst | pH | Electrolyte | kobs (s−1) | η (V) | Faraday Eff. (%) | Ref. |
|---|---|---|---|---|---|---|
| [Cu4(H2Lpa)4]4+ (35) | 12.5 | NaOH/NaOAc, 0.1 M | 0.8 | 500 | 75 | [88] |
| [Cu4(bpy)4(μ2-OH)2 (μ3-OH)(H2O)2]2+ (36) | 7.0 | phosphate, 0.1 M | n.a. | 730 | 98 | [28] |
| [(LGly-Cu)4] (37a) | 12.0 | phosphate, 0.2 M | 257 | 620 1 | 97 | [90] |
| [(LGlu-Cu)4] (37b) | 12.0 | phosphate, 0.2 M | 105 | 760 1 | n.a. | [90] |
| Precursor | pH | Electrolyte | [Precursor] (mM) | η (mV) | j (mA/cm2) | Tafel Slope (mV/pH) | Far. Eff. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| [Cu(MeTPA)(OH2)]2+ (38) | 9–11 | borate, 0.1 M | 0.68 | 600 | 1.0 | 56 | >90 | [77] |
| [Cu(TPA)(OH2)]2+ (28) 1 | 9.2 | borate, 0.1 M | 0.68 | 749 | 1.0 | 85 | 97 | [91] |
| Cu-Bi | 9 | borate, 0.2 M | 1.0 | ~550 | 1.0 | 89 | 95 | [92] |
| [Cu(en)2(OH2)2]2+ (39e) | 12.0 | phosphate, 0.2 M | 1.0 | 540 | 1.0 | 62 | 92 | [94] |
| [Cu(en)2(OH2)2]2+ (39e) | 13.6 | KOH, 1.0 M | 3.0 | 370 | 1.0 | 90 | >95 | [93] |
| [Cu(AA)2(OH2)] (40) | 12.0 | phosphate, 0.2 M | 1.0 | 450 | 1.0 | 64 | 96 | [95] |
| [Cu(TEOA)(H2O)2]2+ (41) | 12.4 | NaOH/NaOAc | 2.0 | 550 | 0.5–2.5 2 | 130 | ~100 | [96] |
| [Cu(Tris)(H2O)2]2+ (42) | 12.0 | phosphate, 0.2 M | 1.0 | 390 | 1.0 | 41 | 97 | [97] |
| [Cu(LM)]2+ (43) | 12.0 | borate, 0.1 M | 0.5 | 400 | 1.0 | 72 | 99 | [99] |
| [Cu2(LR)(Cl)2] (44) | 9.2 | borate, 0.1 M | 0.3 | 630 | 1.0 | 71 | 96 | [101] |
| [Cu(bdmpza)2] (45) | ~13 | NaOH, 0.1 M | 150 nmol/ 0.72 cm 2 | 370 | n.d. | n.d. | n.d. | [102] |
| [Cu(Laza)(ONO2)]3+ (46) | 7.0 | phosphate, 0.1 M | 0.78 | n.d. | n.d. | n.d. | n.d. | [104] |
| [Cu-CMP] (47) | ~14 | KOH, 1.0 M | 0.28 mg/cm 2 | 350 | 1.0 | 62 | 97 | [33] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukács, D.; Szyrwiel, Ł.; Pap, J.S. Copper Containing Molecular Systems in Electrocatalytic Water Oxidation—Trends and Perspectives. Catalysts 2019, 9, 83. https://doi.org/10.3390/catal9010083
Lukács D, Szyrwiel Ł, Pap JS. Copper Containing Molecular Systems in Electrocatalytic Water Oxidation—Trends and Perspectives. Catalysts. 2019; 9(1):83. https://doi.org/10.3390/catal9010083
Chicago/Turabian StyleLukács, Dávid, Łukasz Szyrwiel, and József S. Pap. 2019. "Copper Containing Molecular Systems in Electrocatalytic Water Oxidation—Trends and Perspectives" Catalysts 9, no. 1: 83. https://doi.org/10.3390/catal9010083