Effect of Additives on the Activity of Nickel–Tungsten Sulfide Hydroconversion Catalysts Prepared In Situ from Oil-Soluble Precursors


Abstract
:1. Introduction
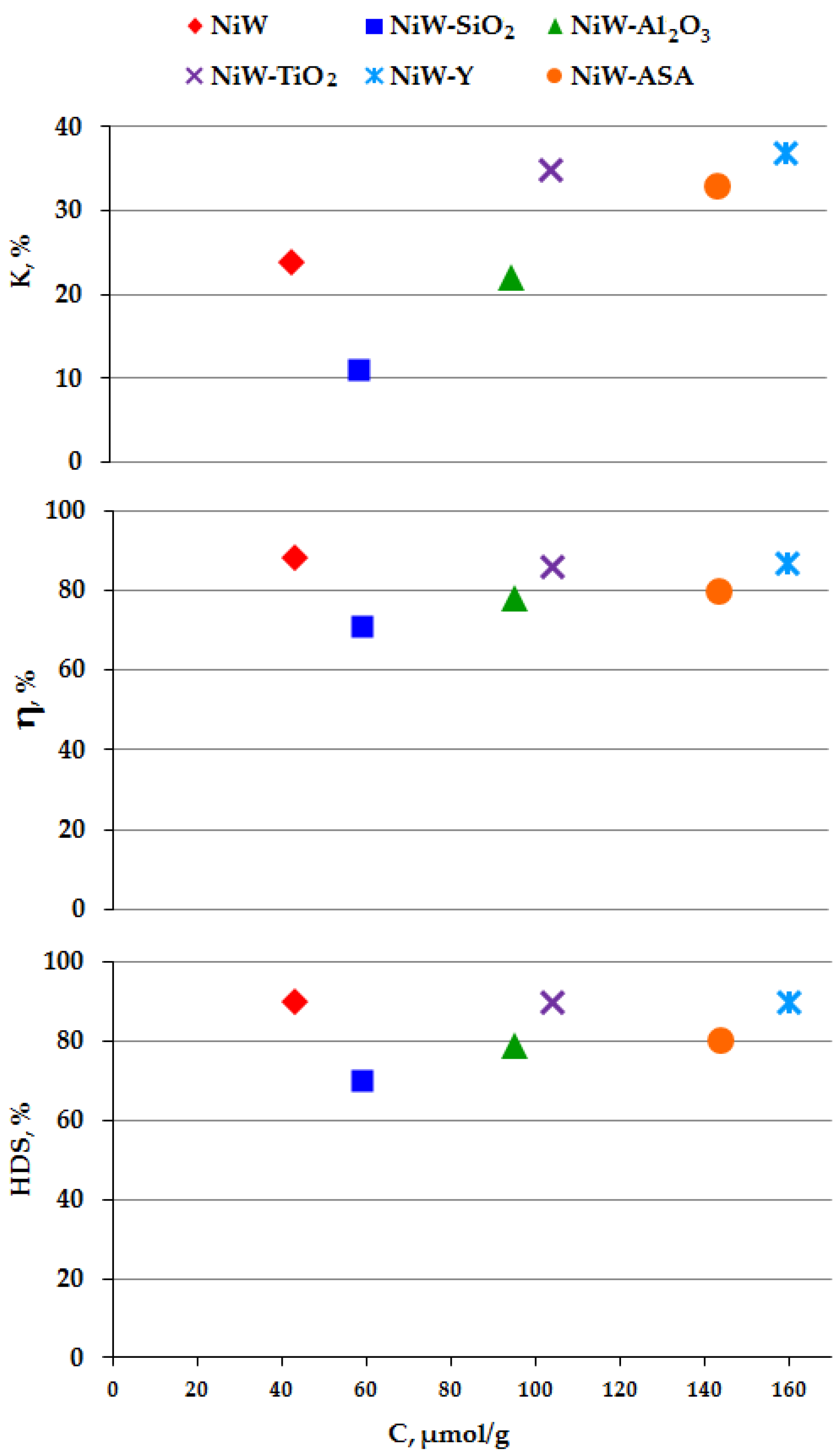
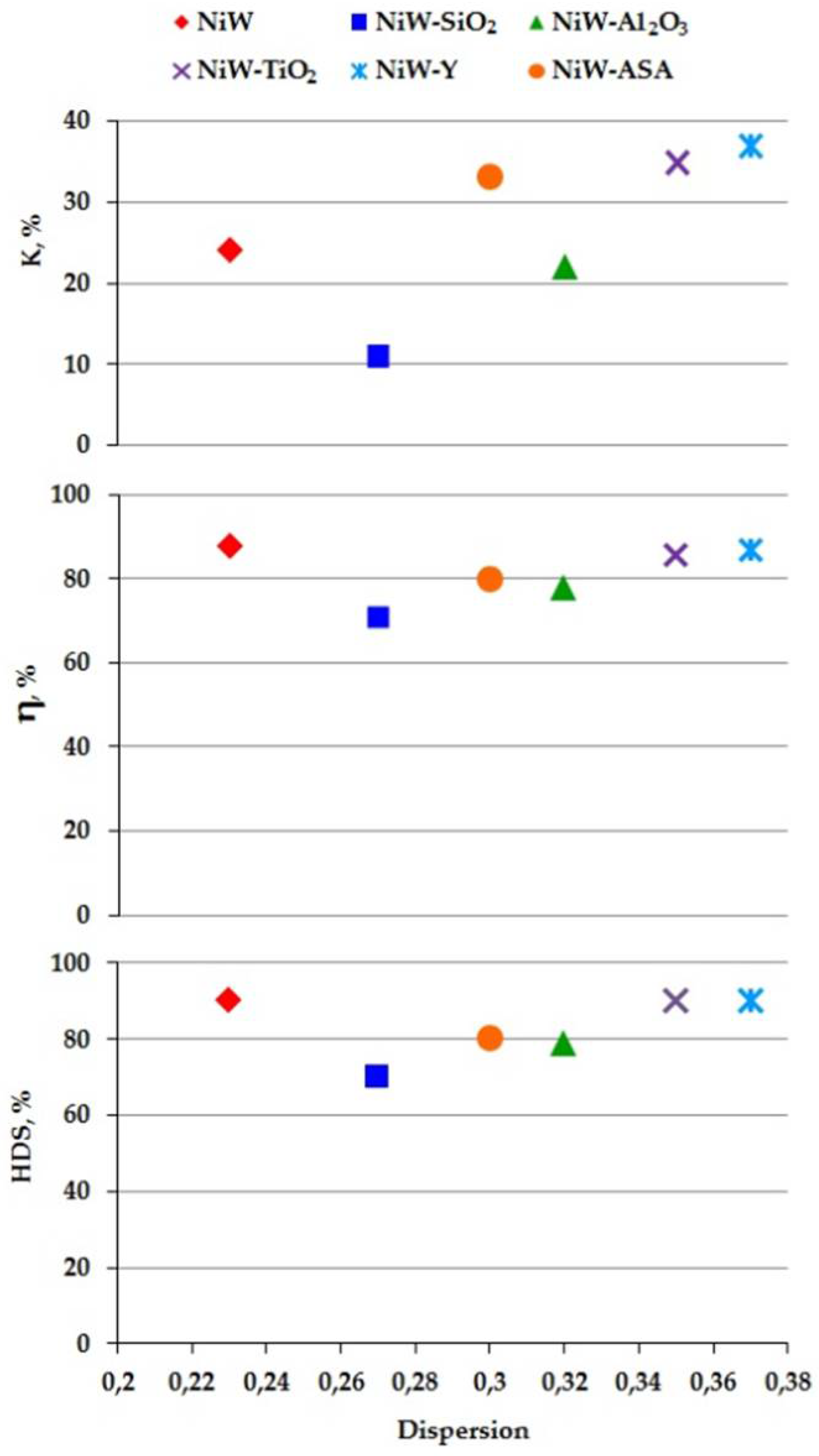
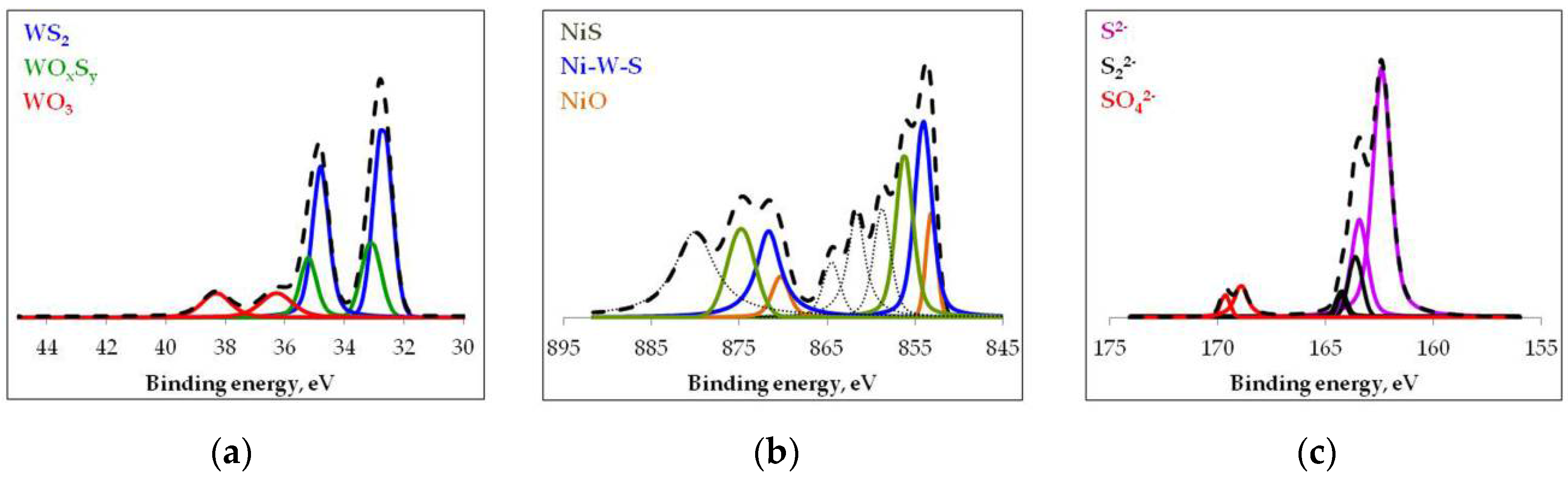
2. Results and Discussion
3. Material and Methods
3.1. Catalyst Preparation Procedure
3.2. Physicochemical Analysis of the Additives and Catalysts
3.3. Catalytic Experiment Procedure
3.4. Analysis of Conversion Products
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Marafi, M.; Stanislaus, A.; Furimsky, E. Hydroprocessing technology. In Handbook of Spent Hydroprocessing Catalysts, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 27–66. ISBN 978-0-444-63881-6. [Google Scholar]
- Furimsky, E. Spent refinery catalysts: Environment, safety and utilization. Catal. Today 1996, 30, 223–286. [Google Scholar] [CrossRef]
- Breysse, M.; Furimsky, E.; Kasztelan, S.; Lacroix, M.; Perot, G. Hydrogen activation by transition metal sulfides. Catal. Rev. Sci. Eng. 2002, 44, 651–735. [Google Scholar] [CrossRef]
- Kasztelan, S.; MacGarvey, B. Hydrogen Content and Hydrogenation Activity of MoS2/γ-Al2O3 and γ-Al2O3 Mechanical Mixtures. J. Catal. 1994, 147, 476–483. [Google Scholar] [CrossRef]
- Raybaud, P. Understanding and predicting improved sulfide catalysts: Insights from first principles modeling. Appl. Catal. A Gen. 2007, 322, 76–91. [Google Scholar] [CrossRef]
- MacGarvey, B.; Kasztelan, S. An Investigation of the Reduction Behavior of MoS2/Al2O3 and the Subsequent Detection of Hydrogen on the Surface. J. Catal. 1994, 148, 149–156. [Google Scholar] [CrossRef]
- Breysse, M.; Bennett, B.A.; Chadwick, D.; Vrinat, M. Structure and HDS Activity of Co-Mo Catalysts: A Comparison of Alumina and Carbon Supports. Bull. Soc. Chim. Belg. 1981, 90, 1271–1278. [Google Scholar] [CrossRef]
- Bouwens, S.M.A.M.; van Zon, F.B.M.; van Dijk, M.P.; van der Kraan, A.M.; de Beer, V.H.J.; van Veen, J.A.R.; Koningsberger, D.C. On the Structural Differences Between Alumina-Supported Comos Type I and Alumina-, Silica-, and Carbon-Supported Comos Type II Phases Studied by XAFS, MES, and XPS. J. Catal. 1994, 146, 375–393. [Google Scholar] [CrossRef] [Green Version]
- Louwers, S.P.A.; Prins, R. Ni EXAFS studies of the Ni–Mo–S structure in carbon-supported and alumina-supported Ni–Mo catalysts. J. Catal. 1992, 133, 94–111. [Google Scholar] [CrossRef]
- Dugulan, A.I.; Craje, M.W.J.; Overweg, A.R.; Kearley, G.J. The evolution of the active phase in CoMo/C hydrodesulfurization catalysts under industrial conditions: A high-pressure Mössbauer emission spectroscopy study. J. Catal. 2005, 229, 276–282. [Google Scholar] [CrossRef]
- Dufresne, P. Hydroprocessing catalysts regeneration and recycling. Appl. Catal. A Gen. 2007, 322, 67–75. [Google Scholar] [CrossRef]
- Brorson, M.; Carlson, A.; Topsoe, H. The morphology of MoS2, WS2, Co–Mo–S, Ni–Mo–S and Ni–W–S nanoclusters in hydrodesulfurization catalysts revealed by HAADF-STEM. Catal. Today 2007, 123, 31–36. [Google Scholar] [CrossRef]
- Topsoe, H. The role of Co–Mo–S type structures in hydrotreating catalysts. Appl. Catal. A Gen. 2007, 322, 3–8. [Google Scholar] [CrossRef]
- Besenbacher, F.; Brorson, M.; Clausen, B.S.; Helveg, S.; Hinnemann, B.; Kibsgard, J.; Lauristen, J.V.; Moses, P.G.; Norskov, J.K.; Topsoe, H. Recent STM, DFT and HAADF-STEM studies of sulfide-based hydrotreating catalysts: Insight into mechanistic, structural and particle size effects. Catal. Today 2008, 130, 86–96. [Google Scholar] [CrossRef]
- Eijsbouts, S.; Mayo, S.W.; Fujita, K. Unsupported transition metal sulfide catalysts: From fundamentals to industrial application. Appl. Catal. A Gen. 2007, 322, 58–66. [Google Scholar] [CrossRef]
- Furimsky, E. Catalyst deactivation. In Catalyst for Upgrading Heavy Petroleum Feed; Elsevier Science: Amsterdam, The Netherlands, 2007; pp. 141–216. ISBN 978-0-444-53084-4. [Google Scholar]
- Nguyen, M.T.; Nguyen, N.T.; Cho, J.; Parka, C.; Parka, S.; Jung, J.; Lee, C.W. A review on the oil-soluble dispersed catalyst for slurry-phase hydrocracking of heavy oil. J. Ind. Eng. Chem. 2016, 43, 1–12. [Google Scholar] [CrossRef]
- Sahu, R.; Song, B.J.; Im, J.S.; Jeon, Y.-P.; Lee, C.W. A review of recent advances in catalytic hydrocracking of heavy residues. J. Ind. Eng. Chem. 2015, 27, 12–24. [Google Scholar] [CrossRef]
- Go, K.S.; Kim, Y.K.; Kwon, E.H.; Nho, N.S. Characteristics of slurry-phase hydrocracking for vacuum residue with reaction temperature and concentrations of MoS2 dispersed catalysts. Catal. Today 2018, 305, 92–101. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, K.-D.; Lee, Y.-K. Effects of dispersed MoS2 catalysts and reaction conditions on slurry phase hydrocracking of vacuum residue. J. Catal. 2017, 347, 127–137. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Jung, J.; Lee, C.W.; Cho, J. Effect of asphaltene dispersion on slurry-phase hydrocracking of heavy residual hydrocarbons. Fuel 2018, 214, 174–186. [Google Scholar] [CrossRef]
- Khadzhiev, S.N.; Kadiev, K.M.; Zhigalina, O.M.; Kadieva, M.K.; Khmelenin, D.N. Structure and Properties of Molybdenum Sulfide Nanoparticles Synthesized In situ in the Hydroconversion Process. Pet. Chem. 2015, 55, 655–662. [Google Scholar] [CrossRef]
- Sizova, I.A.; Maksimov, A.L. Nickel–molybdenum Sulfide Naphthalene Hydrogenation Catalysts Synthesized by the In situ Decomposition of Oil-soluble Precursors. Pet. Chem. 2017, 57, 595–599. [Google Scholar] [CrossRef]
- Khadzhiev, S.N.; Kadiev, K.M.; Kadieva, M.K. Synthesis and Properties of Nanosized Systems as Efficient Catalysts for Hydroconversion of Heavy Petroleum Feedstock. Pet. Chem. 2014, 54, 323–346. [Google Scholar] [CrossRef]
- Onishchenko, M.I.; Kulikov, A.B.; Maksimov, A.L. Application of Zeolite Y-Based Ni–W Supported and In Situ Prepared Catalysts in the Process of Vacuum Gas Oil Hydrocracking. Pet. Chem. 2017, 57, 1287–1294. [Google Scholar] [CrossRef]
- Onishchenko, M.I.; Maksimov, A.L. Activity of Supported and In Situ Synthesized Beta Zeolite Catalysts in the Hydrocracking of Vacuum Gas Oil. Pet. Chem. 2018, 58, 651–658. [Google Scholar] [CrossRef]
- Maximov, A.L.; Sizova, I.A.; Khadzhiev, S.N. Catalysis in a Dispersion Medium for he Hydrogenation of Aromatics and Hydrodearomatization in Oil Refining. Pure Appl. Chem. 2017, 89, 1145–1155. [Google Scholar] [CrossRef]
- Shuyi, Z.; Wenan, D.; Hui, L.; Dong, L.; Guohe, Q. Slurry-phase Residue Hydrocracking with Dispersed Nickel Catalyst. Energy Fuels 2008, 22, 3583–3586. [Google Scholar] [CrossRef]
- Dong, L.; Hui, D.; Jichang, Z.; Guohe, Q. Reverse Microemulsion Synthesis and Characterization of Nano Nickel Sulfide Catalyst for Residue Slurry-Phase Hydrocracking. Energy Fuels 2015, 29, 3353–3358. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, D.; Deng, W.; Que, G. A Review of Slurry-Phase Hydrocracking Heavy Oil Technology. Energy Fuels 2007, 21, 3057–3062. [Google Scholar] [CrossRef]
- Khadzhiev, S.N. Nanoheterogeneous catalysis: A New Sector of Nanotechnologies in Chemistry and Petroleum Chemistry (A Review). Pet. Chem. 2011, 51, 1–15. [Google Scholar] [CrossRef]
- Khadzhiev, S.N. Nanoheterogeneous catalysis: Definition, State, and Research Prospects (Review). Pet. Chem. 2016, 56, 465–479. [Google Scholar] [CrossRef]
- Hur, Y.G.; Lee, D.-W.; Lee, K.-Y. Hydrocracking of Vacuum Residue Using NiWS(x) Dispersed Catalysts. Fuel 2016, 185, 794–803. [Google Scholar] [CrossRef]
- Nguyen, T.S.; Tayakout-Fayolle, M.; Lacroix, M.; Gotteland, D.; Aouine, M.; Bacaud, R.; Afanasiev, P.; Geantet, C. Promotion Effects With Dispersed Catalysts for Residue Slurry Hydroconversion. Fuel 2015, 160, 50–56. [Google Scholar] [CrossRef]
- Maity, S.K.; Ancheyta, J.; Alonso, F.; Rayo, P. Hydrodesulfurization activity of used hydrotreating catalysts. Fuel Proc. Tech. 2013, 106, 453–459. [Google Scholar] [CrossRef]
- Del Bianco, A.; Panariti, N.; Di Carlo, S.; Beltrame, P.L.; Carniti, P. New Developments in Deep Hydroconversion of Heavy Oil Residues with Dispersed Catalysts. 2. Kinetic Aspects of Reaction. Energy Fuels 1994, 8, 593–597. [Google Scholar] [CrossRef]
- Ortiz-Moreno, H.; Ramírez, J.; Cuevas, R.; Marroquín, G.; Ancheyta, J. Heavy oil upgrading at moderate pressure using dispersed catalysts: Effects of temperature, pressure and catalytic precursor. Fuel 2012, 100, 186–192. [Google Scholar] [CrossRef]
- Jeon, S.G.; Na, J.-G.; Ko, C.H.; Lee, K.B.; Rho, N.S.; Park, S.B. A new approach for preparation of oil-soluble bimetallic dispersed catalyst from layered ammonium nickel molybdate. Mater. Sci. Eng. B 2011, 176, 606–610. [Google Scholar] [CrossRef]
- Liu, D.; Kong, X.; Li, M.; Que, G. Study on a Water-Soluble Catalyst for Slurry-Phase Hydrocracking of an Atmospheric Residue. Energy Fuels 2009, 23, 958–961. [Google Scholar] [CrossRef]
- Angeles, M.J.; Leyva, C.; Ancheyta, J.; Ramírez, S. A review of experimental procedures for heavy oil hydrocracking with dispersed catalyst. Catal. Today 2014, 220–222, 274–294. [Google Scholar] [CrossRef]
- Rispoli, G.F.; Bellussi, G. Process for the Refining of Crude Oil. E.P. Patent No. 2,633,002, 4 September 2013. [Google Scholar]
- Bellussi, G.; Rispoli, G.; Landoni, A.; Millini, R.; Molinari, D.; Montanari, E.; Moscotti, D.; Pollesel, P. Hydroconversion of heavy residues in slurry reactors: Developments and perspectives. J. Catal. 2013, 308, 189–200. [Google Scholar] [CrossRef]
- Cyr, T.; Lewkowicz, L.; Ozum, B. Hydrocracking Process Involving Colloidal Catalyst Formed In Situ. U.S. Patent No. 5,578,197, 26 November 1996. [Google Scholar]
- Sizova, I.A.; Kulikov, A.B.; Onishchenko, M.I.; Serdyukov, S.I.; Maksimov, A.L. Synthesis of Nickel–tungsten Sulfide Hydrodearomatization Catalysts by the Decomposition of Oil-soluble Precursors. Pet. Chem. 2016, 56, 44–50. [Google Scholar] [CrossRef]
- Strausz, O.P. Process for Hydrocracking Heavy Oil. U.S. Patent No. 6,068,758, 30 May 2000. [Google Scholar]
- Aldridge, C.L.; Bearden, R.J. Hydroconversion of Heavy Hydrocarbons. U.S. Patent No. 4,066,530, 3 January 1978. [Google Scholar]
- Zhou, B.; Zhou, Z.; Wu, Z. Hydrocarbon-Soluble, Bimetallic Catalyst Precursors and Methods for Making Same. U.S. Patent No. 7,842,635, 30 November 2010. [Google Scholar]
- Panariti, N.; Del Bianco, A.; Del Piero, G.; Marchionna, M. Petroleum residue upgrading with dispersed catalysts: Part 1. Catalysts activity and selectivity. Appl. Catal. A Gen. 2000, 204, 203–213. [Google Scholar] [CrossRef]
- Du, H.; Li, M.; Ren, Y.; Duan, Y. Slurry-phase hydrocracking of heavy oil and model reactant: Effect of dispersed Mo catalyst. Appl. Petrochem. Res. 2015, 5, 89–98. [Google Scholar] [CrossRef]
- Daage, M.; Chianelli, R.R. Structure-Function Relations in Molybdenum Sulfide Catalysts: The “Rim-Edge” Model. J. Catal. 1994, 149, 414–427. [Google Scholar] [CrossRef]
- Kennepohl, D.; Sanford, E. Conversion of Athabasca Bitumen with Dispersed and Supported Mo-Based Catalysts as a Function of Dispersed Catalyst Concentration. Energy Fuels 1996, 10, 229–234. [Google Scholar] [CrossRef]
- Bellussi, G.; Rispoli, G.; Molinari, D.; Landoni, A.; Pollesel, P.; Panariti, N.; Millini, R.; Montanari, E. The role of MoS2 nano-slabs in the protection of solid cracking catalysts for the total conversion of heavy oils to good quality distillates. Catal. Sci. Technol. 2013, 3, 176–182. [Google Scholar] [CrossRef]
- Ferraz, S.G.A.; Santos, B.M.; Zotin, F.M.Z.; Araujo, L.R.R.; Zotin, J.L. Influence of Support Acidity of NiMo Sulfide Catalysts for Hydrogenation and Hydrocracking of Tetralin and Its Reaction Intermediates. Ind. Eng. Chem. Res. 2015, 54, 2646–2656. [Google Scholar] [CrossRef]
- Luck, F. A review of support effects on the activity and selectivity of hydrotreating catalysts. Bull. Soc. Chim. Belg. 1991, 100, 781–800. [Google Scholar] [CrossRef]
- Breysse, M.; Afanasiev, P.; Geantet, C.; Vrinat, M. Overview of support effects in hydrotreating catalysts. Catal. Today. 2003, 86, 5–16. [Google Scholar] [CrossRef]
- Nishijima, A.; Shimada, H.; Sato, T.; Yoshimura, Y.; Hiraishi, J. Support effects on hydrocarcking and hydrogenation activities of molybdenum catlysts used for upgrading coal-derived liquids. Polyhedron 1986, 5, 243–247. [Google Scholar] [CrossRef]
- Shimada, H.; Sato, T.; Yoshimura, Y.; Hiraishi, J.; Nishijima, A. Support effect on the catalytic activity and properties of sulfided molybdenum catalysts. J. Catal. 1988, 110, 275–284. [Google Scholar] [CrossRef]
- Castillo-Villalón, P.; Ramírez, J.; Cuevas, R.; Vázquez, P.; Castañeda, R. Influence of the support on the catalytic performance of Mo, CoMo, and NiMo catalysts supported on Al2O3 and TiO2 during the HDS of thiophene, dibenzothiophene, or 4,6-dimethyldibenzothiophene. Catal. Today 2015, 259, 140–149. [Google Scholar] [CrossRef]
- Ng, K.Y.S.; Gulari, E. Molybdena on titania: II. Thiophene hydrodesulfurization activity and selectivity. J. Catal. 1985, 95, 33–40. [Google Scholar] [CrossRef]
- Vissenberg, M.J.; der Meer, Y.; Hensen, E.J.M.; de Beer, V.H.J.; van der Kraan, A.M.; van Santen, R.A.; van Veen, J.A.R. The Effect of Support In teraction on the Sulfidability of Al2O3- and TiO2-Supported CoW and NiW Hydrodesulfurization Catalysts. J. Catal. 2001, 198, 151–163. [Google Scholar] [CrossRef]
- Shimada, H. Morphology, Dispersion and Catalytic Functions of Supported Molybdenum Sulfide Catalysts for Hydrotreating Petroleum Fractions. J. Jpn. Pet. Inst. 2016, 59, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Kasztelan, S.; Toulhoat, H.; Grimblot, J.; Bonnelle, J.P. A geometrical model of the active phase of hydrotreating catalysts. Appl. Catal. 1984, 13, 127–159. [Google Scholar] [CrossRef]
- Vradman, L.; Landau, M.V. Structure–Function Relations in Supported Ni–W Sulfide Hydrogenation Catalysts. Catal. Lett. 2001, 77, 47–54. [Google Scholar] [CrossRef]
- Stanislaus, A.; Cooper, B.H. Aromatic Hydrogenation Catalysis: A Review. Catal. Rev. Sci. Eng. 1994, 36, 75–123. [Google Scholar] [CrossRef]
- Ichihara, A.; Itoh, T.; Hino, T.; Nomura, M.; Qi, P.; Kabe, T. Effects of Solvents on Deep Hydrodesulfurization of Benzothiophene and Dibenzothiophene. J. Catal. 1993, 140, 184–189. [Google Scholar] [CrossRef]
- Shimada, H. Morphology and orientation of MoS2 clusters on Al2O3and TiO2 supports and their effect on catalytic performance. Catal. Today 2003, 86, 17–29. [Google Scholar] [CrossRef]
- Hensen, E.J.M.; van der Meer, Y.; van Veen, J.A.R.; Niemantsverdriet, J.W. Insight into the formation of the active phases in supported NiW hydrotreating catalysts. Appl. Catal. A Gen. 2007, 322, 16–32. [Google Scholar] [CrossRef]
- Díaz de León, J.N.; Antunes-García, J.; Alonso-Nuñez, G.; Zepeda, T.A.; Galvan, D.H.; de los Reyes, J.A.; Fuentes, S. Support effects of NiW hydrodesulfurization catalysts from experiments and DFT calculations. Appl. Catal. B Environ. 2018, 238, 480–490. [Google Scholar] [CrossRef]
- López-Benítez, A.; Berhault, G.; Burel, L.; Guevara-Lara, A. Novel NiW hydrodesulfurization catalysts supported on Sol-Gel Mn-Al2O3. J. Catal. 2017, 354, 197–212. [Google Scholar] [CrossRef]
- Palcheva, R.; Dimitrov, L.; Tyuliev, G.; Spojakina, A.; Jiratova, K. TiO2 nanotubes supported NiW hydrodesulphurization catalysts: Characterization and activity. App. Surf. Sci. 2013, 265, 309–316. [Google Scholar] [CrossRef]
- Pereyma, V.Y.; Klimov, O.V.; Prosvirin, I.P.; Gerasimov, E.Y.; Yashnik, S.A.; Noskov, A.S. Effect of thermal treatment on morphology and catalytic performance of NiW/Al2O3 catalysts prepared using citric acid as chelating agent. Catal. Today 2018, 305, 162–170. [Google Scholar] [CrossRef]
- Tayeb, K.; Lamonier, C.; Lancelot, C.; Fournier, M.; Bonduelle-Skrzypczak, A.; Bertonici, F. Active phase genesis of NiW hydrocracking catalysts based on nickel salt heteropolytungstate: Comparison with reference catalyst. Appl. Catal. B Environ. 2012, 126, 55–63. [Google Scholar] [CrossRef]
- Ninh, T.K.T.; Laurenti, D.; Leclerc, E.; Vrinat, M. Support effect for CoMoS and CoNiMoS hydrodesulfurization catalysts prepared by controlled method. Appl. Catal. A Gen. 2014, 487, 210–218. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Sepu´lveda-Escribano, A.; Reinoso, F.R. Preparation and characterization of zinc containing MCM-41 spheres. Microporous Mesoporous Mater. 2008, 113, 362–369. [Google Scholar] [CrossRef]
- Chiranjeevi, T.; Kumaran, G.M.; Gupta, J.K.; Murali, D.G. Synthesis and characterization of acidic properties of Al-HMS materials of varying Si/Al ratios. Termochim. Acta 2006, 443, 87–92. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | NiW | NiW-SiO2 | NiW-Al2O3 | NiW-TiO2 | NiW-Y | NiW-ASA |
|---|---|---|---|---|---|---|
| 5.1 | 4.3 | 3.6 | 3.3 | 3.1 | 4.0 | |
| 1.9 | 1.8 | 2.4 | 2.0 | 2.1 | 2.1 | |
| 0.23 | 0.27 | 0.32 | 0.35 | 0.37 | 0.30 |
| Catalyst | Concentration 1, Relative % | Effective Concentration 2, wt % | Effective Content 3 (wt.%) | The Promoter Ratio 4 | |||
|---|---|---|---|---|---|---|---|
| WS2 | Ni–W–S | W | Ni | WS2 | Ni–W–S | ||
| NiW | 60.1 | 47.9 | 9.1 | 5.8 | 5.5 | 2.8 | 0.51 |
| NiW-SiO2 | 52.8 | 56.2 | 6.3 | 2.6 | 3.3 | 1.5 | 0.45 |
| NiW-Al2O3 | 51.8 | 33.7 | 3.0 | 2.6 | 1.5 | 0.9 | 0.60 |
| NiW-TiO2 | 54.3 | 52.5 | 7.9 | 5.1 | 4.3 | 2.7 | 0.63 |
| NiW-ASA | 53.2 | 27.6 | 8.5 | 2.3 | 4.5 | 0.6 | 0.13 |
| NiW-Y | 52.5 | 47.6 | 7.7 | 3.5 | 4.0 | 1.7 | 0.42 |
| Material | Particle Size 1, nm | SBET 2, m2/g | C 3, μmol/g |
|---|---|---|---|
| SiO2 | 900–1000 | 1100 | 13 |
| Al2O3 | 15–25 | 95 | 157 |
| TiO2 | 100–150 | 41 | 77 |
| Y (molar ratio SiO2/Al2O3 = 5.2) | 400–800 | 530 | 532 |
| ASA (molar ratio SiO2/Al2O3 = 20) | 800–1000 | 850 | 170 |
| Parameter | Test Results, °C |
|---|---|
| Fractional composition distillation temperature: | |
| IBP1 | 333 |
| 10% | 360 |
| 50% | 437 |
| EBP2 | 537 |
| Sulfur content, wt % | 1.86 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kniazeva, M.; Maximov, A. Effect of Additives on the Activity of Nickel–Tungsten Sulfide Hydroconversion Catalysts Prepared In Situ from Oil-Soluble Precursors. Catalysts 2018, 8, 644. https://doi.org/10.3390/catal8120644
Kniazeva M, Maximov A. Effect of Additives on the Activity of Nickel–Tungsten Sulfide Hydroconversion Catalysts Prepared In Situ from Oil-Soluble Precursors. Catalysts. 2018; 8(12):644. https://doi.org/10.3390/catal8120644
Chicago/Turabian StyleKniazeva, Mariia, and Anton Maximov. 2018. "Effect of Additives on the Activity of Nickel–Tungsten Sulfide Hydroconversion Catalysts Prepared In Situ from Oil-Soluble Precursors" Catalysts 8, no. 12: 644. https://doi.org/10.3390/catal8120644