Recent Advances in the Gold-Catalysed Low-Temperature Water–Gas Shift Reaction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction and Scope of Review
2. Monometallic Catalysts
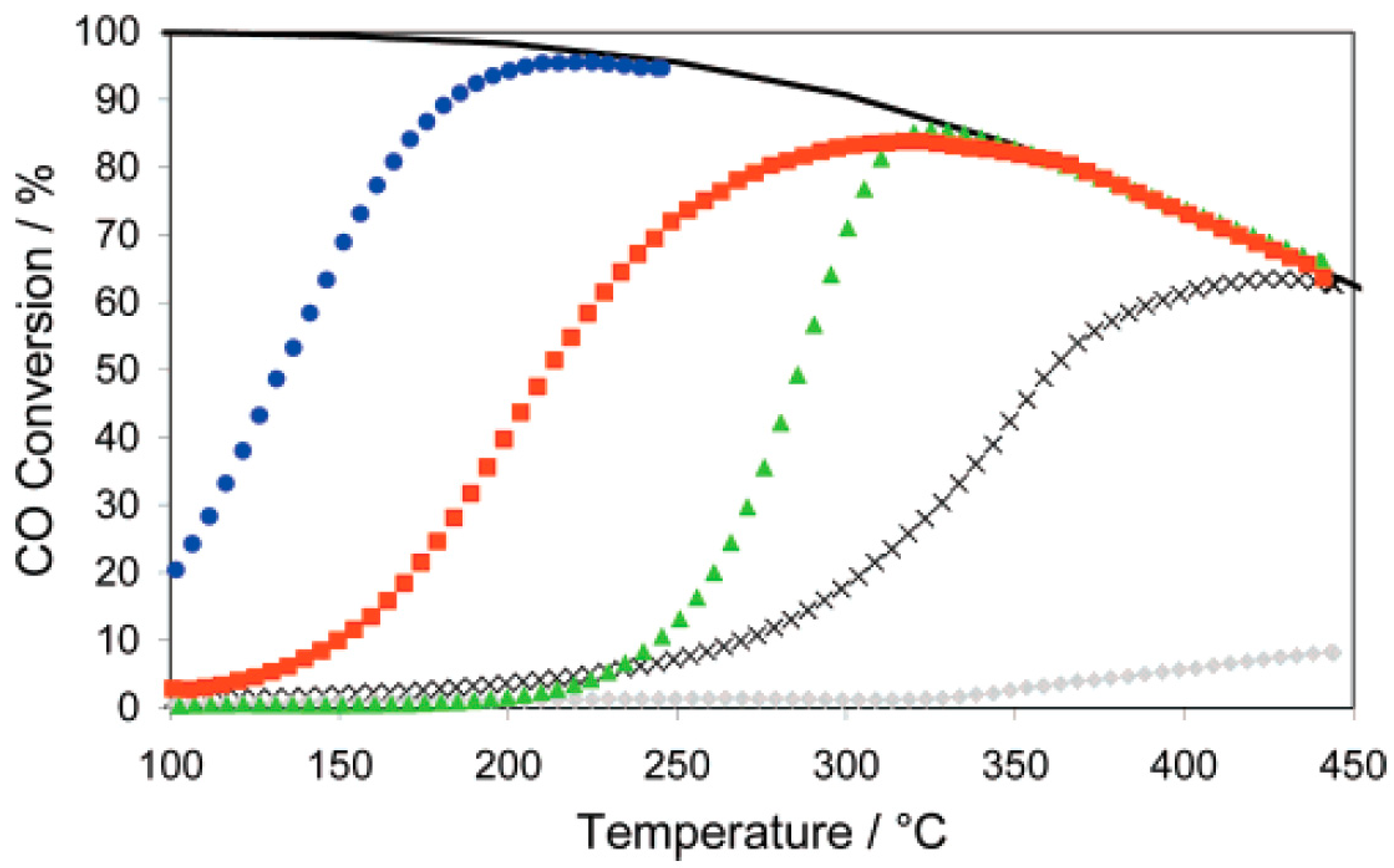
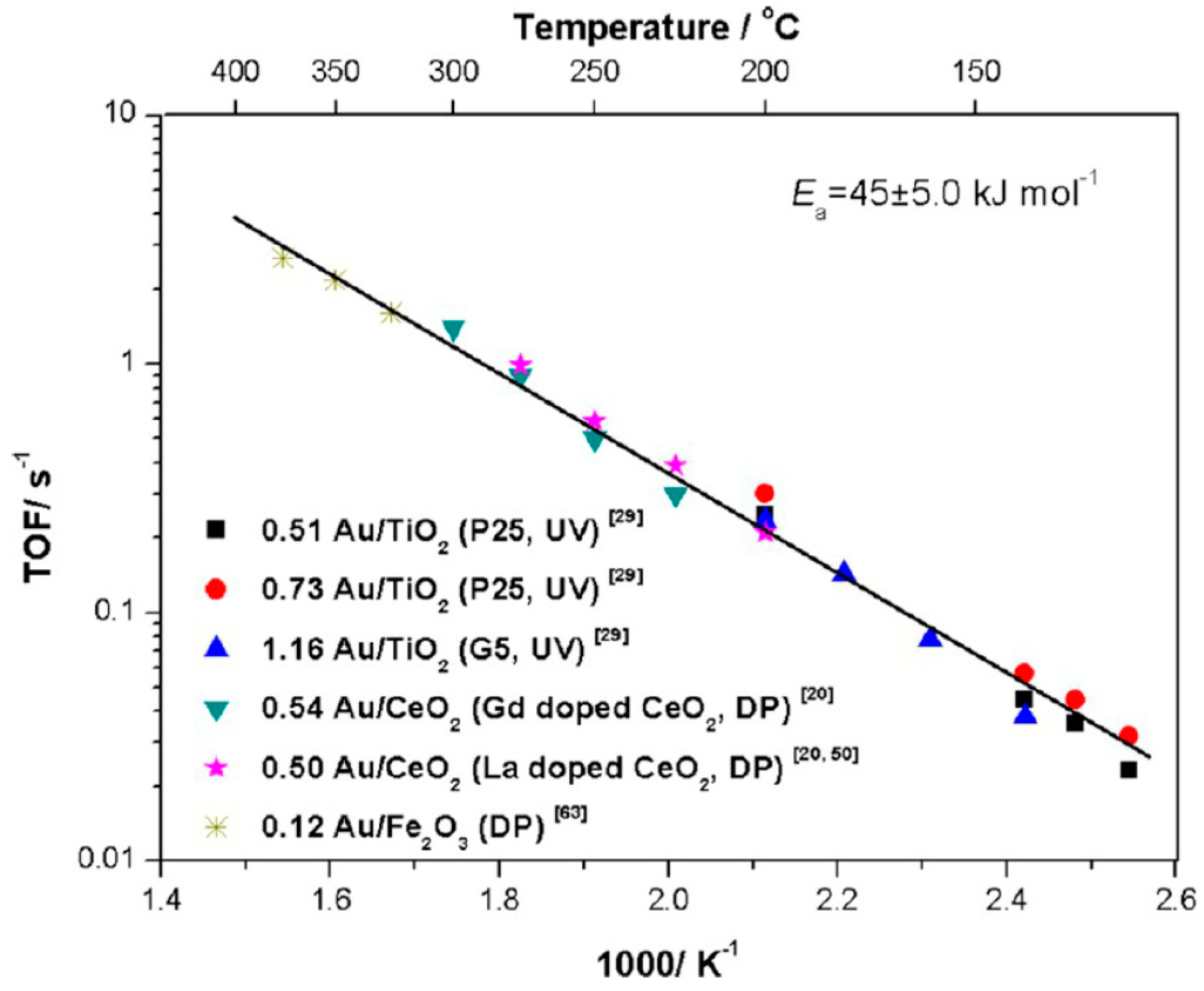
2.1. Gold Supported on Reducible Supports
2.2. Gold Supported on Non-Reducible Supports
3. Bimetallic Catalysts
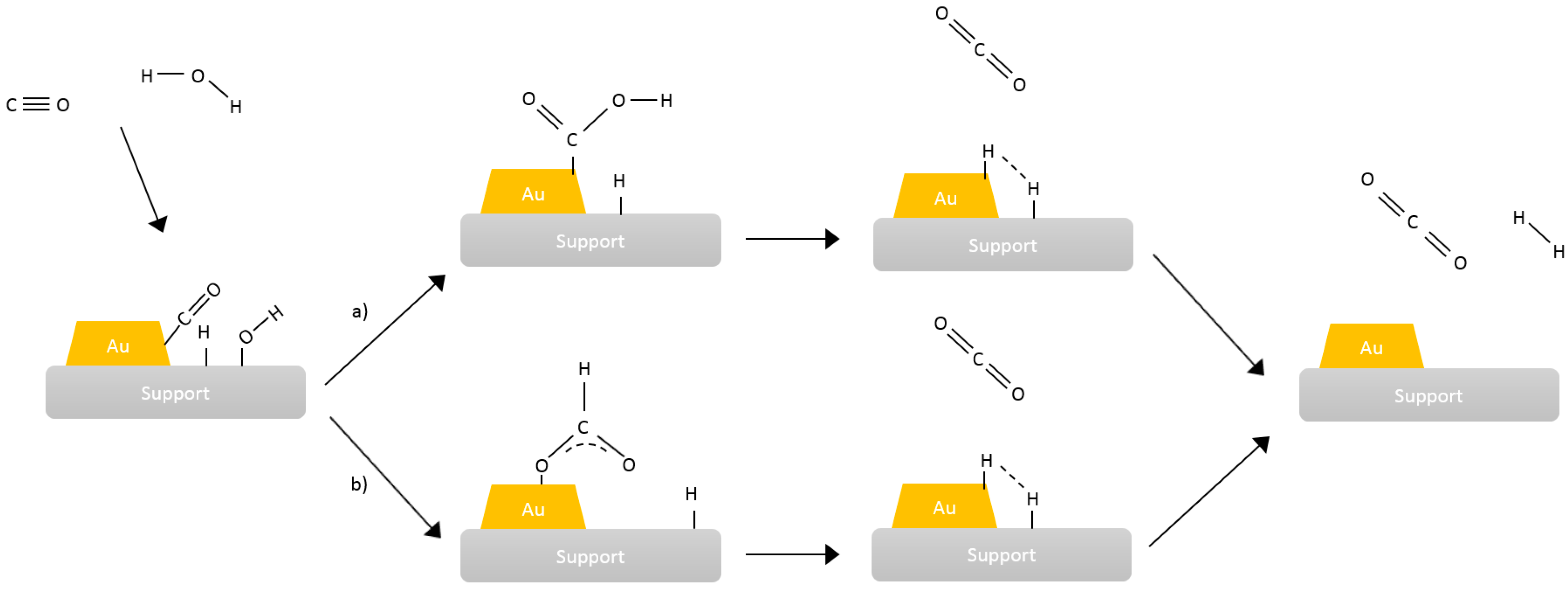
4. Mechanistic and Fundamental Studies
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Rodriguez, J.A.; Grinter, D.C.; Liu, Z.Y.; Palomino, R.M.; Senanayake, S.D. Ceria-based model catalysts: Fundamental studies on the importance of the metal-ceria interface in CO oxidation, the water-gas shift, CO2 hydrogenation, and methane and alcohol reforming. Chem. Soc. Rev. 2017, 46, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, L.; Petrova, P.; Ivanov, I.; Munteanu, G.; Boutonnet, M.; Sobczac, J.W.; Lisowski, W.; Kaszkur, Z.; Markov, P.; Venezia, A.M.; et al. Nanosized gold catalysts on Pr-modified ceria for pure hydrogen production via WGS reaction. Mater. Chem. Phys. 2015, 157, 138–146. [Google Scholar] [CrossRef]
- Ramirez Reina, T.; Ivanova, S.; Jose Delgado, J.; Ivanov, I.; Idakiev, V.; Tabakova, T.; Angel Centeno, M.; Antonio Odriozola, J. Viability of Au/CeO2-ZnO/Al2O3 Catalysts for Pure Hydrogen Production by the Water-Gas Shift Reaction. Chemcatchem 2014, 6, 1401–1409. [Google Scholar] [CrossRef]
- Barakat, T.; Rooke, J.C.; Genty, E.; Cousin, R.; Siffert, S.; Su, B.L. Gold catalysts in environmental remediation and water-gas shift technologies. Energy Environ. Sci. 2013, 6, 371–391. [Google Scholar] [CrossRef]
- Maciel, C.G.; Silva, T.d.F.; Assaf, E.M.; Assaf, J.M. Hydrogen production and purification from the water-gas shift reaction on CuO/CeO2-TiO2 catalysts. Appl. Energy 2013, 112, 52–59. [Google Scholar] [CrossRef]
- Ralph, T.R.; Hogarth, M.P. Catalysis for Low Temperature Fuel Cells. Platin. Met. Rev. 2002, 46, 146. [Google Scholar]
- Yao, S.Y.; Zhang, X.; Zhou, W.; Gao, R.; Xu, W.Q.; Ye, Y.F.; Lin, L.L.; Wen, X.D.; Liu, P.; Chen, B.B.; et al. Atomic-layered Au clusters on alpha-MoC as catalysts for the low-temperature water-gas shift reaction. Science 2017, 357, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ratnasamy, C.; Wagner, J.P. Water Gas Shift Catalysis. Catal. Rev. Sci. Eng. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Ramirez Reina, T.; González, M.; Sandra, P.; Ivanova, S.; Odriozola, J.A. Twenty years of golden future in the Water Gas Shift Reaction. In Heterogeneous Gold Catalysts and Catalysis; Ma, Z., Dai, S., Eds.; Royal Society of Chemistry: London, UK, 2014; pp. 111–139. [Google Scholar]
- Andreeva, D.; Idakiev, V.; Tabakova, T.; Andreev, A.; Giovanoli, R. Low-temperature water-gas shift reaction on Au/alpha-Fe2O3 catalyst. Appl. Catal. A Gen. 1996, 134, 275–283. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Andreeva, D.; Tabakova, T. FTIR study of the low-temperature water-gas shift reaction on Au/Fe2O3 and Au/TiO2 catalysts. J. Catal. 1999, 188, 176–185. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Low-temperature water-gas shift reaction over Cu- and Ni-loaded cerium oxide catalysts. Appl. Catal. B Environ. 2000, 27, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Q.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martinez-Arias, A.; Fernandez-Garcia, M. In situ studies of the active sites for the water gas shift reaction over Cu-CeO2 catalysts: Complex interaction between metallic copper and oxygen vacancies of ceria. J. Phys. Chem. B 2006, 110, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gorte, R.J. The effect of Fe and other promoters on the activity of Pd/ceria for the water-gas shift reaction. Appl. Catal. A Gen. 2003, 247, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au-CeO2 catalysts for low-temperature water-gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar] [CrossRef]
- Karpenko, A.; Leppelt, R.; Cai, J.; Plzak, V.; Chuvilin, A.; Kaiser, U.; Behm, R.J. Deactivation of a Au/CeO2 catalyst during the low-temperature water-gas shift reaction and its reactivation: A combined TEM, XRD, XPS, DRIFTS, and activity study. J. Catal. 2007, 250, 139–150. [Google Scholar] [CrossRef]
- Andreeva, D. Low temperature water gas shift over gold catalysts. Gold Bull. 2002, 35, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Mageed, A.M.; Kučerová, G.; Bansmann, J.; Behm, R.J. Active Au Species During the Low-Temperature Water Gas Shift Reaction on Au/CeO2: A Time-Resolved Operando XAS and DRIFTS Study. ACS Catal. 2017, 7, 6471–6484. [Google Scholar] [CrossRef]
- Tibiletti, D.; Amieiro-Fonseca, A.; Burch, R.; Chen, Y.; Fisher, J.M.; Goguet, A.; Hardacre, C.; Hu, P.; Thompsett, A. DFT and in situ EXAFS investigation of gold/ceria-zirconia low-temperature water gas shift catalysts: Identification of the nature of the active form of gold. J. Phys. Chem. B 2005, 109, 22553–22559. [Google Scholar] [CrossRef]
- Goguet, A.; Burch, R.; Chen, Y.; Hardacre, C.; Hu, P.; Joyner, R.W.; Meunier, F.C.; Mun, B.S.; Thompsett, A.; Tibiletti, D. Deactivation mechanism of a Au/CeZrO4 catalyst during a low-temperature water gas shift reaction. J. Phys. Chem. C 2007, 111, 16927–16933. [Google Scholar] [CrossRef]
- Daly, H.; Goguet, A.; Hardacre, C.; Meunier, F.C.; Pilasombat, R.; Thompsett, D. The effect of reaction conditions on the stability of Au/CeZrO4 catalysts in the low-temperature water-gas shift reaction. J. Catal. 2010, 273, 257–265. [Google Scholar] [CrossRef]
- Carter, J.; Liu, X.; He, Q.; Althahban, S.; Nowicka, E.; Freakley, S.; Niu, L.; Morgan, D.; Li, Y.; Niemantsverdriet, H.; et al. Activation and Deactivation of gold/ceria-zirconia in the Low-Temperature Water-Gas Shift Reaction. Angew. Chem. Int. Ed. 2017, 56, 16037–16041. [Google Scholar] [CrossRef] [PubMed]
- Stere, C.E.; Anderson, J.A.; Chansai, S.; Delgado, J.J.; Goguet, A.; Graham, W.G.; Hardacre, C.; Taylor, S.F.R.; Tu, X.; Wang, Z.Y.; et al. Non-Thermal Plasma Activation of Gold-Based Catalysts for Low-Temperature Water-Gas Shift Catalysis. Angew. Chem. Int. Ed. 2017, 56, 5579–5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Davies, C.J.; Lu, L.; Dawson, S.; Thetford, A.; Gibson, E.K.; Morgan, D.J.; Jones, W.; et al. Identification of single-site gold catalysis in acetylene hydrochlorination. Science 2017, 355, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, D.; Ivanov, I.; Ilieva, L.; Sobczak, J.W.; Avdeev, G.; Tabakova, T. Nanosized gold catalysts supported on ceria and ceria-alumina for WGS reaction: Influence of the preparation method. Appl. Catal. A Gen. 2007, 333, 153–160. [Google Scholar] [CrossRef]
- Andreeva, D.; Ivanova, I.; Ilieva, L.; Abrashev, M.V. Gold catalysts supported on ceria and ceria-alumina for water-gas shift reaction. Appl. Catal. A Gen. 2006, 302, 127–132. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Boosting the activity of a Au/CeO2/Al2O3 catalyst for the WGS reaction. Catal. Today 2015, 253, 149–154. [Google Scholar] [CrossRef]
- Reina, T.R.; Xu, W.Q.; Ivanova, S.; Centeno, M.A.; Hanson, J.; Rodriguez, J.A.; Odriozola, J.A. In situ characterization of iron-promoted ceria-alumina gold catalysts during the water-gas shift reaction. Catal. Today 2013, 205, 41–48. [Google Scholar] [CrossRef]
- Andreeva, D.; Kantcheva, M.; Ivanov, I.; Ilieva, L.; Sobczak, J.W.; Lisowski, W. Gold supported on ceria doped by Me3+ (Me = Al and Sm) for water gas shift reaction: Influence of dopant and preparation method. Catal. Today 2010, 158, 69–77. [Google Scholar] [CrossRef]
- Andreeva, D.; Ivanov, I.; Ilieva, L.; Abrashev, M.V.; Zanella, R.; Sobczak, J.W.; Lisowski, W.; Kantcheva, M.; Avdeev, G.; Petrov, K. Gold catalysts supported on ceria doped by rare earth metals for water gas shift reaction: Influence of the preparation method. Appl. Catal. A Gen. 2009, 357, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Vecchietti, J.; Collins, S.; Jose Delgado, J.; Malecka, M.; del Rio, E.; Chen, X.; Bernal, S.; Bonivardi, A. Gold Catalysts Supported on Cerium-Gallium Mixed Oxide for the Carbon Monoxide Oxidation and Water Gas Shift Reaction. Top. Catal. 2011, 54, 201–209. [Google Scholar] [CrossRef]
- Tabakova, T.; Ilieva, L.; Ivanov, I.; Zanella, R.; Sobczak, J.W.; Lisowski, W.; Kaszkur, Z.; Andreeva, D. Influence of the preparation method and dopants nature on the WGS activity of gold catalysts supported on doped by transition metals ceria. Appl. Catal. B Environ. 2013, 136, 70–80. [Google Scholar] [CrossRef]
- Kondrat, S.A.; Smith, P.J.; Wells, P.P.; Chater, P.A.; Carter, J.H.; Morgan, D.J.; Fiordaliso, E.M.; Wagner, J.B.; Davies, T.E.; Lu, L.; et al. Stable amorphous georgeite as a precursor to a high-activity catalyst. Nature 2016, 531, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.L.; Reina, T.R.; Ivanov, I.; Penkova, A.; Ivanova, S.; Tabakova, T.; Centeno, M.A.; Idakiev, V.; Odriozola, J.A. Multicomponent Au/Cu-ZnO-Al2O3 catalysts: Robust materials for clean hydrogen production. Appl. Catal. A Gen. 2018, 558, 91–98. [Google Scholar] [CrossRef]
- Santos, J.L.; Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Gold promoted Cu/ZnO/Al2O3 catalysts prepared from hydrotalcite precursors: Advanced materials for the WGS reaction. Appl. Catal. B Environ. 2017, 201, 310–317. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. The role of Au, Cu & CeO2 and their interactions for an enhanced WGS performance. Appl. Catal. B Environ. 2016, 187, 98–107. [Google Scholar] [CrossRef]
- Si, R.; Flytzani-Stephanopoulos, M. Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2 catalysts for the water-gas shift reaction. Angew. Chem. Int. Ed. 2008, 47, 2884–2887. [Google Scholar] [CrossRef]
- Ta, N.; Liu, J.; Chenna, S.; Crozier, P.A.; Li, Y.; Chen, A.; Shen, W. Stabilized Gold Nanoparticles on Ceria Nanorods by Strong Interfacial Anchoring. J. Am. Chem. Soc. 2012, 134, 20585–20588. [Google Scholar] [CrossRef]
- Yuan, Z.-Y.; Idakiev, V.; Vantomme, A.; Tabakova, T.; Ren, T.-Z.; Su, B.-L. Mesoporous and nanostructured CeO2 as supports of nano-sized gold catalysts for low-temperature water-gas shift reaction. Catal. Today 2008, 131, 203–210. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Gold Atoms Stabilized on Various Supports Catalyze the Water-Gas Shift Reaction. Accounts Chem. Res. 2014, 47, 783–792. [Google Scholar] [CrossRef]
- Sandoval, A.; Gomez-Cortes, A.; Zanella, R.; Diaz, G.; Saniger, J.M. Gold nanoparticles: Support effects for the WGS reaction. J. Mol. Catal. A Chem. 2007, 278, 200–208. [Google Scholar] [CrossRef]
- Shekhar, M.; Wang, J.; Lee, W.S.; Williams, W.D.; Kim, S.M.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Size and Support Effects for the Water-Gas Shift Catalysis over Gold Nanoparticles Supported on Model Al2O3 and TiO2. J. Am. Chem. Soc. 2012, 134, 4700–4708. [Google Scholar] [CrossRef] [PubMed]
- Gil, S.; Romero, A.; de Lucas, A.; Sanchez, P.; Dorado, F.; Raquel de la Osa, A.; Manuel Garcia-Vargas, J.; Luis Valverde, J. Nano-Scale Au Supported on Carbon Materials for the Low Temperature Water Gas Shift (WGS) Reaction. Catalysts 2011, 1, 155–174. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, S.; Wang, Y.; Herron, J.A.; Xu, Y.; Allard, L.F.; Lee, S.; Huang, J.; Mavrikakis, M.; Flytzani-Stephanopoulos, M. Catalytically active Au-O(OH)(x)-species stabilized by alkali ions on zeolites and mesoporous oxides. Science 2014, 346, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.F.; Guan, G.Q.; Hao, X.G.; Cao, J.; Abudula, A. Molybdenum carbide as alternative catalyst for hydrogen production—A review. Renew. Sustain. Energy Rev. 2017, 75, 1101–1129. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Water-gas-shift reaction on molybdenum carbide surfaces: Essential role of the oxycarbide. J. Phys. Chem. B 2006, 110, 19418–19425. [Google Scholar] [CrossRef]
- Patt, J.; Moon, D.J.; Phillips, C.; Thompson, L. Molybdenum carbide catalysts for water-gas shift. Catal. Lett. 2000, 65, 193–195. [Google Scholar] [CrossRef]
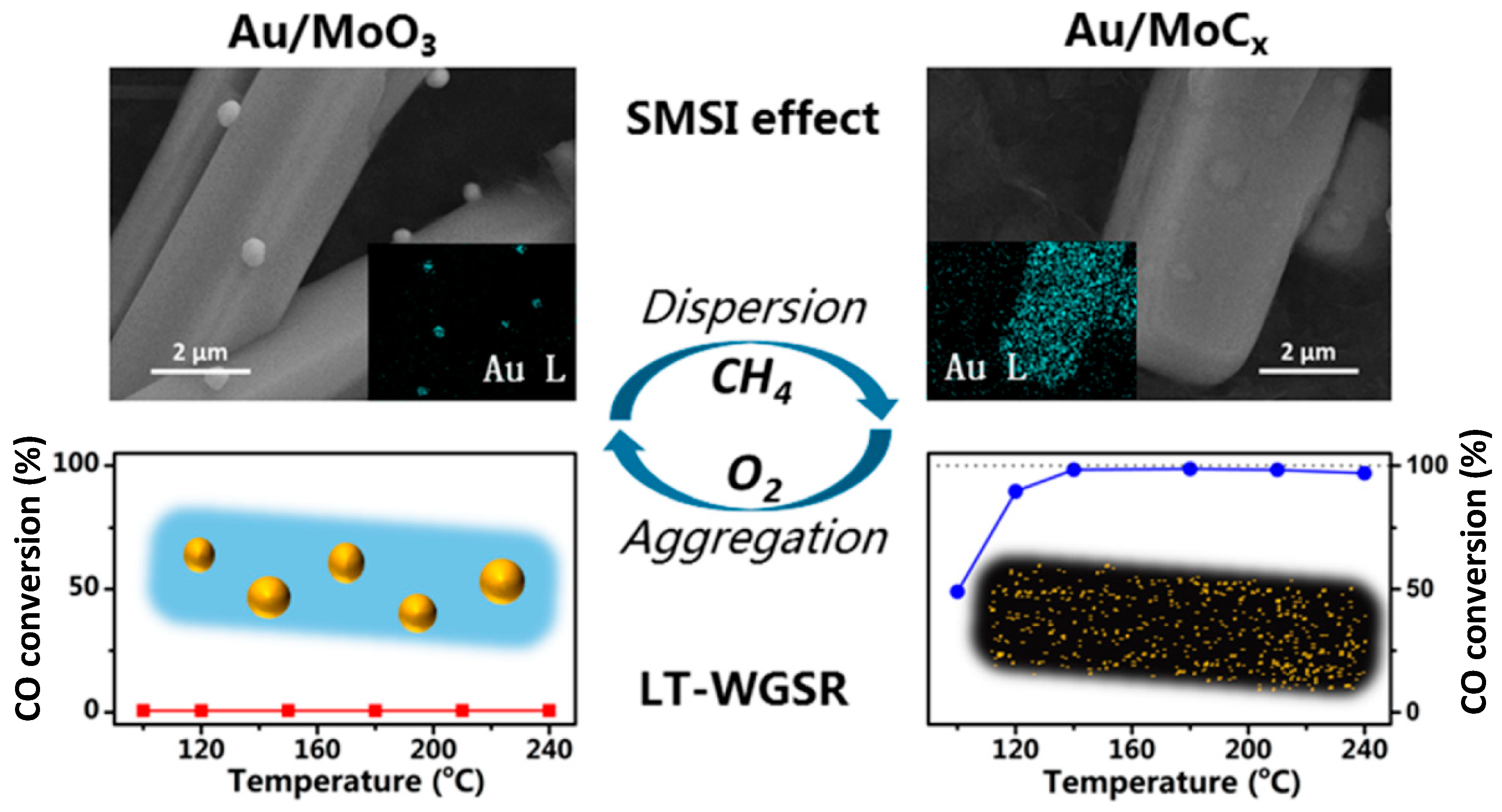
- Dong, J.; Fu, Q.; Jiang, Z.; Mei, B.; Bao, X. Carbide-Supported Au Catalysts for Water–Gas Shift Reactions: A New Territory for the Strong Metal–Support Interaction Effect. J. Am. Chem. Soc. 2018, 140, 13808–13816. [Google Scholar] [CrossRef]
- Tang, H.L.; Su, Y.; Zhang, B.S.; Lee, A.F.; Isaacs, M.A.; Wilson, K.; Li, L.; Ren, Y.G.; Huang, J.H.; Haruta, M.; et al. Classical strong metal-support interactions between gold nanoparticles and titanium dioxide. Sci. Adv. 2017, 3, e1700231. [Google Scholar] [CrossRef]
- Fu, Q.; Wagner, T.; Olliges, S.; Carstanjen, H.D. Metal-oxide interfacial reactions: Encapsulation of Pd on TiO2 (110). J. Phys. Chem. B 2005, 109, 944–951. [Google Scholar] [CrossRef]
- Song, L.; Cao, X.; Li, L. Engineering Stable Surface Oxygen Vacancies on ZrO2 by Hydrogen-Etching Technology: An Efficient Support of Gold Catalysts for Water-Gas Shift Reaction. ACS Appl. Mater. Interfaces 2018, 10, 31249–31259. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Song, L.; Zhu, L.F.; Yan, Z.; Cao, X.B. Black TiO2-x with stable surface oxygen vacancies as the support of efficient gold catalysts for water-gas shift reaction. Catal. Sci. Technol. 2018, 8, 1277–1287. [Google Scholar] [CrossRef]
- Song, L.; Lu, Z.F.; Zhang, Y.T.; Su, Q.; Li, L. Hydrogen-Etched TiO2-x as Efficient Support of Gold Catalysts for Water-Gas Shift Reaction. Catalysts 2018, 8, 26. [Google Scholar] [CrossRef]
- Kesavan, L.; Tiruvalam, R.; Ab Rahim, M.H.; bin Saiman, M.I.; Enache, D.I.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Knight, D.W.; et al. Solvent-Free Oxidation of Primary Carbon-Hydrogen Bonds in Toluene Using Au-Pd Alloy Nanoparticles. Science 2011, 331, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold-Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Sankar, M.; Nowicka, E.; Tiruvalam, R.; He, Q.; Taylor, S.H.; Kiely, C.J.; Bethell, D.; Knight, D.W.; Hutchings, G.J. Controlling the Duality of the Mechanism in Liquid-Phase Oxidation of Benzyl Alcohol Catalysed by Supported Au-Pd Nanoparticles. Chem. A Eur. J. 2011, 17, 6524–6532. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, A.; Aluha, J.; Scurrell, M.S. The water-gas shift reaction over Au-based, bimetallic catalysts. The Au-M (M = Ag, Bi, Co, Cu, Mn, Ni, Pb, Ru, Sn, Tl) on iron(III) oxide system. Catal. Lett. 2003, 90, 1–6. [Google Scholar] [CrossRef]
- Venugopal, A.; Aluha, J.; Mogano, D.; Scurrell, M.S. The gold-ruthenium-iron oxide catalytic system for the low temperature water-gas-shift reaction—The examination of gold-ruthenium interactions. Appl. Catal. A Gen. 2003, 245, 149–158. [Google Scholar] [CrossRef]
- Hurtado-Juan, M.-A.; Yeung, C.M.Y.; Tsang, S.C. A study of co-precipitated bimetallic gold catalysts for water-gas shift reaction. Catal. Commun. 2008, 9, 1551–1557. [Google Scholar] [CrossRef]
- Burch, R.; Goguet, A.; Meunier, F.C. A critical analysis of the experimental evidence for and against a formate mechanism for high activity water-gas shift catalysts. Appl. Catal. A Gen. 2011, 409, 3–12. [Google Scholar] [CrossRef]
- Castano, M.G.; Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Pt vs. Au in water-gas shift reaction. J. Catal. 2014, 314, 1–9. [Google Scholar] [CrossRef]
- Carter, J.H.; Althahban, S.; Nowicka, E.; Freakley, S.J.; Morgan, D.J.; Shah, P.M.; Golunski, S.; Kiely, C.J.; Hutchings, G.J. Synergy and Anti-Synergy between Palladium and Gold in Nanoparticles Dispersed on a Reducible Support. ACS Catal. 2016, 6, 6623–6633. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.K.; Beale, A.M.; Catlow, C.R.A.; Chutia, A.; Gianolio, D.; Gould, A.; Kroner, A.; Mohammed, K.M.H.; Perdjon, M.; Rogers, S.M.; et al. Restructuring of AuPd Nanoparticles Studied by a Combined XAFS/DRIFTS Approach. Chem. Mater. 2015, 27, 3714–3720. [Google Scholar] [CrossRef]
- Zhu, B.; Thrimurthulu, G.; Delannoy, L.; Louis, C.; Mottet, C.; Creuze, J.; Legrand, B.; Guesmi, H. Evidence of Pd segregation and stabilization at edges of AuPd nano-clusters in the presence of CO: A combined DFT and DRIFTS study. J. Catal. 2013, 308, 272–281. [Google Scholar] [CrossRef]
- Saqlain, M.A.; Hussain, A.; Siddiq, M.; Leitao, A.A. Synergy between Pd and Au in a Pd-Au(100) bimetallic surface for the water gas shift reaction: A DFT study. RSC Adv. 2015, 5, 47066–47073. [Google Scholar] [CrossRef]
- Rodriguez, J.A. Gold-based catalysts for the water-gas shift reaction: Active sites and reaction mechanism. Catal. Today 2011, 160, 3–10. [Google Scholar] [CrossRef]
- Bond, G. Mechanisms of the gold-catalysed water-gas shift. Gold Bull. 2009, 42, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Tao, F.; Ma, Z. Water-gas shift on gold catalysts: Catalyst systems and fundamental studies. Phys. Chem. Chem. Phys. 2013, 15, 15260–15270. [Google Scholar] [CrossRef] [PubMed]
- Tabakova, T.; Boccuzzi, F.; Manzoli, M.; Sobczak, J.W.; Idakiev, V.; Andreeva, D. A comparative study of nanosized IB/ceria catalysts for low-temperature water-gas shift reaction. Appl. Catal. A Gen. 2006, 298, 127–143. [Google Scholar] [CrossRef]
- Burch, R. Gold catalysts for pure hydrogen production in the water-gas shift reaction: Activity, structure and reaction mechanism. Phys. Chem. Chem. Phys. 2006, 8, 5483–5500. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Akita, T.; Tsubota, S.; Kiuchi, M.; Haruta, M. Low-temperature activity of Au/CeO2 for water gas shift reaction, and characterization by ADF-STEM, temperature-programmed reaction, and pulse reaction. Appl. Catal. A Gen. 2005, 291, 179–187. [Google Scholar] [CrossRef]
- Jacobs, G.; Chenu, E.; Patterson, P.M.; Williams, L.; Sparks, D.; Thomas, G.; Davis, B.H. Water-gas shift: Comparative screening of metal promoters for metal/ceria systems and role of the metal. Appl. Catal. A Gen. 2004, 258, 203–214. [Google Scholar] [CrossRef]
- Leppelt, R.; Schumacher, B.; Plzak, V.; Kinne, M.; Behm, R.J. Kinetics and mechanism of the low-temperature water-gas shift reaction on Au/CeO2 catalysts in an idealized reaction atmosphere. J. Catal. 2006, 244, 137–152. [Google Scholar] [CrossRef]
- Meunier, F.C.; Reid, D.; Goguet, A.; Shekhtman, S.; Hardacre, C.; Burch, R.; Deng, W.; Flytzani-Stephanopoulos, M. Quantitative analysis of the reactivity of formate species seen by DRIFTS over a Au/Ce(La)O-2 water-gas shift catalyst: First unambiguous evidence of the minority role of formates as reaction intermediates. J. Catal. 2007, 247, 277–287. [Google Scholar] [CrossRef]
- Jacobs, G.; Patterson, P.M.; Graham, U.M.; Crawford, A.C.; Davis, B.H. Low temperature water gas shift: The link between the catalysis of WGS and formic acid decomposition over Pt/ceria. Int. J. Hydrogen Energy 2005, 30, 1265–1276. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Senanayake, S.D.; Stacchiola, D.; Liu, P.; Hrbek, J. The Activation of Gold and the Water-Gas Shift Reaction: Insights from Studies with Model Catalysts. Accounts Chem. Res. 2014, 47, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.Y.; Guo, L.; Cao, Z.R.; Li, A.X.; An, X.Y. Density functional theory study of water-gas shift reaction on TM@Cu-12 core-shell nanoclusters. Prot. Met. Phys. Chem. Surf. 2016, 52, 387–398. [Google Scholar] [CrossRef]
- Saqlain, M.A.; Hussain, A.; Siddiq, M.; Leenaerts, O.; Leitao, A.A. DFT Study of Synergistic Catalysis of the Water-Gas-Shift Reaction on Cu-Au Bimetallic Surfaces. Chemcatchem 2016, 8, 1208–1217. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carter, J.H.; Hutchings, G.J. Recent Advances in the Gold-Catalysed Low-Temperature Water–Gas Shift Reaction. Catalysts 2018, 8, 627. https://doi.org/10.3390/catal8120627
Carter JH, Hutchings GJ. Recent Advances in the Gold-Catalysed Low-Temperature Water–Gas Shift Reaction. Catalysts. 2018; 8(12):627. https://doi.org/10.3390/catal8120627
Chicago/Turabian StyleCarter, James H., and Graham J. Hutchings. 2018. "Recent Advances in the Gold-Catalysed Low-Temperature Water–Gas Shift Reaction" Catalysts 8, no. 12: 627. https://doi.org/10.3390/catal8120627