
Gold-Based Catalysts for Complete Formaldehyde Oxidation: Insights into the Role of Support Composition

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
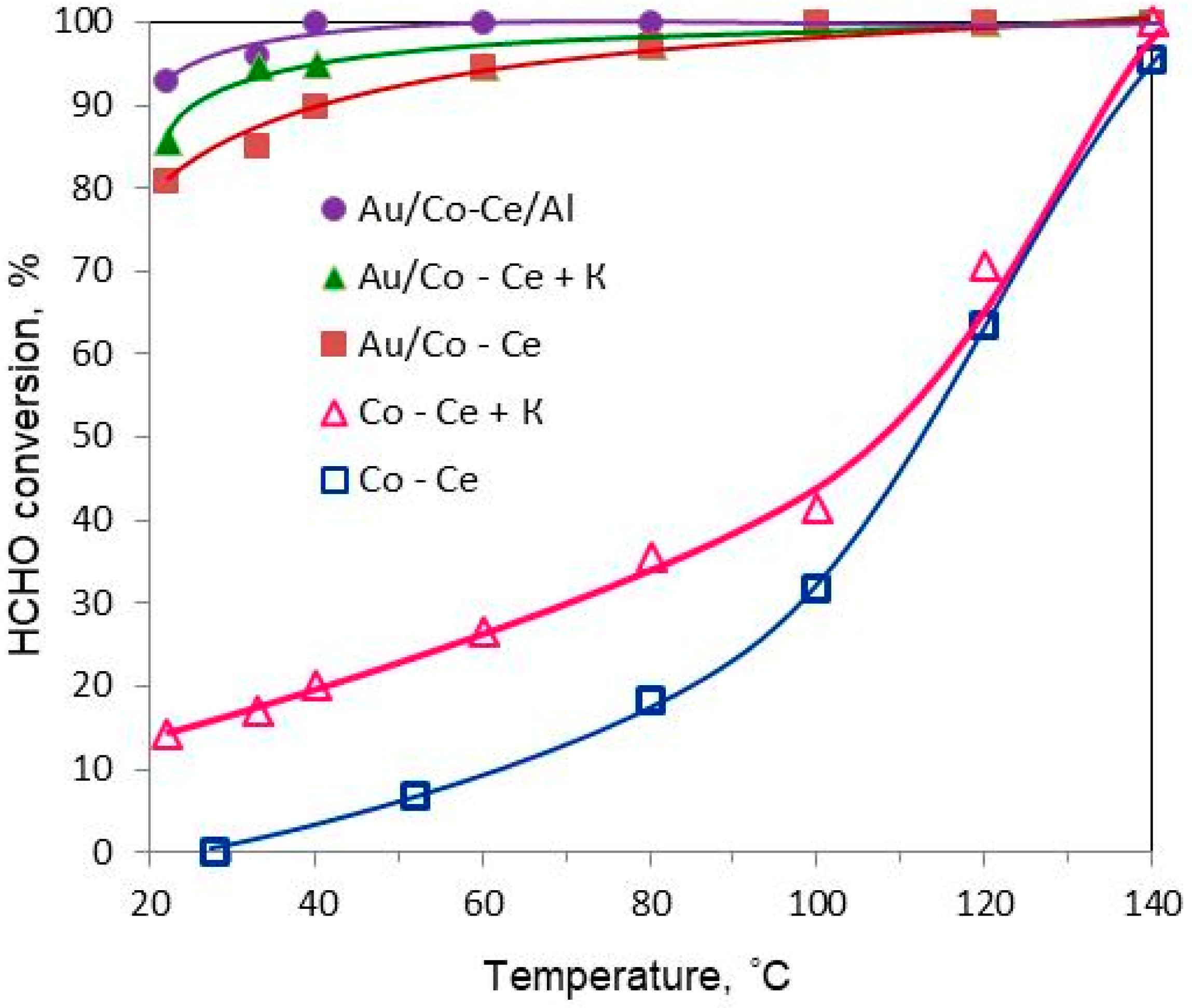
2.1. Catalytic Activity in HCHO Oxidation
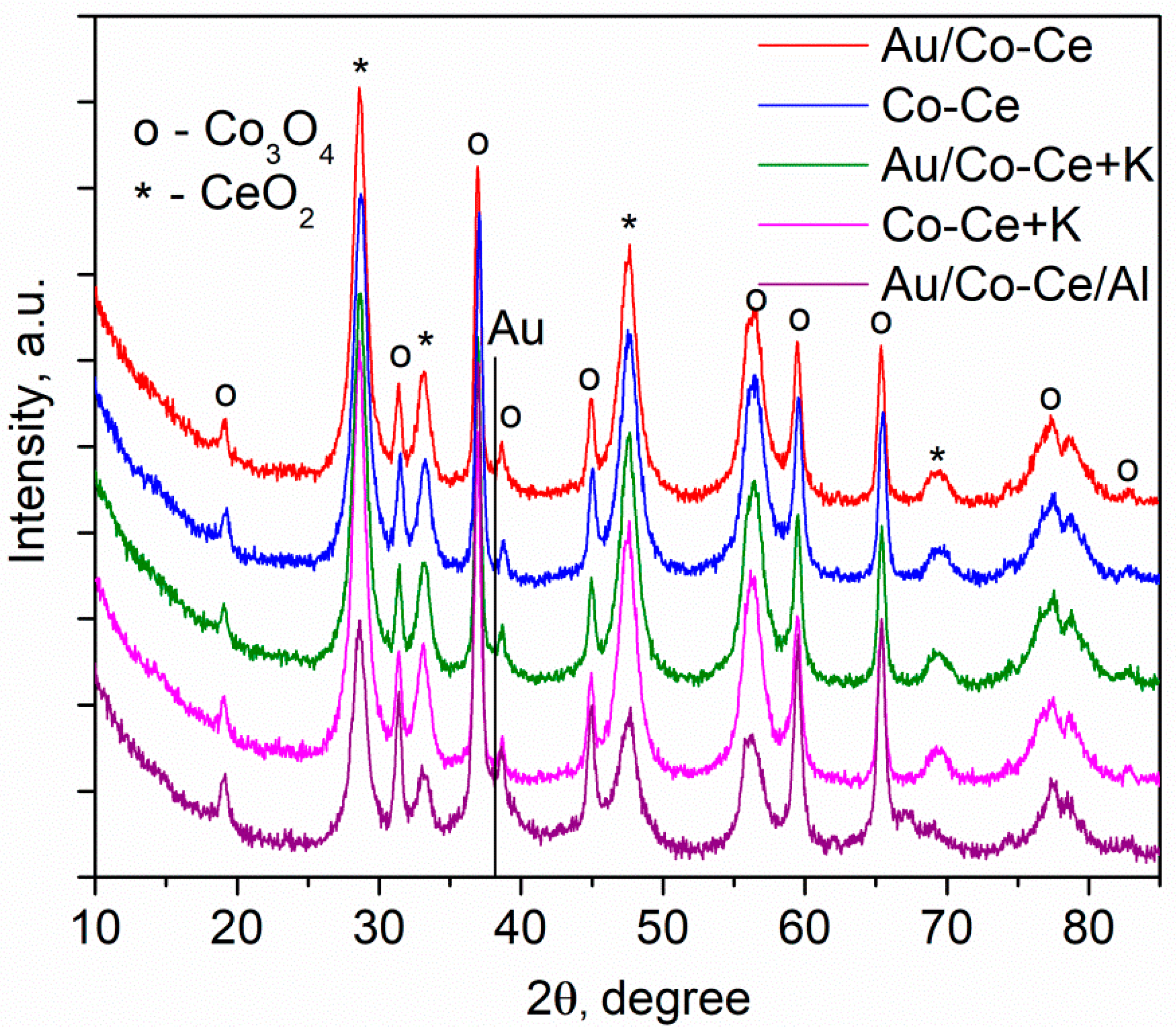
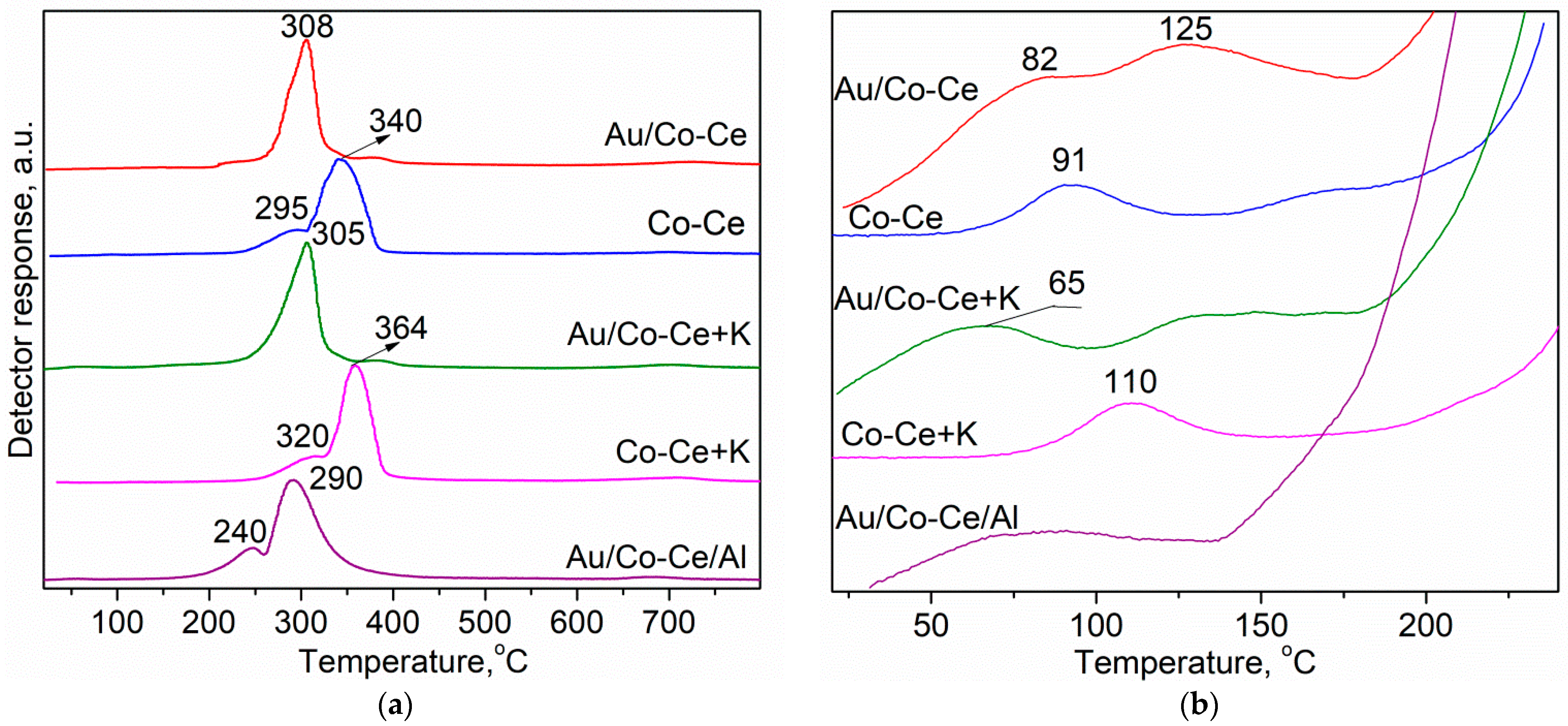
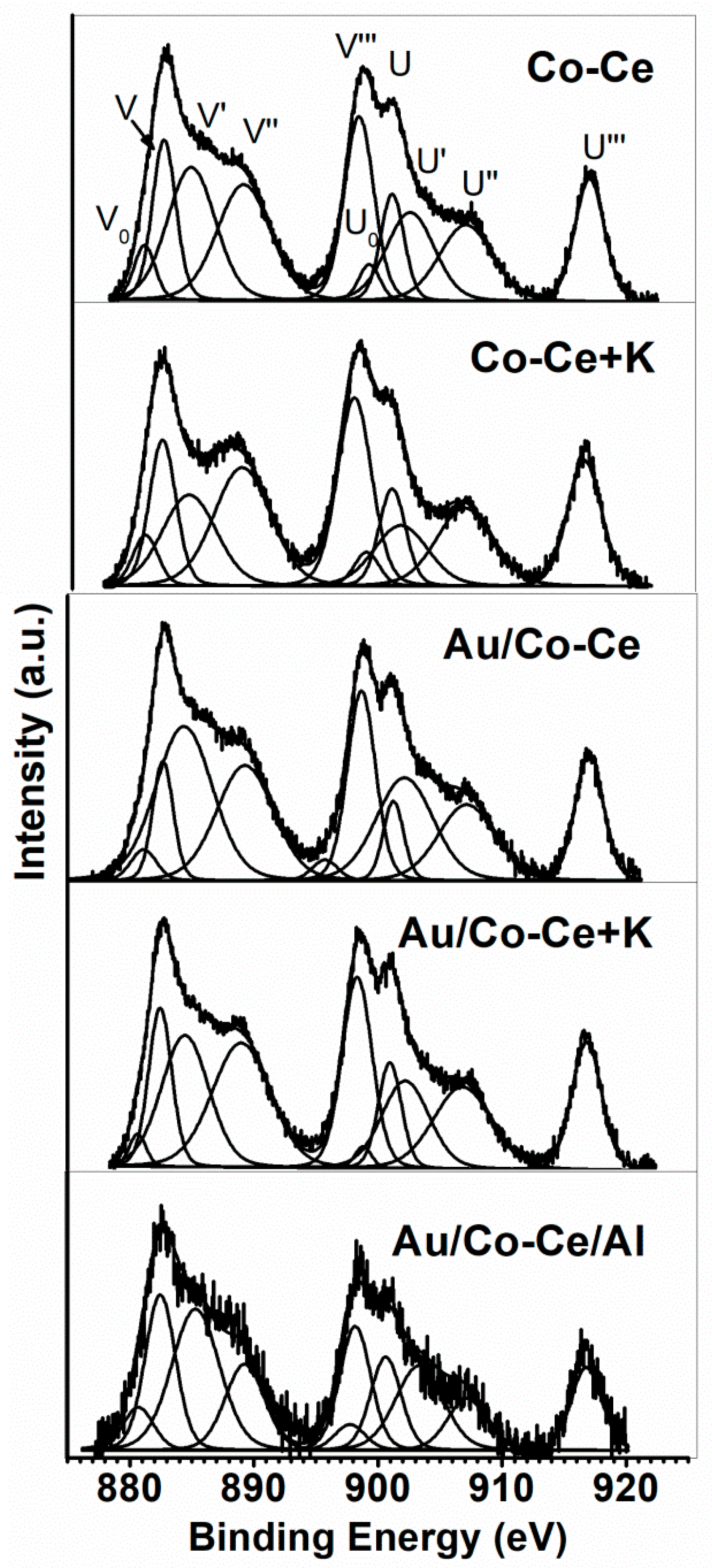
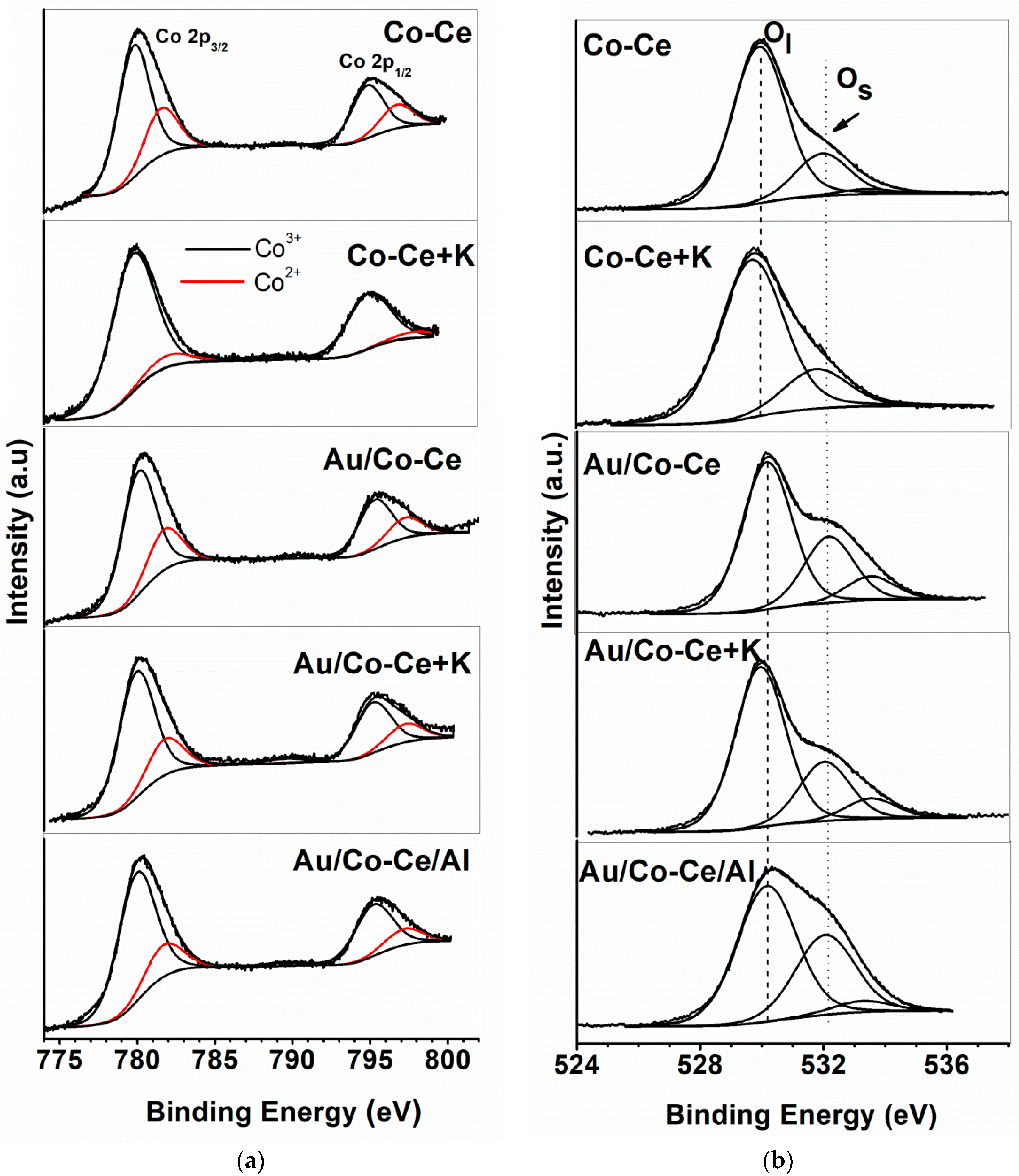
2.2. Sample Characterization
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalytic Activity Measurements
3.3. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bai, B.; Qiao, Q.; Li, J.; Hao, J. Progress in research on catalysts for catalytic oxidation of formaldehyde. Chin. J. Catal. 2016, 37, 102–122. [Google Scholar] [CrossRef]
- Guo, J.; Lin, C.; Jiang, C.; Zhang, P. Review on noble metal-based catalysts for formaldehyde oxidation at room temperature. Appl. Surf. Sci. 2019, 475, 237–255. [Google Scholar] [CrossRef]
- Yusuf, C.; Snape, J.; He, H.; Xu, C.; Liu, M.; Zhao, G.Z.; Chen, B.; Tang, C.; Wang, J.; Behera, S.N. Advances on transition metal oxides catalysts for formaldehyde oxidation: A review. Catal. Rev. Sci. Eng. 2017, 59, 189–233. [Google Scholar] [CrossRef]
- Li, R.; Huang, Y.; Zhu, D.; Ho, W.; Lee, S.; Cao, J. A Review of Co3O4-based catalysts for formaldehyde oxidation at low temperature: Effect parameters and reaction mechanism. Aerosol Sci. Eng. 2020, 4, 147–168. [Google Scholar] [CrossRef]
- Ma, C.; Wang, D.; Xue, W.; Dou, B.; Wang, H.; Hao, Z. Investigation of formaldehyde oxidation over Co3O4–CeO2 and Au/Co3O4–CeO2 catalysts at room temperature: Effective removal and determination of reaction mechanism. Environ. Sci. Technol. 2011, 45, 3628–3634. [Google Scholar] [CrossRef]
- Bai, B.; Arandiyan, H.; Li, J. Comparison of the performance for oxidation of formaldehyde on nano-Co3O4, 2D-Co3O4, and 3D-Co3O4 catalysts. Appl. Catal. B Environ. 2013, 142–143, 677–683. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, M.; Zhang, B.; Gao, Y.; Lu, W.; Guo, Y. Controlled synthesis of porous Co3O4 nanofibers by spiral electrospinning and their application for formaldehyde oxidation. RSC Adv. 2016, 6, 102127–102133. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Hu, H.; Wang, S.; Ying, L.; Huang, Y. Achieving low temperature formaldehyde oxidation: A case study of NaBH4 reduced cobalt oxide nanowires. Inorg. Chem. Commun. 2017, 82, 20–23. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Zhang, L.; Jiang, D. Surface oxygen vacancies on Co3O4 mediated catalytic formaldehyde oxidation at room temperature. Catal. Sci. Technol. 2016, 6, 3845–3853. [Google Scholar] [CrossRef]
- Huang, F.; Chen, C.; Wang, F.; Wang, B.; Zhang, L.; Lu, S.; Li, K. Effect of calcination temperature on the catalytic oxidation of formaldehyde over Co3O4-CeO2 catalysts. Catal. Surv. Asia 2017, 21, 143–149. [Google Scholar] [CrossRef]
- Xie, S.; Dai, H.; Deng, J.; Liu, Y.; Yang, H.; Jiang, Y.; Tan, W.; Ao, A.; Guo, G. Au/3DOM Co3O4: Highly active nanocatalysts for the oxidation of carbon monoxide and toluene. Nanoscale 2013, 5, 11207–11219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Jin, Y.; Li, C.Y.; Shen, Y.N.; Han, L.; Hu, Z.X.; Di, X.W.; Liu, Z.L. Creation of three-dimensionally ordered macroporous Au/CeO2 catalysts with controlled pore sizes and their enhanced catalytic performance for formaldehyde oxidation. Appl. Catal. B Environ. 2009, 91, 11–20. [Google Scholar] [CrossRef]
- Huang, Y.; Fan, W.; Long, B.; Li, H.; Qiu, W.; Zhao, F.; Tong, Y.; Ji, H. Alkali-modified non-precious metal 3D-NiCo2O4 nanosheets for efficient formaldehyde oxidation at low temperature. J. Mater. Chem. A 2016, 4, 3648–3654. [Google Scholar] [CrossRef]
- Xie, J.; Meng, M.; Tang, Y.; Yang, P.; Kang, C.; Zhou, Z.; Huang, S. Investigation of removal of HCHO by Zn modifed Co3O4 catalyst at room temperature. Res. Chem. Intermed. 2019, 45, 3879–3893. [Google Scholar] [CrossRef]
- Xie, J.; Meng, M.; Lin, Z.; Ding, H.; Chen, J.; Huang, S.; Zhou, Z. Exploring removal of formaldehyde at room temperature over Cr and Zn modified Co3O4 catalyst prepared by hydrothermal method. Res. Chem. Intermed. 2020, 46, 1789–1804. [Google Scholar] [CrossRef]
- Baidya, T.; Murayama, T.; Bera, P.; Safonova, O.V.; Steiger, P.; Katiyar, N.K.; Biswas, K.; Haruta, M. Low-temperature CO oxidation over combustion made Fe- and Cr-doped Co3O4 catalysts: Role of dopant’s nature toward achieving superior catalytic activity and stability. J. Phys. Chem. C 2017, 121, 15256–15265. [Google Scholar] [CrossRef]
- Bai, B.; Li, J. Positive effects of K+ ions on three-dimensional mesoporous Ag/Co3O4 catalyst for HCHO oxidation. ACS Catal. 2014, 4, 2753–2762. [Google Scholar] [CrossRef]
- Fan, Z.; Zhang, Z.; Fang, W.; Yao, X.; Zou, G.; Shangguan, W. Low-temperature catalytic oxidation of formaldehyde over Co3O4 catalysts prepared using various precipitants. Chin. J. Catal. 2016, 37, 947–954. [Google Scholar] [CrossRef]
- Fan, Z.; Shi, J.; Zhang, Z.; Chen, M.; Shangguan, W. Promotion effect of potassium carbonate on catalytic activity of Co3O4 for formaldehyde removal. J. Chem. Technol. Biotechnol. 2018, 93, 3562–3568. [Google Scholar] [CrossRef]
- Tang, W.; Weng, J.; Lu, X.; Wen, L.; Suburamanian, A.; Nam, C.Y.; Gao, P.X. Alkali-metal poisoning effect of total CO and propane oxidation over Co3O4 nanocatalysts. Appl. Catal. B Environ. 2019, 256, 117859. [Google Scholar] [CrossRef]
- Scirè, S.; Liotta, L.F. Supported gold catalysts for the total oxidation of volatile organic compounds. Appl. Catal. B Environ. 2012, 125, 222–246. [Google Scholar] [CrossRef]
- Takei, T.; Akita, T.; Nakamura, I.; Fujitani, T.; Okumura, M.; Okazaki, K.; Huang, J.; Ishida, T.; Haruta, M. Heterogeneous catalysis by gold. In Advances in Catalysis; Gates, B.C., Jentoft, F.C., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2012; Volume 55, pp. 1–124. [Google Scholar]
- Gaálová, J.; Topka, P. Gold and ceria as catalysts for VOC abatement: A review. Catalysts 2021, 11, 789. [Google Scholar] [CrossRef]
- Jing, M.; Song, W.; Chen, L.; Ma, S.; Deng, J.; Zheng, H.; Li, Y.; Liu, J.; Zhao, Z. Density functional theory study of the formaldehyde catalytic oxidation mechanism on a Au-doped CeO2(111) surface. J. Phys. Chem. C 2018, 122, 438–448. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Y.; Li, C.; Hu, W.; Jing, P.; Wang, Q.; Zhang, J. Three dimensionally ordered macroporous Au/CeO2–Co3O4 catalysts with nanoporous walls for enhanced catalytic oxidation of formaldehyde. Appl. Catal. B Environ. 2012, 127, 47–58. [Google Scholar] [CrossRef]
- Lu, S.; Wang, F.; Chen, C.; Huang, F.; Li, K. Catalytic oxidation of formaldehyde over CeO2-Co3O4 catalysts. J. Rare Earths 2017, 35, 867–874. [Google Scholar] [CrossRef]
- Qu, J.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. 3D gold-modified cerium and cobalt oxide catalyst on a graphene aerogel for highly efficient catalytic formaldehyde oxidation. Small 2018, 15, 1804415. [Google Scholar] [CrossRef] [PubMed]
- Rochard, G.; Giraudon, J.M.; Liotta, L.F.; La Parola, V.; Lamonier, J.F. Au/Co promoted CeO2 catalysts for formaldehyde total oxidation at ambient temperature: Role of oxygen vacancies. Catal. Sci. Technol. 2019, 9, 3203–3213. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Venezia, A.M.; Anghel, E.M.; State, R.; Avdeev, G.; Tabakova, T. Mechanochemically prepared Co3O4-CeO2 catalysts for complete benzene oxidation. Catalysts 2021, 11, 1316. [Google Scholar] [CrossRef]
- Haneda, M.; Kintaichi, Y.; Bion, N.; Hamada, H. Alkali metal-doped cobalt oxide catalysts for NO decomposition. Appl. Catal. B Environ. 2003, 46, 473–482. [Google Scholar] [CrossRef]
- Ma, L.; Seo, C.Y.; Chen, X.; Li, J.; Schwank, J.W. Sodium-promoted Ag/CeO2 nanospheres for catalytic oxidation of formaldehyde. Chem. Eng. J. 2018, 350, 419–428. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Delgado, J.J.; Ivanov, I.; Idakiev, V.; Tabakova, T.; Centeno, M.A.; Odriozola, J.A. Viability of Au/CeO2-ZnO/Al2O3 catalysts for pure hydrogen production by the water-gas shift reaction. ChemCatChem 2014, 6, 1401–1409. [Google Scholar]
- Gabrovska, M.; Ivanov, I.; Nikolova, D.; Kovacheva, D.; Tabakova, T. Hydrogen production via water-gas shift reaction over gold supported on Ni-based layered double hydroxides. Int. J. Hydrogen Energy 2021, 46, 458–473. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, L.C.; Chen, M.; Cao, Y.; He, H.Y.; Fan, K.N. Dry citrate-precursor synthesized nanocrystalline cobalt oxide as highly active catalyst for total oxidation of propane. J. Catal. 2009, 263, 104–113. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, H.; Deng, J.; Xie, S.; Yang, H.; Tan, W.; Han, W.; Jiang, Y.; Guo, G. Mesoporous Co3O4-supported gold nanocatalysts: Highly active for the oxidation of carbon monoxide, benzene, toluene, and o-xylene. J. Catal. 2014, 309, 408–418. [Google Scholar] [CrossRef]
- Laguna, O.H.; Sarria, F.R.; Centeno, M.A.; Odriozola, J.A. Gold supported on metal-doped ceria catalysts (M = Zr, Zn and Fe) for the preferential oxidation of CO (PROX). J. Catal. 2010, 276, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Shoko, E.; Smith, M.F.; McKenzie, R.H. Charge Distribution near bulk oxygen vacancies in cerium oxides. J. Phys. Condens. Matter 2010, 22, 223201. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, H.; Qin, Z.; Wang, G.; Liang, F.; Wang, J. Preferential oxidation of CO in H2 rich stream over Au/CeO2-Co3O4 catalysts. Catal. Commun. 2008, 9, 1487–1492. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Pantaleo, G.; Zanella, R.; Sobczak, J.W.; Lisowski, W.; Kaszkur, Z.; Munteanu, G.; Yordanova, I.; Liotta, L.F.; et al. Alumina supported Au/Y-doped ceria catalysts for pure hydrogen production via PROX. Int. J. Hydrogen Energy 2019, 44, 233–245. [Google Scholar] [CrossRef]
- Chen, B.; Zhu, X.; Wang, Y.; Yu, L.; Shi, C. Gold stabilized on various oxide supports catalyzing formaldehyde oxidation at room temperature. Chin. J. Catal. 2016, 37, 1729–1737. [Google Scholar] [CrossRef]
- Yao, Y.F.Y. The oxidation of hydrocarbons and CO over metal oxides: III. Co3O4. J. Catal. 1974, 33, 108–122. [Google Scholar] [CrossRef]
- Yao, H.C.; Yao, Y.F.Y. Ceria in automotive exhaust catalysts: I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Zou, G.; Xu, Y.; Wang, S.; Chen, M.; Shangguan, W. The synergistic effect in Co-Ce oxides for catalytic oxidation of diesel soot. Catal. Sci. Technol. 2015, 5, 1084–1092. [Google Scholar] [CrossRef]
- Deng, J.; Song, W.; Chen, L.; Wang, L.; Jing, M.; Ren, Y.; Zhao, Z.; Liu, J. The effect of oxygen vacancies and water on HCHO catalytic oxidation over Co3O4 catalyst: A combination of density functional theory and microkinetic study. Chem. Eng. J. 2019, 355, 540–550. [Google Scholar] [CrossRef]
- Andreeva, D.; Idakiev, V.; Tabakova, T.; Ilieva, L.; Falaras, P.; Bourlinos, A.; Travlos, A. Low-temperature water-gas shift reaction over Au/CeO2 catalysts. Catal. Today 2002, 72, 51–57. [Google Scholar] [CrossRef]
- Liotta, L.F.; Pantaleo, G.; Puleo, F.; Venezia, A.M. Au/CeO2-SBA-15 catalysts for CO oxidation: Effect of ceria loading on physicochemical properties and catalytic performances. Catal. Today 2012, 187, 10–19. [Google Scholar] [CrossRef]
- Balzer, R.; Probst, L.F.D.; Drago, V.; Schreiner, W.H.; Fajardo, H.V. Catalytic oxidation of volatile organic compounds (n-hexane, benzene, toluene, o-xylene) promoted by cobalt catalysts supported on γ-Al2O3-CeO2. Braz. J. Chem. Eng. 2014, 31, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Wang, J.; Yoshida, A.; Wang, P.; Yu, T.; Wang, Z.; Hao, X.; Abudula, A.; Guan, G. Catalytic oxidation of volatile organic compound over cerium modified cobalt-based mixed oxide catalysts synthesized by electrodeposition method. Appl. Catal. B Environ. 2020, 271, 118941. [Google Scholar] [CrossRef]
- Kang, M.; Song, M.W.; Lee, C.H. Catalytic carbon monoxide oxidation over CoOx/CeO2 composite catalysts. Appl. Catal. A Gen. 2003, 251, 143–156. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl. Catal. B Environ. 2007, 75, 167–174. [Google Scholar] [CrossRef]
- Sun, M.; Wang, L.; Feng, B.; Zhang, Z.; Lu, G.; Guo, Y. The role of potassium in K/Co3O4 for soot combustion under loose contact. Catal. Today 2011, 175, 100–105. [Google Scholar] [CrossRef]
- Ismail, A.; Li, M.; Zahid, M.; Fan, L.; Zhang, C.; Li, Z.; Zhu, Y. Effect of strong interaction between Co and Ce oxides in CoxCe1-xO2-δ oxides on its catalytic oxidation of toluene. Mol. Catal. 2021, 502, 111356. [Google Scholar] [CrossRef]
- Dang-Bao, T.; Anh, N.P.; Phuong, P.H.; Van, N.T.T.; Nghia, T.N.D.; Tien, H.V.; Tri, N. Fabrication of cobalt-doped ceria nanorods for p-xylene deep oxidation: Effects of cobalt precursor and loading. Mater. Trans. 2020, 61, 1294–1300. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Zeng, X.; Zhu, T. Correlation between the physicochemical properties and catalytic performances of micro/mesoporous CoCeOx mixed oxides for propane combustion. Appl. Catal. A Gen. 2019, 572, 61–70. [Google Scholar] [CrossRef]
- Ma, C.; Mu, Z.; He, C.; Li, P.; Li, J.; Hao, Z. Catalytic oxidation of benzene over nanostructured porous Co3O4-CeO2 composite catalysts. J. Environ. Sci. 2011, 23, 2078–2086. [Google Scholar] [CrossRef]
- Casaletto, M.P.; Longo, A.; Martorana, A.; Prestianni, A.; Venezia, A.M. XPS study of supported gold catalysts: The role of Au0 and Au+δ species as active sites. Surf. Interface Anal. 2006, 38, 215–218. [Google Scholar] [CrossRef]
- Jia, M.L.; Bai, H.F.; Zhaorigetu; Shen, Y.N.; Li, Y.F. Preparation of Au/CeO2 catalyst and its catalytic performance for HCHO oxidation. J. Rare Earths 2008, 26, 528–531. [Google Scholar] [CrossRef]
- Li, H.F.; Zhang, N.; Chen, P.; Luo, M.F.; Lu, J.Q. High surface area Au/CeO2 catalysts for low temperature formaldehyde oxidation. Appl. Catal. B Environ. 2011, 110, 279–285. [Google Scholar] [CrossRef]
- Chen, B.B.; Shi, C.A.; Crocker, M.; Wang, Y.; Zhu, A.M. Catalytic removal of formaldehyde at room temperature over supported gold catalysts. Appl. Catal. B Environ. 2013, 132, 245–255. [Google Scholar] [CrossRef]
- Bu, Y.; Chen, Y.; Jiang, G.; Hou, X.; Li, S.; Zhang, Z. Understanding of Au-CeO2 interface and its role in catalytic oxidation of formaldehyde. Appl. Catal. B Environ. 2020, 260, 118138. [Google Scholar] [CrossRef]
- Lou, Y.; Ma, J.; Cao, X.; Wang, L.; Dai, Q.; Zhao, Z.; Cai, Y.; Zhan, W.; Guo, Y.; Hu, P.; et al. Promoting effects of In2O3 on Co3O4 for CO oxidation: Tuning O2 activation and CO adsorption strength simultaneously. ACS Catal. 2014, 4, 4143–4152. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, S.; Lei, F.; Liu, J.; Liang, L.; Xie, Y. Atomically-thin nonlayered cobalt oxide porous sheets for highly efficient oxygen evolving electrocatalysts. Chem. Sci. 2014, 5, 3976–3982. [Google Scholar] [CrossRef]
- Yu, J.; Li, X.; Xu, Z.; Xiao, W. NaOH-modified ceramic honeycomb with enhanced formaldehyde adsorption and removal performance. Environ. Sci. Technol. 2013, 47, 9928–9933. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.B.; Zhu, X.B.; Crocker, M.; Wang, Y.; Shi, C. Complete oxidation of formaldehyde at ambient temperature over γ-Al2O3 supported Au catalyst. Catal. Commun. 2013, 42, 93–97. [Google Scholar] [CrossRef]
- Bortolotti, M.; Lutterotti, L.; Lonardelli, I. ReX: A computer program for structural analysis using powder diffraction data. J. Appl. Cryst. 2009, 42, 538–539. [Google Scholar] [CrossRef]
- Kotzev, N.; Shopov, D. A thermodesorption study of the system olefin—NiO. J. Catal. 1971, 22, 297–301. [Google Scholar] [CrossRef]
- Monti, D.A.M.; Baiker, A. Temperature-programmed reduction, parametric sensitivity and estimation of kinetic parameters. J. Catal. 1983, 83, 323–335. [Google Scholar] [CrossRef]
- Venezia, A.M.; Murania, R.; Pantaleo, G.; Deganello, G. Pd and PdAu on mesoporous silica for methane oxidation: Effect of SO2. J. Catal. 2007, 251, 94–102. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSA (m2 g−1) | Vpore (cm3 g−1) | Dpore (nm) | DCo3O4 (nm) | aCo3O4 (Å) | DCeO2 (nm) | aCeO2 (Å) |
|---|---|---|---|---|---|---|---|
| Co-Ce | 72 | 0.24 | 13.3 | 20.20 | 8.074 (5) | 8.16 | 5.410 (7) |
| Сo-Се+К | 43 | 0.19 | 17.9 | 18.95 | 8.078 (1) | 7.13 | 5.422 (1) |
| Au/Сo-Се | 68 | 0.25 | 14.9 | 19.46 | 8.081 (1) | 6.96 | 5.418 (1) |
| Au/Сo-Се+К | 65 | 0.24 | 13.1 | 20.32 | 8.080 (1) | 6.96 | 5.418 (1) |
| Al2O3-b | 219 | 0.40 | 7.4 | ||||
| Au/Co-Ce/Al | 158 | 0.34 | 8.5 | 17.66 | 8.081 (2) | 7.64 | 5.420 (2) |
| Sample | Au (at.%) | Ce (at.%) | Co (at.%) | Ce/Co 0.10 1 | O (at.%) | K (at.%) | Al (at.%) |
|---|---|---|---|---|---|---|---|
| Co-Ce | - | 13.1 | 19.0 | 0.69 | 67.9 | - | - |
| Co-Ce+K | - | 11.4 | 16.6 | 0.69 | 68.5 | 3.5 | - |
| Au/Co-Ce | 0.4 | 15.1 | 18.2 | 0.83 | 66.3 | - | - |
| Au/Co-Ce+K | 0.3 | 16.3 | 19.3 | 0.84 | 64.1 | - | - |
| Au/Co-Ce/Al | 0.4 | 2.9 | 16.1 | 0.19 | 64.1 | - | 16.4 |
| Sample | Au 4f7/2 (eV) | Ce 3d5/2 (eV) | Ce3+/Cet | Co 2p3/2 (eV) | Co3+/Cot | O 1s (eV) | Os/Ol | K 2p3/2 (eV) | Al 2p (eV) |
|---|---|---|---|---|---|---|---|---|---|
| Co-Ce | 779.8 (73) | 529.9 (77) | |||||||
| - | 882.3 | 0.33 | 781.7 (27) | 0.73 | 531.9 (21) | 0.27 | - | - | |
| 533.4 (02) | |||||||||
| Co-Ce+K | - | 882.2 | 0.26 | 779.7 (84) | 0.84 | 529.7 (79) | 0.26 | 293.2 | - |
| 781.9 (16) | 531.8 (21) | ||||||||
| Au/Co-Ce | 780.1 (74) | 530.2 (62) | |||||||
| 84.6 | 882.6 | 0.40 | 781.9 (26) | 0.74 | 532.2 (28) | 0.45 | - | - | |
| 533.5 (10) | |||||||||
| Au/Co-Ce+K | 779.9 (75) | 0.75 | 529.9 (67) | ||||||
| 84.5 | 882.4 | 0.29 | 781.8 (25) | - | 532.0 (25) | 0.37 | n.d. | - | |
| 533.5 (08) | |||||||||
| Au/Co-Ce/Al | 780.0 (78) | 530.2 (60) | |||||||
| 84.3 | 882.3 | 0.40 | 781.7 (22) | 0.78 | 532.1 (35) | 0.58 | - | 74.6 | |
| 533.0 (05) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilieva, L.; Dimitrov, D.; Kolentsova, E.; Venezia, A.M.; Karashanova, D.; Avdeev, G.; Petrova, P.; State, R.; Tabakova, T. Gold-Based Catalysts for Complete Formaldehyde Oxidation: Insights into the Role of Support Composition. Catalysts 2022, 12, 705. https://doi.org/10.3390/catal12070705
Ilieva L, Dimitrov D, Kolentsova E, Venezia AM, Karashanova D, Avdeev G, Petrova P, State R, Tabakova T. Gold-Based Catalysts for Complete Formaldehyde Oxidation: Insights into the Role of Support Composition. Catalysts. 2022; 12(7):705. https://doi.org/10.3390/catal12070705
Chicago/Turabian StyleIlieva, Lyuba, Dimitar Dimitrov, Elitsa Kolentsova, Anna Maria Venezia, Daniela Karashanova, Georgi Avdeev, Petya Petrova, Razvan State, and Tatyana Tabakova. 2022. "Gold-Based Catalysts for Complete Formaldehyde Oxidation: Insights into the Role of Support Composition" Catalysts 12, no. 7: 705. https://doi.org/10.3390/catal12070705