
Effect of an Sb-Doped SnO2 Support on the CO-Tolerance of Pt2Ru3 Nanocatalysts for Residential Fuel Cells


Abstract
:
1. Introduction
2. Results and Discussion
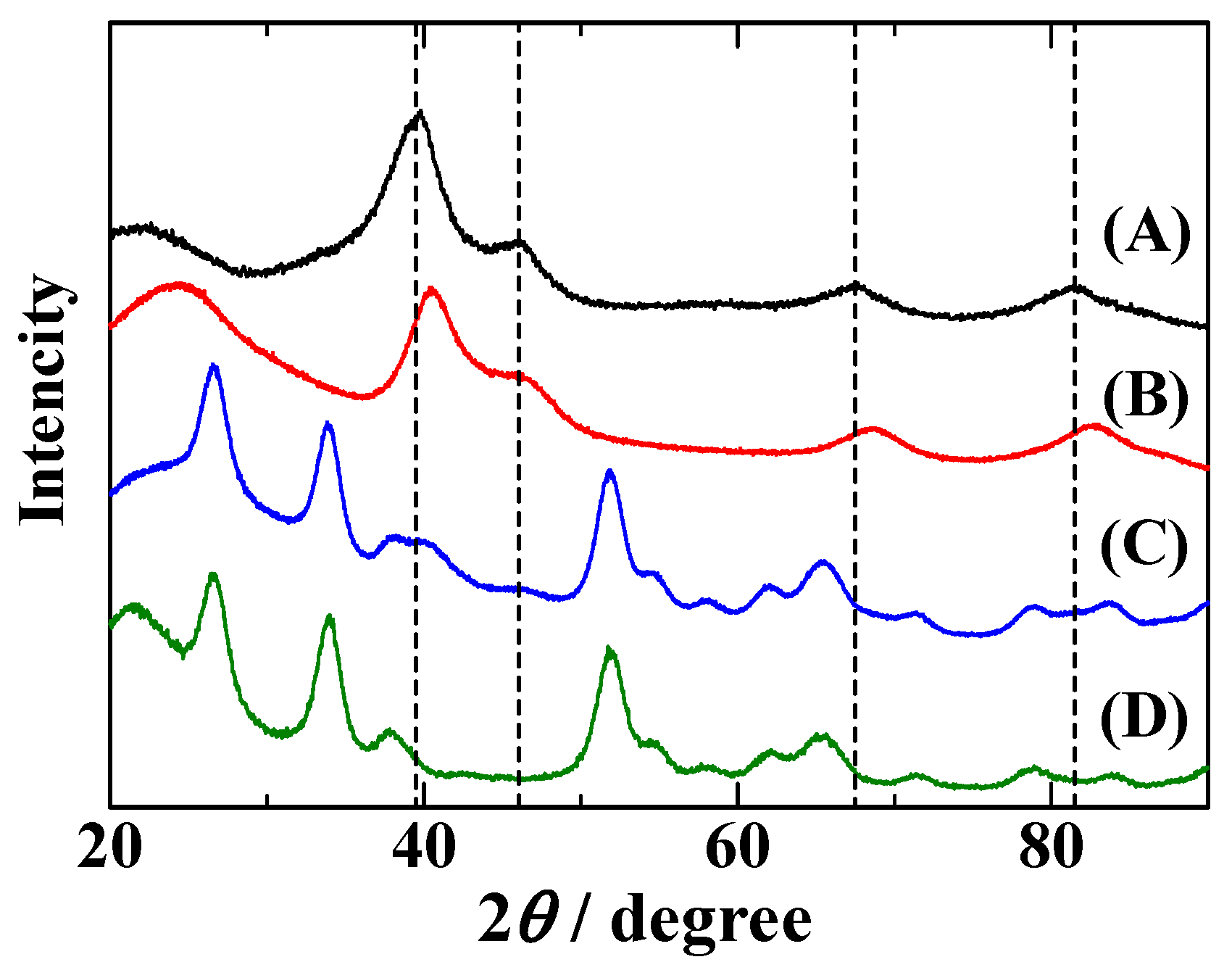
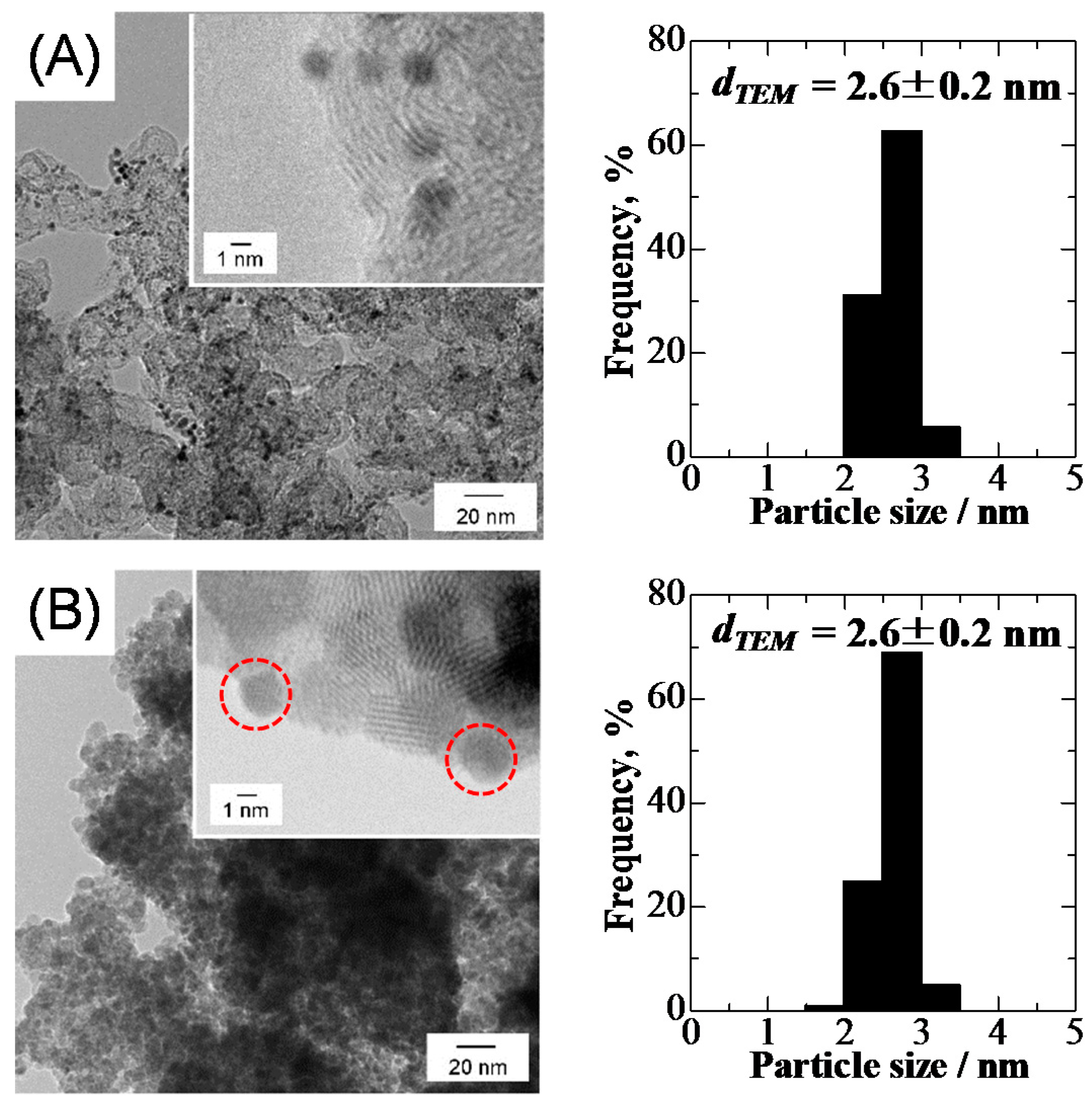
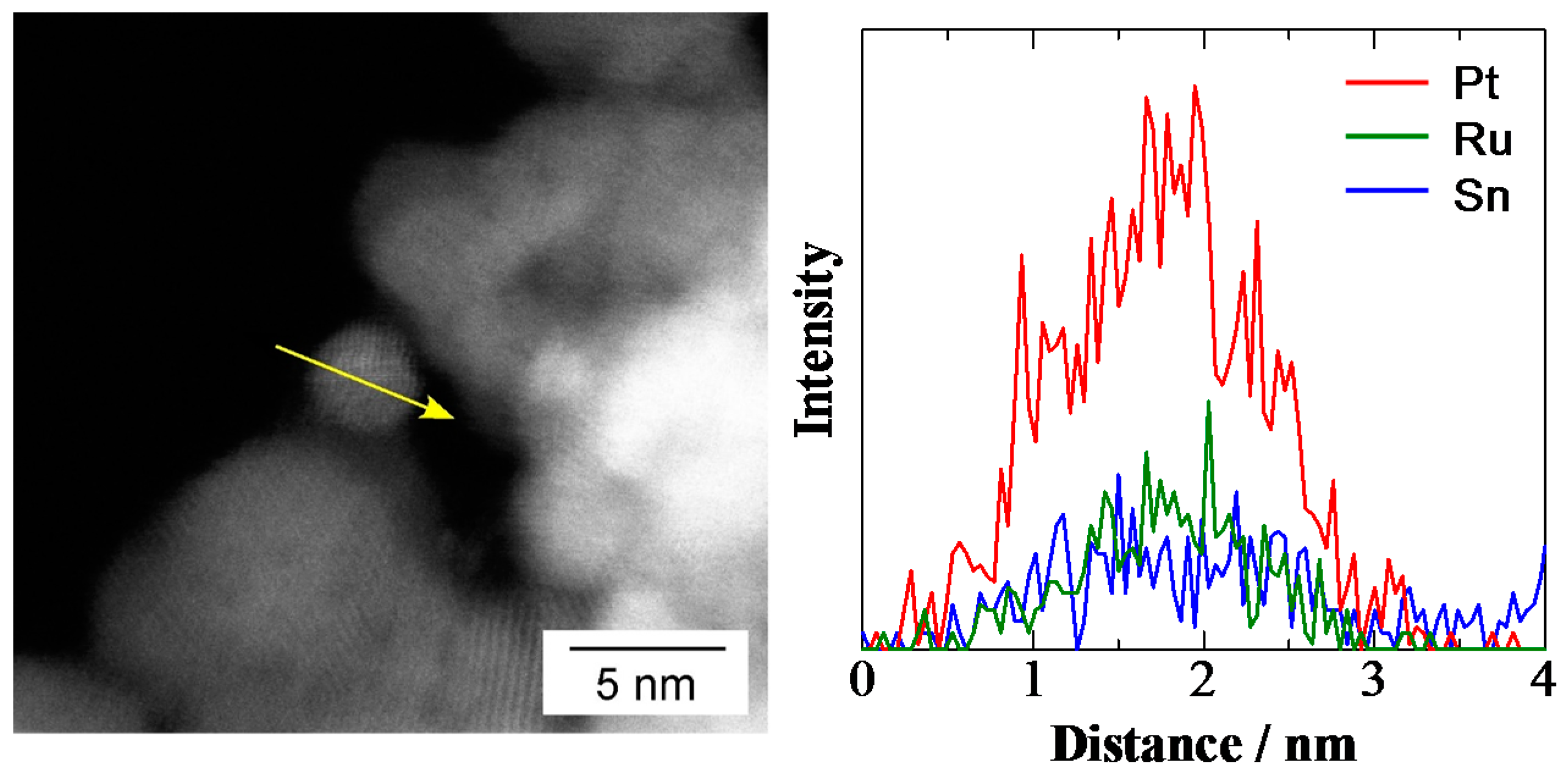
2.1. Characterization of Pt2Ru3/CB and Pt2Ru3/Sb-SnO2
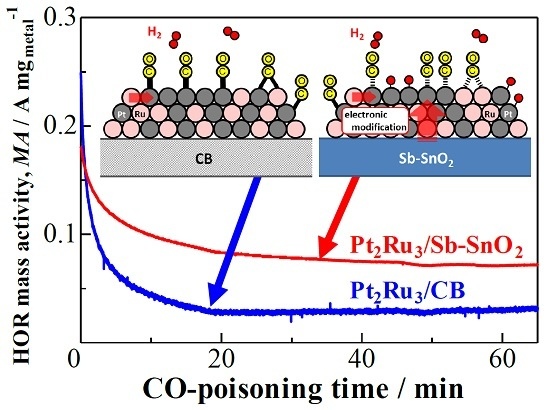
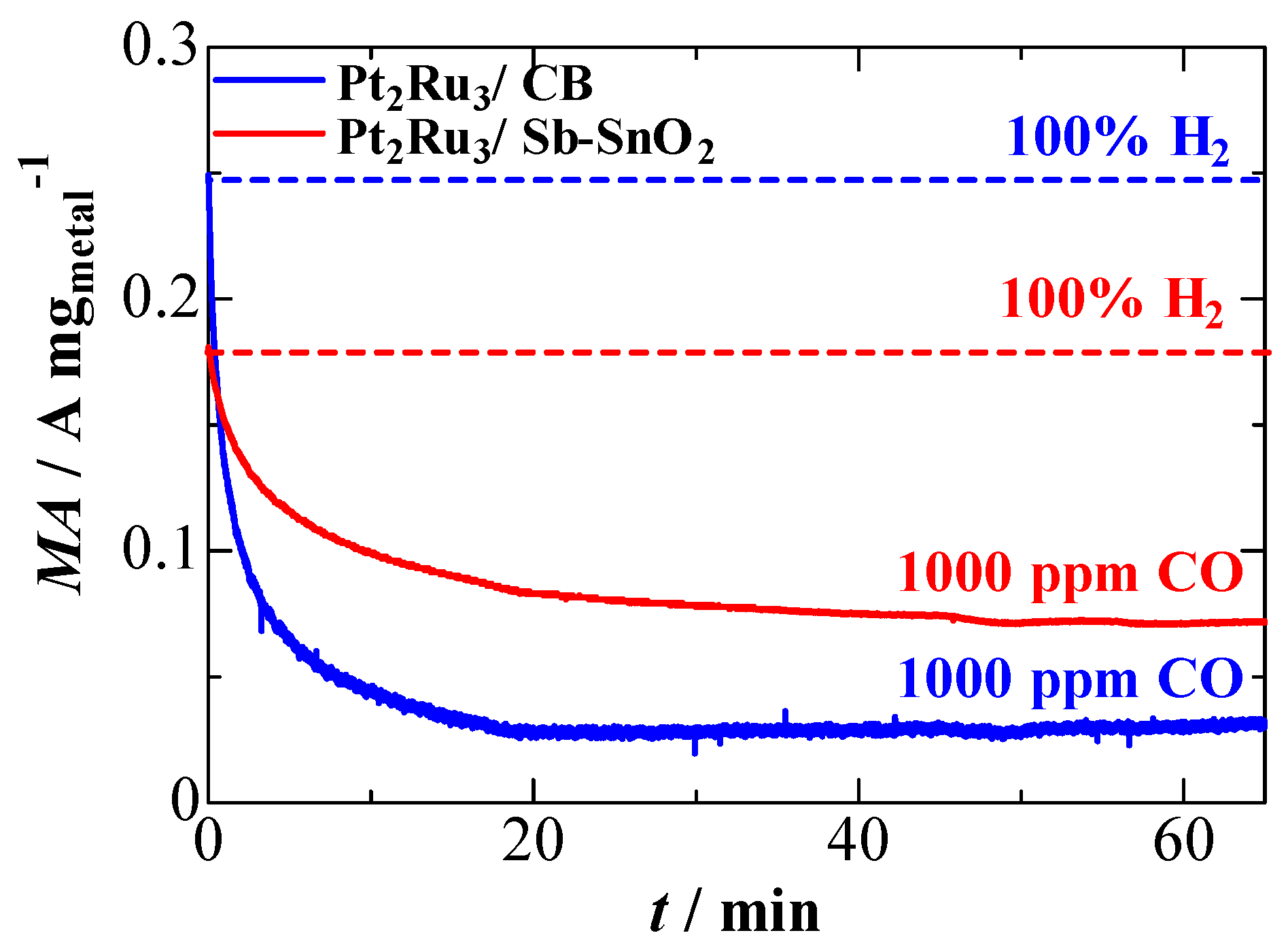
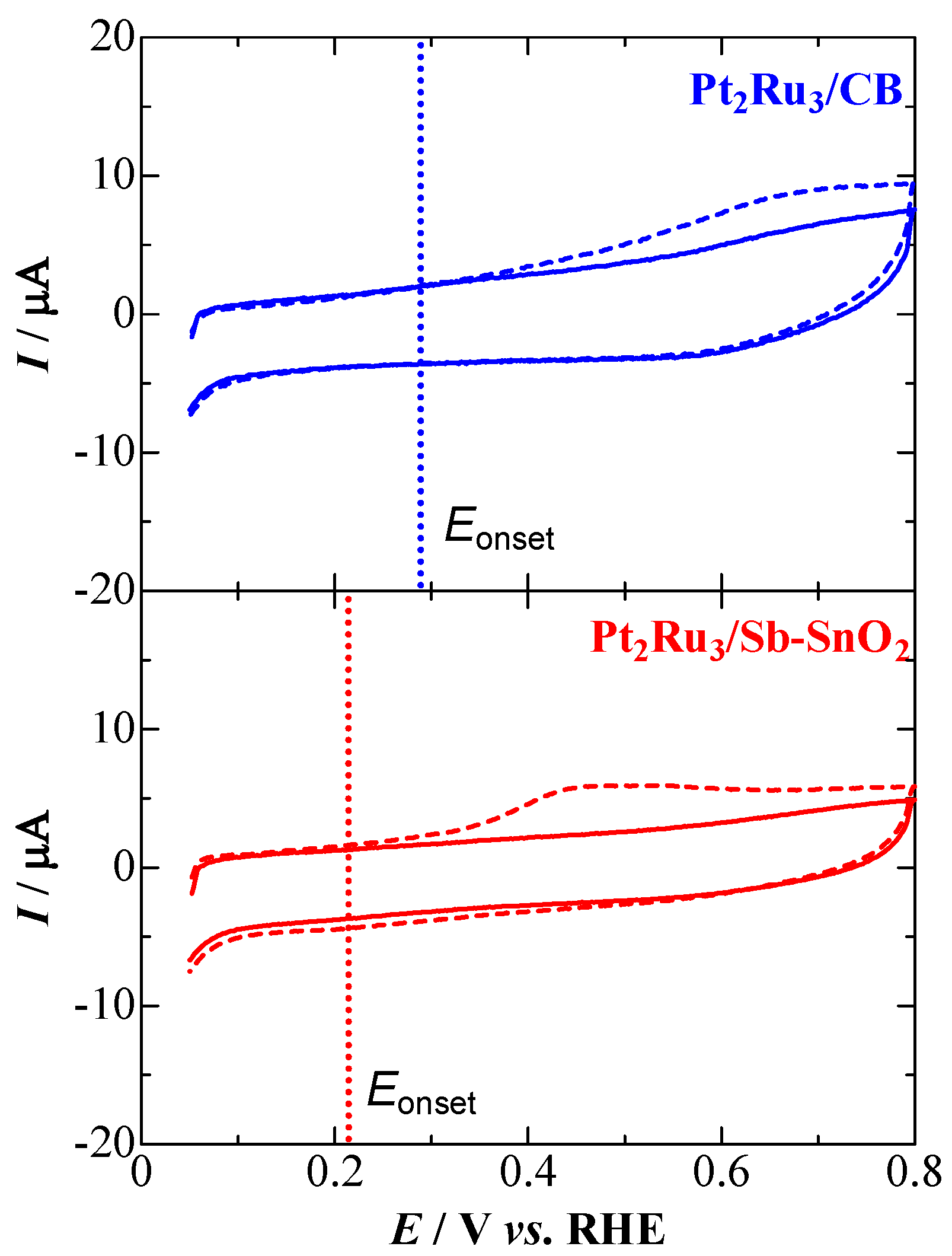
2.2. CO Tolerance for the Hydrogen Oxidation Reaction (HOR)
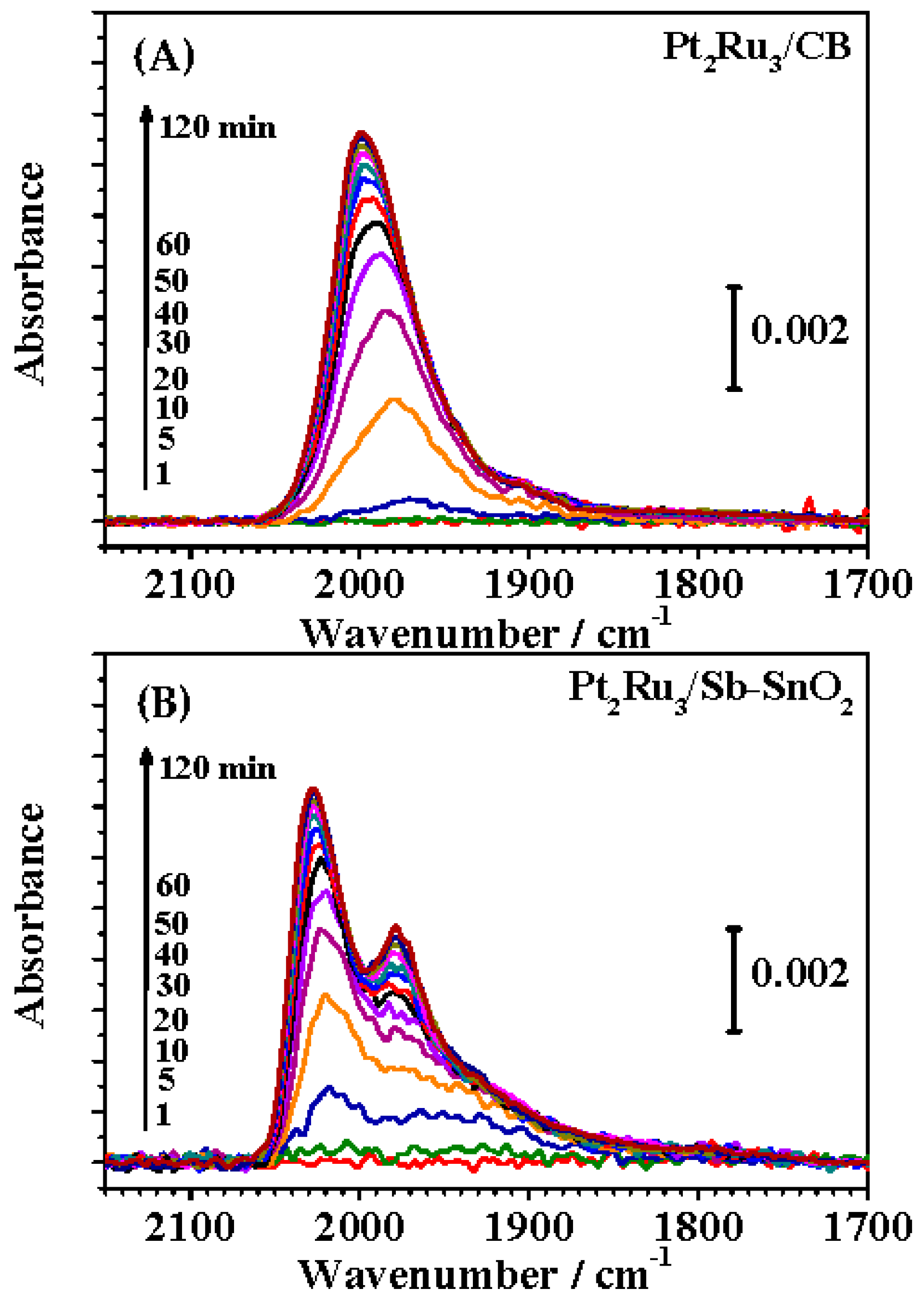
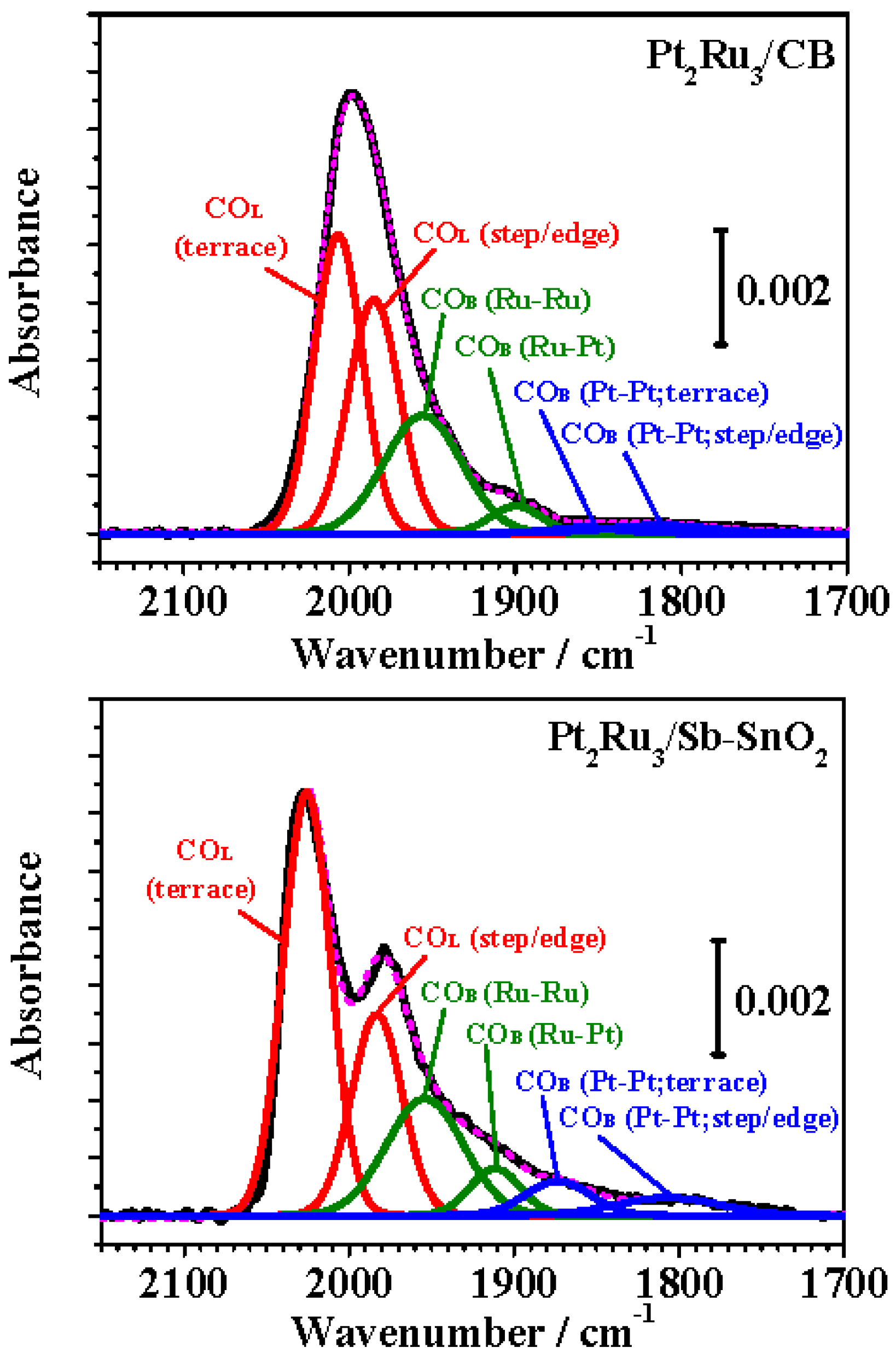
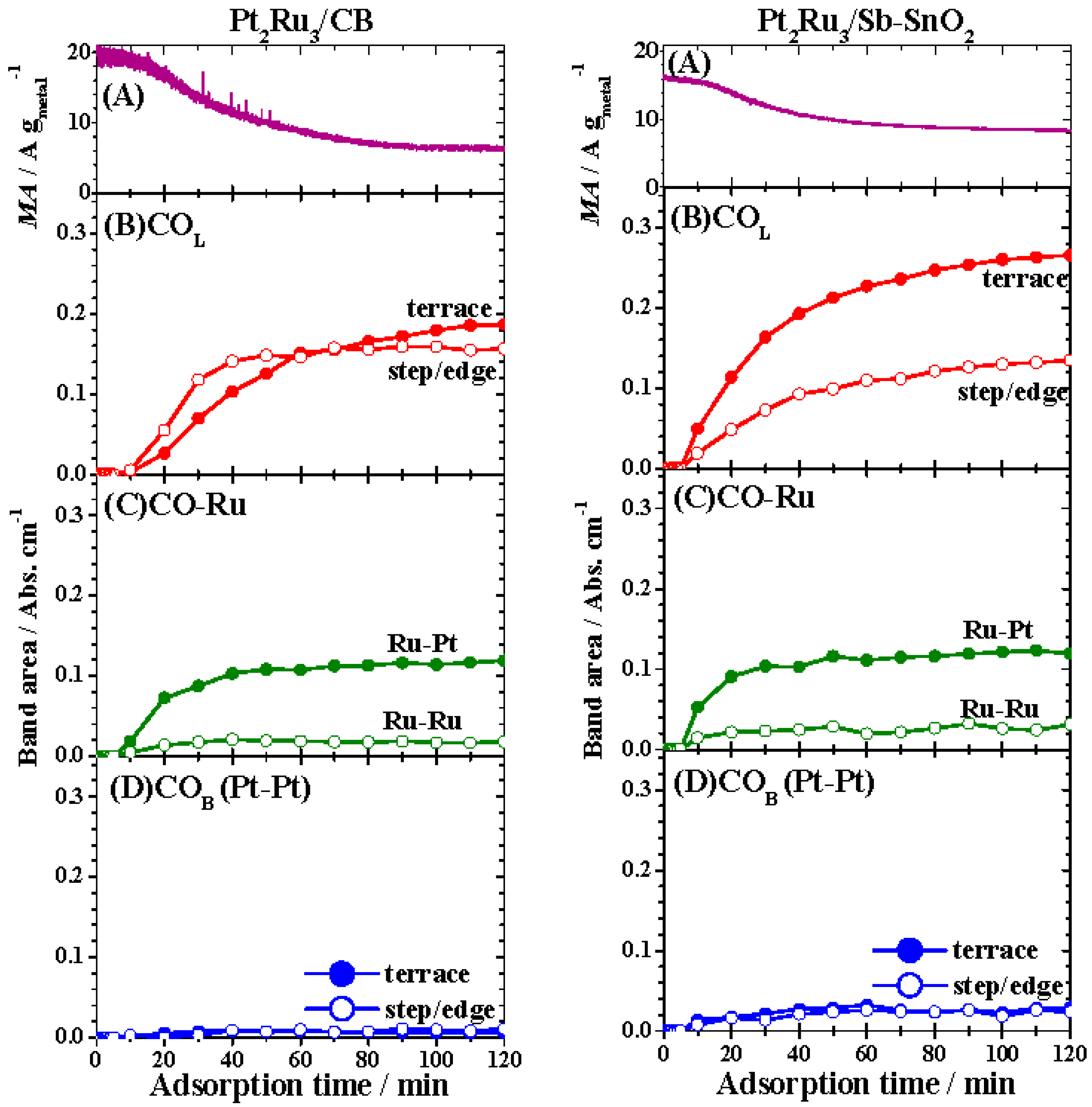
2.3. FTIR Analysis of CO Adsorption on Pt-Ru Alloys
3. Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lemons, R.A. Fuel Cells for Transportation. J. Power Sources 1990, 29, 251–264. [Google Scholar] [CrossRef]
- Igarashi, H.; Fujino, T.; Watanabe, M. Hydrogen Electro-Oxidation on Platinum Catalysts in the Presence of Trace Carbon Monoxide. J. Electroanal. Chem. 1995, 391, 119–123. [Google Scholar] [CrossRef]
- Igarashi, H.; Fujino, T.; Zhu, Y.; Uchida, H.; Watanabe, M. CO Tolerance of Pt Alloy Electrocatalysts for Polymer Electrolyte Fuel Cells and the Detoxification Mechanism. Phys. Chem. Phys. Chem. 2001, 3, 306–314. [Google Scholar] [CrossRef]
- Kimura, M.; Miyao, T.; Komori, S.; Chen, A.; Higashiyama, K.; Yamashita, H.; Watanabe, M. Selective Methanation of CO in Hydrogen-Rich Gases Involving Large Amounts of CO2 over Ru-Modified Ni-Al Mixed Oxide Catalysts. Appl. Catal. A: Gen. 2010, 379, 182–187. [Google Scholar] [CrossRef]
- Martínez-Huerta, M.V.; Rodríguez, J.L.; Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.G.; Pastor, E. Novel Synthesis Method of CO-Tolerant PtRu-MoOx Nanoparticles: Structural Characteristics and Performance for Methanol Electrooxidation. Chem. Mater. 2008, 20, 4249–4259. [Google Scholar] [CrossRef]
- Lee, K.-S.; Park, I.-S.; Cho, Y.-H.; Jung, D.-S.; Jung, N.; Park, H.-Y.; Sung, Y.-E. Electrocatalytic Activity and Stability of Pt Supported on Sb-Doped SnO2 Nanoparticles for Direct Alcohol Fuel Cells. J. Catal. 2008, 258, 143–152. [Google Scholar] [CrossRef]
- Santos, A.L.; Profeti, D.; Olivi, P. Electrooxidation of Methanol on Pt Microparticles Dispersed on SnO2 Thin Films. Electrochim. Acta 2005, 50, 2615–2621. [Google Scholar] [CrossRef]
- Takeguchi, T.; Anzai, Y.; Kikuchi, R.; Eguchi, K.; Ueda, W. Preparation and Characterization of CO-Tolerant Pt and Pd Anodes Modified with SnO2 Nanoparticles for PEFC. J. Electrochem. Soc. 2007, 154, B1132–B1137. [Google Scholar] [CrossRef]
- Takeguchi, T.; Kunifuji, A.; Narischat, N.; Ito, M.; Noguchi, H.; Uosaki, K.; Mukai, S.R. Ligand Effect of SnO2 on a Pt-Ru Catalyst and the Relationship Between Bond Strength and CO Tolerance. Catal. Sci. Technol. 2016, 6, 3214–3219. [Google Scholar] [CrossRef]
- Wang, G.; Takeguchi, T.; Muhamad, E.N.; Yamanaka, T.; Ueda, W. Effect of Addition of SnOx to the Pt2Ru3/C Catalyst on CO Tolerance for the Polymer Electrolyte Fuel Cell. J. Electrochem. Soc. 2011, 158, B448–B453. [Google Scholar] [CrossRef]
- Devaux, D.; Yano, H.; Uchida, H.; Dobois, J.-L.; Watanabe, M. Electro-Oxidation of Hydrolysed Ploy-Oxymethylene-Dimethylether ono PtRu Supported Catalysts. Electrochim. Acta 2001, 56, 1460–1465. [Google Scholar] [CrossRef]
- Shen, P.K.; Tseung, A.C.C. Anodic Oxidation of Methanol on Pt/WO3 in Acidic Media. J. Electrochem. Soc. 1994, 141, 3082–3090. [Google Scholar] [CrossRef]
- Villullas, H.M.; Mattos-Costa, F.I.; Bulhoes, L.O.S. Electrochemical Oxidation of Methanol on Pt Nanoparticles Dispersed on RuO2. J. Phys. Chem. B 2004, 108, 12898–12903. [Google Scholar] [CrossRef]
- Uchida, H.; Izumi, K.; Watanabe, M. Temperature Dependence of CO-Tolerant Hydrogen Oxidation Reaction Activity at Pt, Pt-Co, and Pt-Ru Electrodes. J. Phys. Chem. B 2006, 110, 21924–21930. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Kataoka, M.; Yamashita, H.; Uchida, H.; Watanabe, M. Oxygen Reduction Activity of Carbon-Supported Pt-M (M = V, Ni, Cr, Co, and Fe) Alloys Prepared by Nanocapsule Method. Langmuir 2007, 23, 6438–6445. [Google Scholar] [CrossRef] [PubMed]
- Okaya, K.; Yano, H.; Uchida, H.; Watanabe, M. Control of Particle Size of Pt and Pt Alloy Electrocatalysts Supported on Carbon Black by the Nanocapsule Method. ACS Appl. Mater. Interfaces 2010, 2, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Motoo, S. Electrocatalysis by Ad-Atoms: Part II. Enhancement of the Oxidation of Methanol on Platinum by Ruthenium Ad-Atoms. J. Electroanal. Chem. 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by Ad-Atoms: Part III. Enhancement of the Oxidation of Carbon Monoxide on Platinum by Ruthenium Ad-Atoms. J. Electroanal. Chem. 1975, 60, 275–283. [Google Scholar] [CrossRef]
- Sato, T.; Kunimatsu, K.; Uchida, H.; Watanabe, M. Adsorption/oxidation of CO on Highly Dispersed Pt Catalyst Studied by Combined Electrochemical and ATR-FTIRAS Methods Part 1. ATR-FTIRAS Spectra of CO Adsorbed on Highly Dispersed Pt Catalyst on Carbon Black and Carbon Un-Supported Pt Black. Electrochim. Acta 2007, 53, 1265–1278. [Google Scholar] [CrossRef]
- Sato, T.; Kunimatsu, K.; Okaya, K.; Yano, H.; Watanabe, M.; Uchida, H. In situ ATR-FTIR Analysis of the CO-Tolerance Mechanism on Pt2Ru3/C Catalysts Prepared by the Nanocapsule Method. Energy Environ. Sci. 2011, 4, 433–438. [Google Scholar] [CrossRef]
- Sato, T.; Okaya, K.; Kunimatsu, K.; Yano, H.; Watanabe, M.; Uchida, H. Effect of Particle Size and Composition on CO-Tolerance at Pt-Ru/C Catalysts Analyzed by in Situ Attenuated Total Reflection FTIR Spectroscopy. ACS Catal. 2012, 2, 450–455. [Google Scholar] [CrossRef]
- Kunimatsu, K.; Sato, T.; Uchida, H.; Watanabe, M. Role of Terrace/Step Edge Sites in CO Adsorption/Oxidation on a Polycrystalline Pt Electrode Studied by in Situ ATR-FTIR Method. Electrochim. Acta 2008, 53, 6104–6110. [Google Scholar] [CrossRef]
- Kunimatsu, K.; Sato, T.; Uchida, H.; Watanabe, M. Adsorption/Oxidation of CO on Highly Dispersed Pt Catalyst Studied by Combined Electrochemical and ATR-FTIRAS Methods: Oxidation of CO Adsorbed on Carbon-Supported Pt Catalyst and Unsupported Pt Black. Langmuir 2008, 24, 3590–3601. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.P.; Rodes, A.; Feliu, J.M.; Koper, M.T.M.; van Santen, R.A. Role of Crystalline Defects in Electrocatalysis: CO Adsorption and Oxidation on Stepped Platinum Electrodes As Studied by in situ Infrared Spectroscopy. J. Phys. Chem. B 2002, 106, 9863–9872. [Google Scholar] [CrossRef]
- Benfield, R.E. Mean Coordination Numbers and the Non-Metal Transition in Clusters. J. Chem. Soc. Faraday Trans. 1992, 88, 1107–1110. [Google Scholar] [CrossRef]
- Okaya, K.; Yano, H.; Kakinuma, K.; Watanabe, M.; Uchida, H. Temperature Dependence of Oxygen Reduction Reaction Activity at Stabilized Pt Skin-PtCo Alloy/Graphitized Carbon Black Catalysts Prepared by a Modified Nanocapsule Method. ACS Appl. Mater. Interfaces 2012, 4, 6982–6991. [Google Scholar] [CrossRef] [PubMed]
- Markovic, N.M.; Grgur, B.N.; Ross, P.N. Temperature-Dependent Hydrogen Electrochemistry on Platinum Low-Index Single-Crystal Surfaces in Acid Solutions. J. Phys. Chem. B 1997, 101, 5405–5413. [Google Scholar] [CrossRef]
- Motoo, D.; Watanabe, M. Electrocatalysis by Sn and Ge Ad-Atoms. J. Electroanal. Chem. 1976, 69, 429–431. [Google Scholar] [CrossRef]
- Arenz, M.; Stamenkovic, V.; Blizanac, B.B.; Mayrhofer, K.J.; Markovic, N.M.; Ross, P.N. Carbon-Supported Pt-Sn electrocatalysts for the Anodic Oxidation of H2, CO, and H2/CO Mixtures : Part II: The Structure–Activity Relationship. J. Catal. 2005, 232, 402–410. [Google Scholar] [CrossRef]
- Godoi, D.R.M.; Villullas, H.M.; Zhu, F.-C.; Jiang, Y.-X.; Sun, S.-G.; Guo, J.; Sun, L.; Chen, R. A Comparative Investigation of Metal-Support Interactions on the Catalytic Activity of Pt Nanoparticles for Ethanol Oxidation in Alkaline Medium. J. Power Sources 2016, 311, 81–90. [Google Scholar] [CrossRef]
- Wakisaka, M.; Mitsui, S.; Hirose, Y.; Kawashima, K.; Uchida, H.; Watanabe, M. Electronic Structure of Pt-Co and Pt-Ru Alloy for CO-Tolerant Anode Catalysts in Polymer Electrolyte Fuel Cells Studied by EC-XPS. J. Phys. Chem. B 2006, 110, 23489–23496. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Ikeda, N.; Watanabe, M. Electrochemical Quartz Crystal Microbalance Study of Copper Ad-Atoms on Gold Electrodes. Part II. Further Discussion on the Specific Adsorption of Anions from Solutions of Perchloric and Sulfuric Acid. J. Electroanal. Chem. 1997, 424, 5–12. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | dTEM (nm) b | Metal Loaded (wt %) c | Composition d | |
|---|---|---|---|---|
| Pt (atom %) | Ru (atom %) | |||
| Pt2Ru3/CB | 2.6 ± 0.2 | 20.0 | 40.8 | 59.2 |
| Pt2Ru3/Sb-SnO2 | 2.6 ± 0.2 | 11.2 | 42.1 | 57.9 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogihara, Y.; Yano, H.; Watanabe, M.; Iiyama, A.; Uchida, H. Effect of an Sb-Doped SnO2 Support on the CO-Tolerance of Pt2Ru3 Nanocatalysts for Residential Fuel Cells. Catalysts 2016, 6, 139. https://doi.org/10.3390/catal6090139
Ogihara Y, Yano H, Watanabe M, Iiyama A, Uchida H. Effect of an Sb-Doped SnO2 Support on the CO-Tolerance of Pt2Ru3 Nanocatalysts for Residential Fuel Cells. Catalysts. 2016; 6(9):139. https://doi.org/10.3390/catal6090139
Chicago/Turabian StyleOgihara, Yoshiyuki, Hiroshi Yano, Masahiro Watanabe, Akihiro Iiyama, and Hiroyuki Uchida. 2016. "Effect of an Sb-Doped SnO2 Support on the CO-Tolerance of Pt2Ru3 Nanocatalysts for Residential Fuel Cells" Catalysts 6, no. 9: 139. https://doi.org/10.3390/catal6090139