
Brønsted Acid-Catalyzed Direct Substitution of 2-Ethoxytetrahydrofuran with Trifluoroborate Salts

Abstract
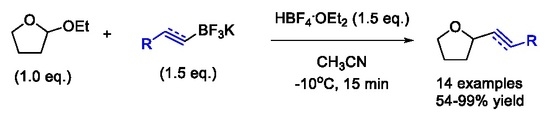
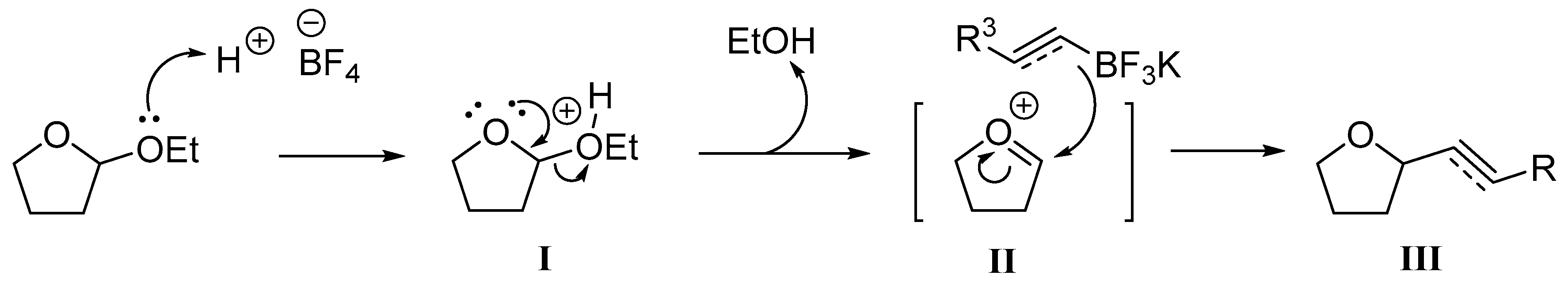
:
1. Introduction
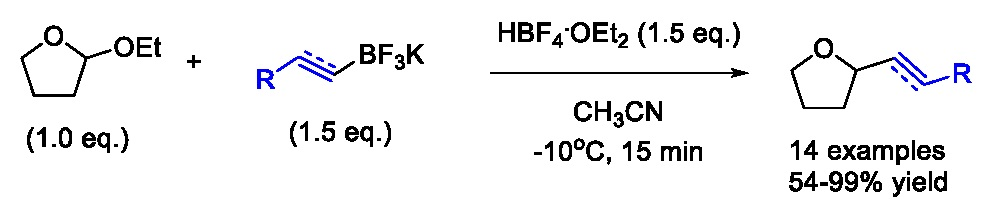
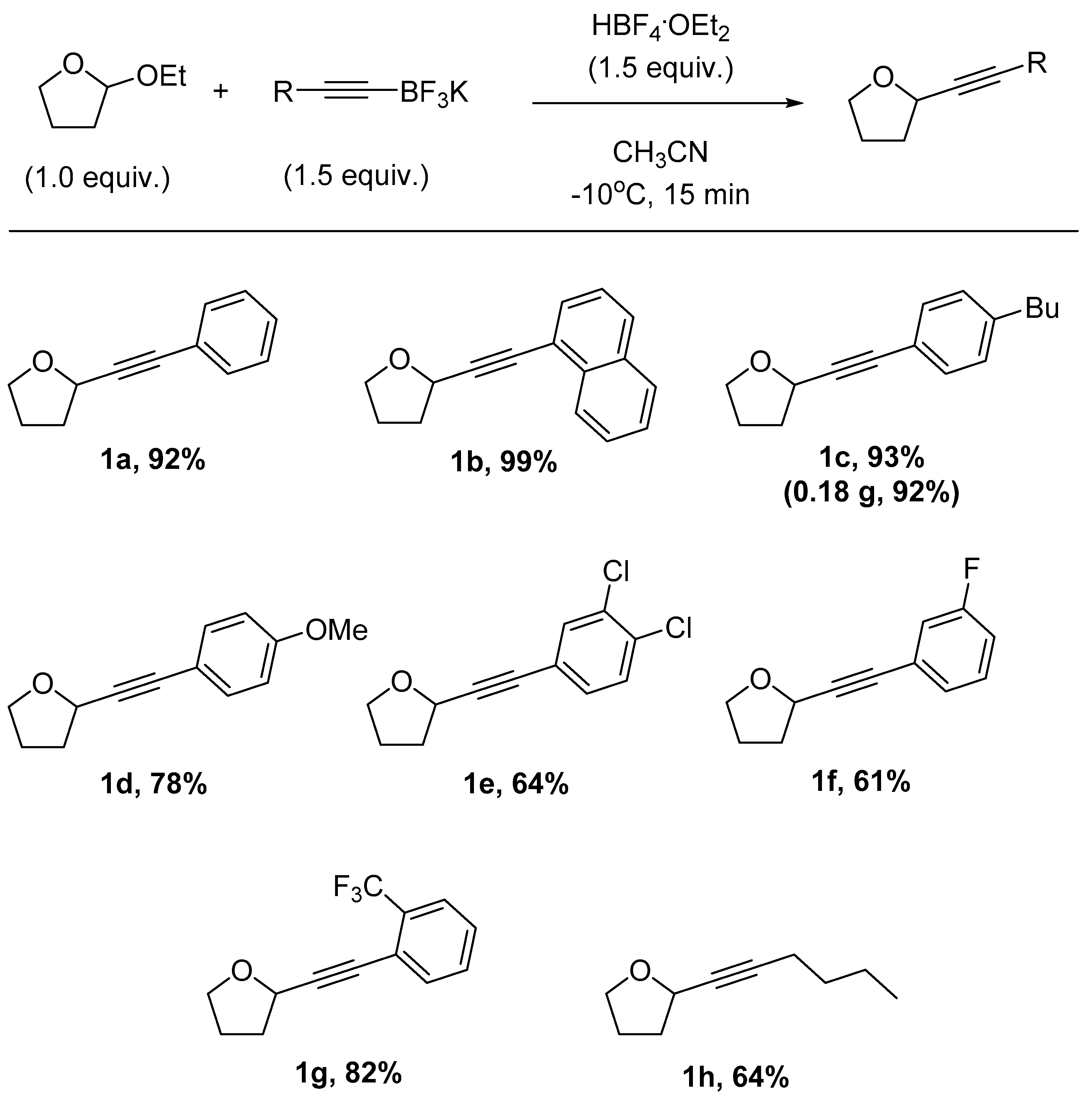
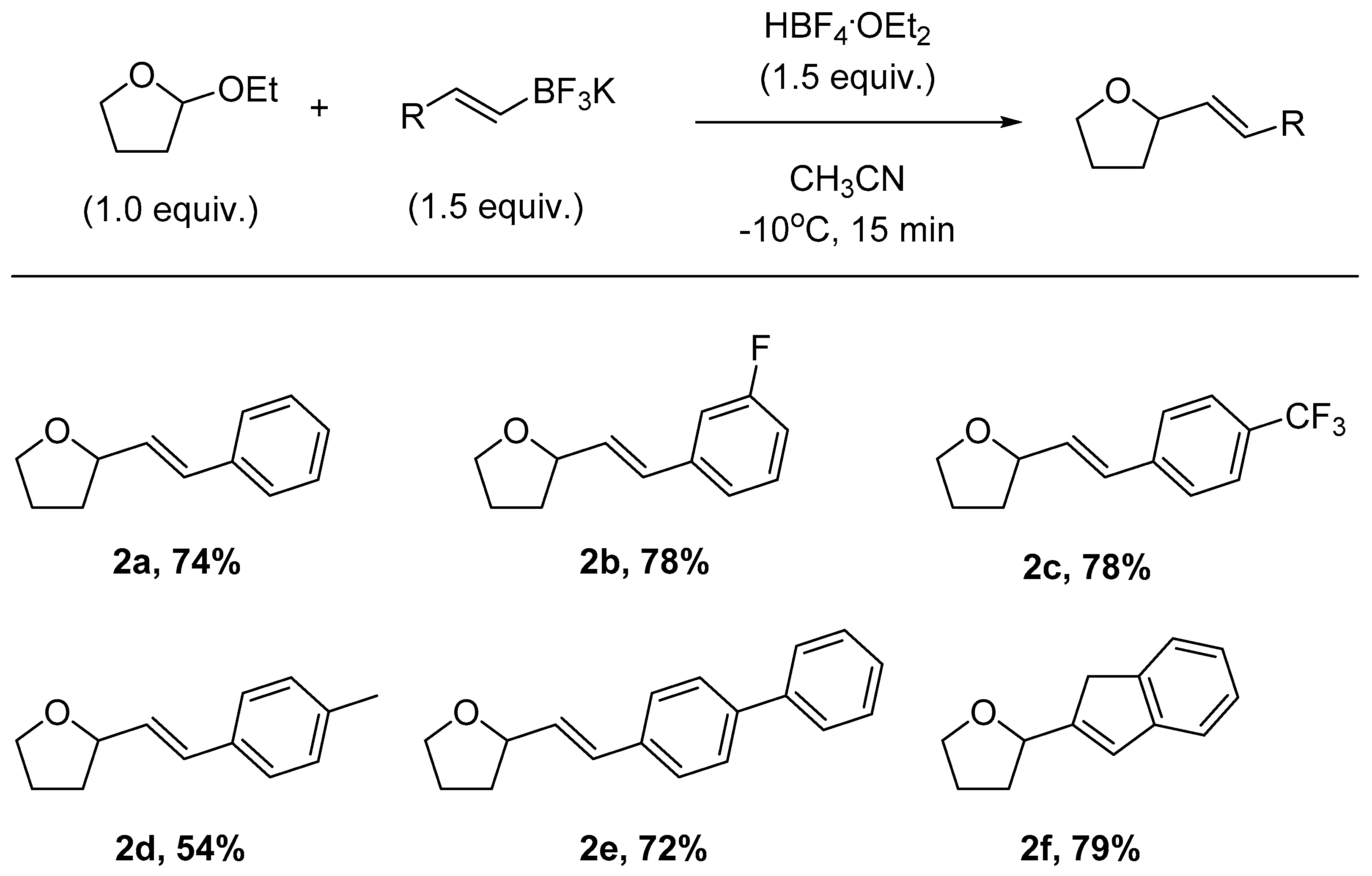
2. Results and Discussion
3. Experimental Section
3.1. General Synthetic Methods
3.2. General Procedure for the Synthesis of Alkynyl Potassium Organotrifluoroborate Salts
3.3. General Procedure for the Synthesis of Alkenyl Potassium Organotrifluoroborate Salts
3.4. General Procedure for the Synthesis of Tetrahydrofurans
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Nasir, N.M.; Ermanis, K.; Clarke, P.A. Strategies for the construction of tetrahydropyran rings in the synthesis of natural products. Org. Biomol. Chem. 2014, 12, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Lorente, A.; Lamariano-Merketegi, J.; Albericio, F.; Álvarez, M. Tetrahydrofuran-Containing Macrolides: A Fascinating Gift from the Deep Sea. Chem. Rev. 2013, 113, 4567–4610. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.M.; Bolshan, Y. Brønsted Acid-Catalyzed Reactions of Trifluoroborate Salts with Benzhydryl Alcohols. J. Org. Chem. 2015, 80, 12676–12685. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.; Bolshan, Y. A General Access to Propargylic Ethers through Brønsted Acid Catalyzed Alkynylation of Acetals and Ketals with Trifluoroborates. Chem. Eur. J. 2015, 21, 13535–13538. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Ellis, N. Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res. 2007, 40, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Darses, S.; Genêt, J.-P. Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis. Chem. Rev. 2008, 108, 288–325. [Google Scholar] [CrossRef] [PubMed]
- Stefani, H.A.; Cella, R.; Vieira, A.S. Recent advances in organotrifluoroborate chemistry. Tetrahedron 2007, 63, 3623–3658. [Google Scholar] [CrossRef]
- Molander, G.A.; Figueroa, R. Organotrifluoroborates: Expanding Organoboron Chemistry. Aldrichim. Acta 2005, 38, 49–56. [Google Scholar] [CrossRef]
- Darses, S.; Genêt, J.P. Potassium Trifluoro(organo)borates: New Perspectives in Organic Chemistry. Eur. J. Org. Chem. 2003, 2003, 4313–4327. [Google Scholar] [CrossRef]
- Roscales, S.; Csákÿ, A.G. Transition-metal-free C-C bond forming reactions of aryl, alkenyl and alkynylboronic acids and their derivatives. Chem. Soc. Rev. 2014, 43, 8215–8225. [Google Scholar] [CrossRef] [PubMed]
- Candeias, N.R.; Montalbano, F.; Cal, P.M.S.D.; Gois, P.M.P. Boronic Acids and Esters in the Petasis-Borono Mannich Multicomponent Reaction. Chem. Rev. 2010, 110, 6169–6193. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.A.; Bode, J.W. Synthesis of Dialkyl Ethers from Organotrifluoroborates and Acetals. J. Am. Chem. Soc. 2009, 131, 18057–18059. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.-V.T.; Mitchell, T.A.; Bode, J.W. Expanded Substrate Scope and Improved Reactivity of Ether-Forming Cross-Coupling Reactions of Organotrifluoroborates and Acetals. J. Am. Chem. Soc. 2011, 133, 14082–14089. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Vedachalam, S.; Xiang, S.; Liu, X.-W. Direct C-Glycosylation of Organotrifluoroborates with Glycosyl Fluorides and Its Application to the Total Synthesis of (+)-Varitriol. Org. Lett. 2011, 13, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.S.; Fiorante, P.F.; Hough, T.L.S.; Ferreira, F.P.; Lüdtke, D.S.; Stefani, H.A. Nucleophilic Addition of Potassium Alkynyltrifluoroborates to ᴅ-Glucal Mediated by BF3·OEt2: Highly Stereoselective Synthesis of α-C-glycosides. Org. Lett. 2008, 10, 5215–5218. [Google Scholar] [CrossRef] [PubMed]
- Postema, M.H.D. Recent Developments in the Synthesis of C-Glycosides. Tetrahedron 1992, 48, 8545–8599. [Google Scholar] [CrossRef]
- Postema, M.H.D. C-Glycoside Synthesis; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Levy, D.E.; Tang, C. The Chemistry of C-Glycosides; Pergamon: Oxford, UK, 1995. [Google Scholar]
- Sinaÿ, P. Synthesis of oligosaccharide mimetics. Pure Appl. Chem. 1997, 69, 459–463. [Google Scholar] [CrossRef]
- Beau, J.-M.; Gallagher, T. Nucleophilic C-Glycosyl Donors for C-Glycoside Synthesis. Topics Curr. Chem. 1997, 187, 1–54. [Google Scholar]
- Nicotra, F. Synthesis of C-Glycosides of Biological Interest. Topics Curr. Chem. 1997, 187, 55–83. [Google Scholar]
- Du, Y.; Linhardt, R.J. Recent Advances in Stereoselective C-Glycoside Synthesis. Tetrahedron 1998, 54, 9913–9959. [Google Scholar] [CrossRef]
- Bililign, T.; Griffith, B.R.; Thorson, J.S. Structure, activity, synthesis and biosynthesis of aryl-C-glycosides. Nat. Prod. Rep. 2005, 22, 742–760. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.W.; He, M.S. Recent advances in aryl C-glycoside synthesis. Curr. Top. Med. Chem. 2005, 5, 1333–1350. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.J.; Nguyen, H.M. Recent Advances in Transition Metal-Catalyzed Glycosylation. ACS Catal. 2012, 2, 1563–1595. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.S.; Ley, S.V. Direct Substitution of 2-Benzenesulfonyl Cyclic Ethers Using Organozinc Reagents. Tetrahedron Lett. 1988, 29, 4869–4872. [Google Scholar] [CrossRef]
- Brown, D.S.; Ley, S.V. Substitution reactions of 2-benzenesulfonyl cyclic ethers: tetrahydro-2-(phenylethynyl)-2H-pyran. Org. Synth. 1991, 70, 157. [Google Scholar]
- Buffet, M.F.; Dixon, D.J.; Ley, S.V.; Reynolds, D.J.; Storer, R.I. Anomeric oxygen to carbon rearrangements of alkynyl tributylstannane derivatives of furanyl (γ)- and pyranyl (δ)-lactols. Org. Biomol. Chem. 2004, 2, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Zhai, W.; Williams, R.M. Alkynylation of mixed acetals with organotin acetylides. J. Am. Chem. Soc. 1988, 110, 2501–2505. [Google Scholar] [CrossRef]
- Gong, J.; Fuchs, P.L. Alkynylation of C-H Bonds via Reaction with Acetylenic Triflones. J. Am. Chem. Soc. 1996, 118, 4486–4487. [Google Scholar] [CrossRef]
- Xiang, J.; Fuchs, P.L. Alkenylation of C-H Bonds via Reaction with Vinyl and Dienyl Triflones. Stereospecific Synthesis of Trisubstituted Vinyl Triflones via Organocopper Addition to Acetylenic Triflones. J. Am. Chem. Soc. 1996, 118, 11986–11987. [Google Scholar] [CrossRef]
- Daniels, D.S.B.; Thompson, A.L.; Anderson, E.A. Palladium-Catalyzed Asymmetric Synthesis of 2-Alkynyl Oxacycles. Angew. Chem. Int. Ed. 2011, 50, 11506–11510. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ghanbari, S.; Nakamura, S.; Hall, D.G. Boronic Acid Catalysis as a Mild and Versatile Strategy for Direct Carbo- and Heterocyclizations of Free Allylic Alcohols. Angew. Chem. Int. Ed. 2012, 51, 6187–6190. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Rooke, S.; Sparey, T.J.; Taylor, P.C. Synthesis of Styryl Tetrahydrofurans and Tetrahydropyrans via Addition of Radicals to Unsaturated Sulfimides. Tetrahedron Lett. 1996, 37, 909–912. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Y.-X.; An, Y.; Song, X.-L.; Wang, Y.-H.; Zhu, L.-L.; Guo, L. Radical Addition of Ethers to Terminal Alkynes with High E-Selectivity. Eur. J. Org. Chem. 2009, 2009, 5146–5152. [Google Scholar] [CrossRef]
- Li, J.; Zhang, J.; Tan, H.; Wang, D.Z. Visible-Light Promoted Vinylation of Tetrahydrofurans with Alkynes through Direct C-H Bond Functionalization. Org. Lett. 2015, 17, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, H.; Zhang, X.; Zeng, W.; Liang, Y. Synthesis of 2-Alkynyltetrahydrofuran Derivatives from Tetrahydrofuran and Alkynyl Bromides. Synthesis 2013, 45, 3137–3146. [Google Scholar]
- Zhang, J.; Li, P.; Wang, L. KOAc-promoted alkynylation of α-C-H bonds of ethers with alkynyl bromides under transition-metal-free conditions. Org. Biomol. Chem. 2014, 12, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhou, W.; Han, J.; Li, G.; Pan, Y. Iron-catalyzed alkenylation of cyclic ethers via decarboxylative sp3(C)-sp2(C) coupling. Tetrahedron Lett. 2013, 54, 6507–6510. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Xi, L.-Y.; Zhang, L.; Liang, S.; Chen, S.-Y.; Yu, X.-Q. Metal-free alkynylation of α-C-H bonds of ethers with ethynylbenziodoxolones. RSC Adv. 2014, 4, 54349–54353. [Google Scholar] [CrossRef]
- Sølvhøj, A.; Ahlburg, A.; Madsen, R. Dimethylzinc-Initiated Radical Coupling of β-Bromostyrenes with Ethers and Amines. Chem. Eur. J. 2015, 21, 16272–16279. [Google Scholar] [CrossRef] [PubMed]
- Kadota, I.; Lutete, L.M.; Shibuya, A.; Yamamoto, Y. Palladium/benzoic acid-catalyzed hydroalkoxylation of alkynes. Tetrahedron Lett. 2001, 42, 6207–6210. [Google Scholar] [CrossRef]
- Patil, N.T.; Lutete, L.M.; Wu, H.; Pahadi, N.K.; Gridnev, I.D.; Yamamoto, Y. Palladium-Catalyzed Intramolecular Asymmetric Hydroamination, Hydroalkoxylation, and Hydrocarbonation of Alkynes. J. Org. Chem. 2006, 71, 4270–4279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-Z.; Tian, Y.; Li, G.-X.; Qu, J. Intramolecular Etherification and Polyene Cyclization of π-Activated Alcohols Promoted by Hot Water. J. Org. Chem. 2015, 80, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Yasuda, A.; Shibata, M.; Ban, S.; Hashimoto, Y.; Okamoto, I.; Tamura, O. Gold(I)/(III)-Catalyzed Synthesis of Cyclic Ethers; Valency-Controlled Cyclization Modes. Org. Lett. 2015, 17, 2668–2671. [Google Scholar] [CrossRef] [PubMed]
- Haidzinskaya, T.; Kerchner, H.; Liu, J.; Watson, M.P. Diastereoselective, Zinc-Catalyzed Alkynylation of α-Bromo Oxocarbenium Ions. Org. Lett. 2015, 17, 3857–3859. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Meng, Z.; Lou, H.; Liu, L. Practical and Highly Selective C-H Functionalization of Structurally Diverse Ethers. Angew. Chem. Int. Ed. 2014, 53, 13845–13849. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; MacMillan, D.W.C. Organocatalytic Vinyl and Friedel-Crafts Alkylations with Trifluoroborates Salts. J. Am. Chem. Soc. 2007, 129, 15438–15439. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Felix, L.A. Stereoselective Suzuki-Miyaura Cross-Coupling Reactions of Potassium Alkenyltrifluoroborates with Alkenyl Bromides. J. Org. Chem. 2005, 70, 3950–3956. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Bolshan, Y. Metal-Free Synthesis of Ynones from Acyl Chlorides and Potassium Alkynyltrifluoroborate Salts. Org. Lett. 2014, 16, 488–491. [Google Scholar] [CrossRef] [PubMed]




| Entry | BF3K (Equiv.) | Brønsted Acid | Brønsted Acid (Equiv.) | Yield (%) |
|---|---|---|---|---|
| 1 | 1.1 | HBF4·OEt2 | 1.1 | 75 |
| 2 | 1.1 | CF3COOH | 1.1 | trace |
| 3 | 1.5 | HBF4·OEt2 | 1.5 | 92 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, K.M.; Bolshan, Y. Brønsted Acid-Catalyzed Direct Substitution of 2-Ethoxytetrahydrofuran with Trifluoroborate Salts. Catalysts 2016, 6, 94. https://doi.org/10.3390/catal6070094
Fisher KM, Bolshan Y. Brønsted Acid-Catalyzed Direct Substitution of 2-Ethoxytetrahydrofuran with Trifluoroborate Salts. Catalysts. 2016; 6(7):94. https://doi.org/10.3390/catal6070094
Chicago/Turabian StyleFisher, Kayla M., and Yuri Bolshan. 2016. "Brønsted Acid-Catalyzed Direct Substitution of 2-Ethoxytetrahydrofuran with Trifluoroborate Salts" Catalysts 6, no. 7: 94. https://doi.org/10.3390/catal6070094