
Mixed-Metal Semiconductor Anodes for Electrochemical Water Splitting and Reactive Chlorine Species Generation: Implications for Electrochemical Wastewater Treatment

Abstract
:
1. Introduction
2. Results and Discussion
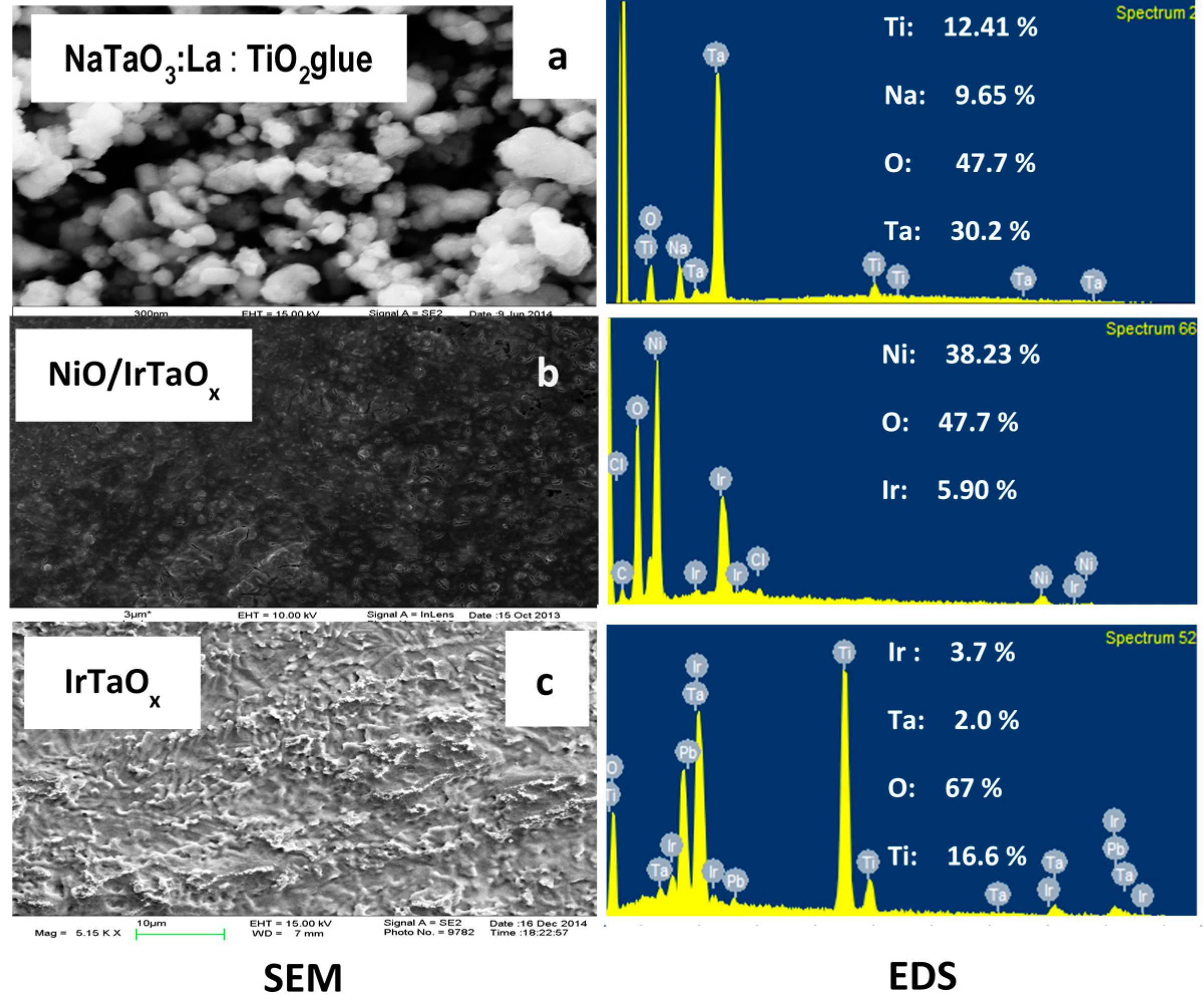
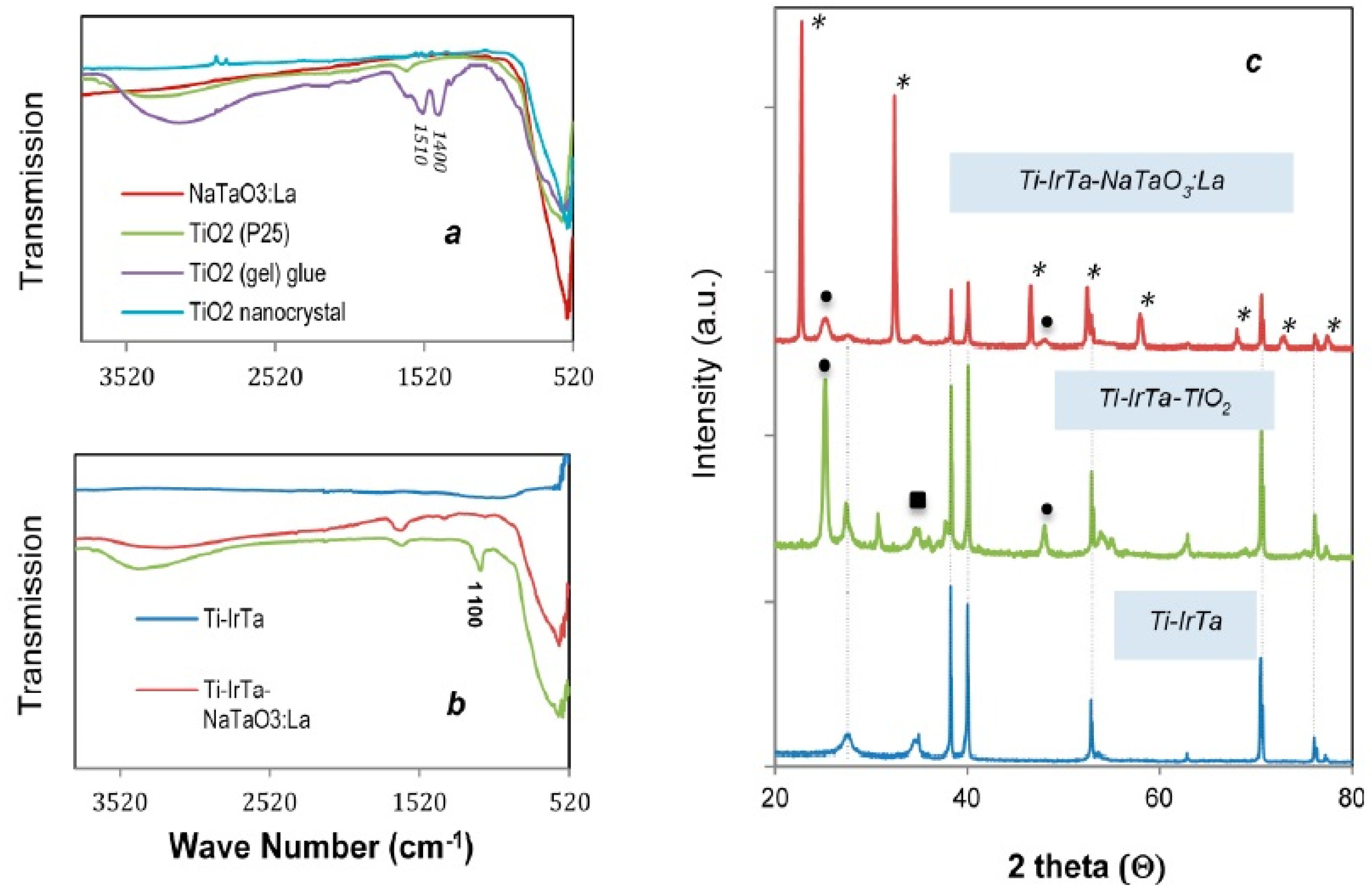
2.1. Anode Characterization
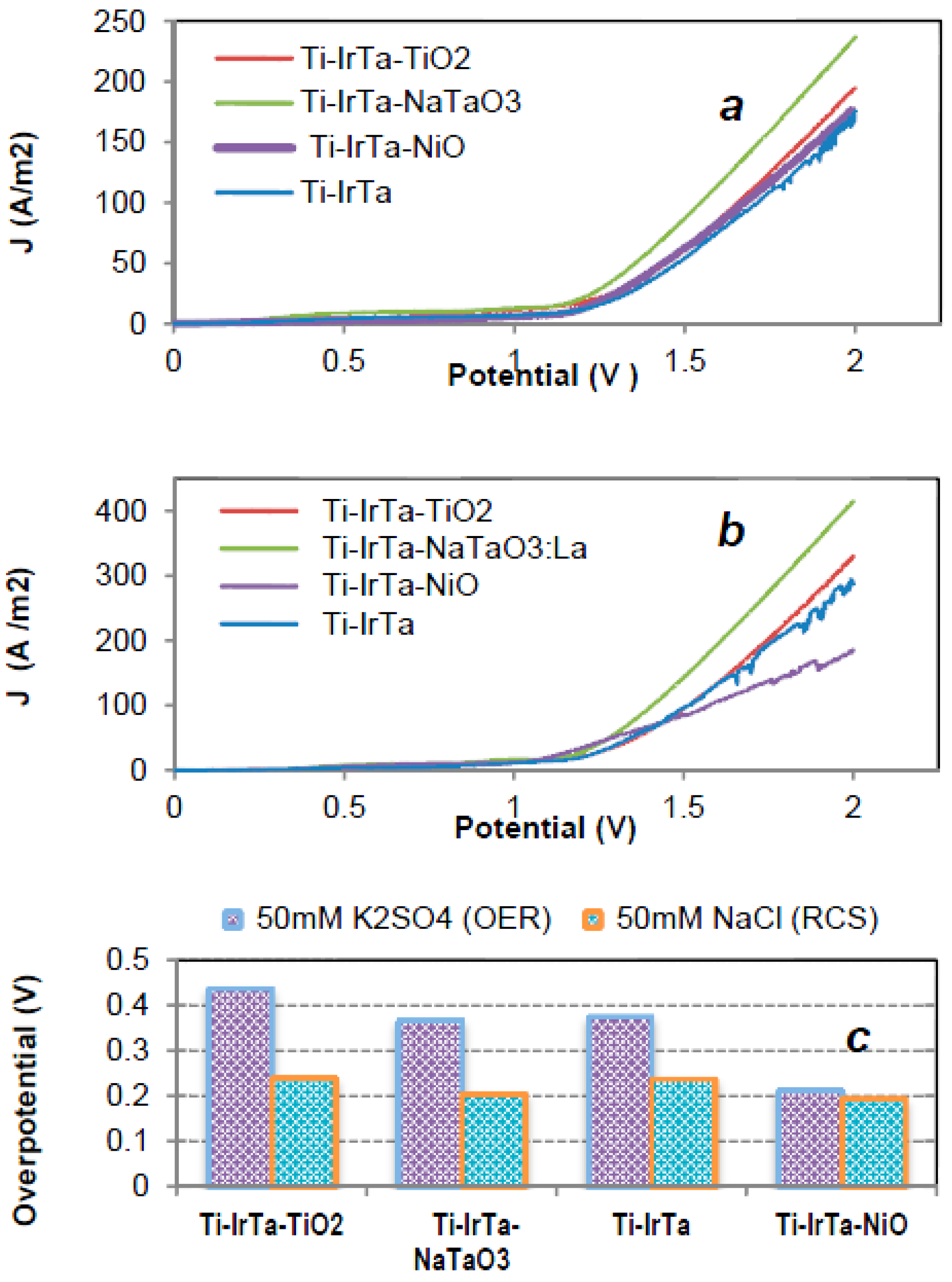
2.2. Electrochemical Characterization
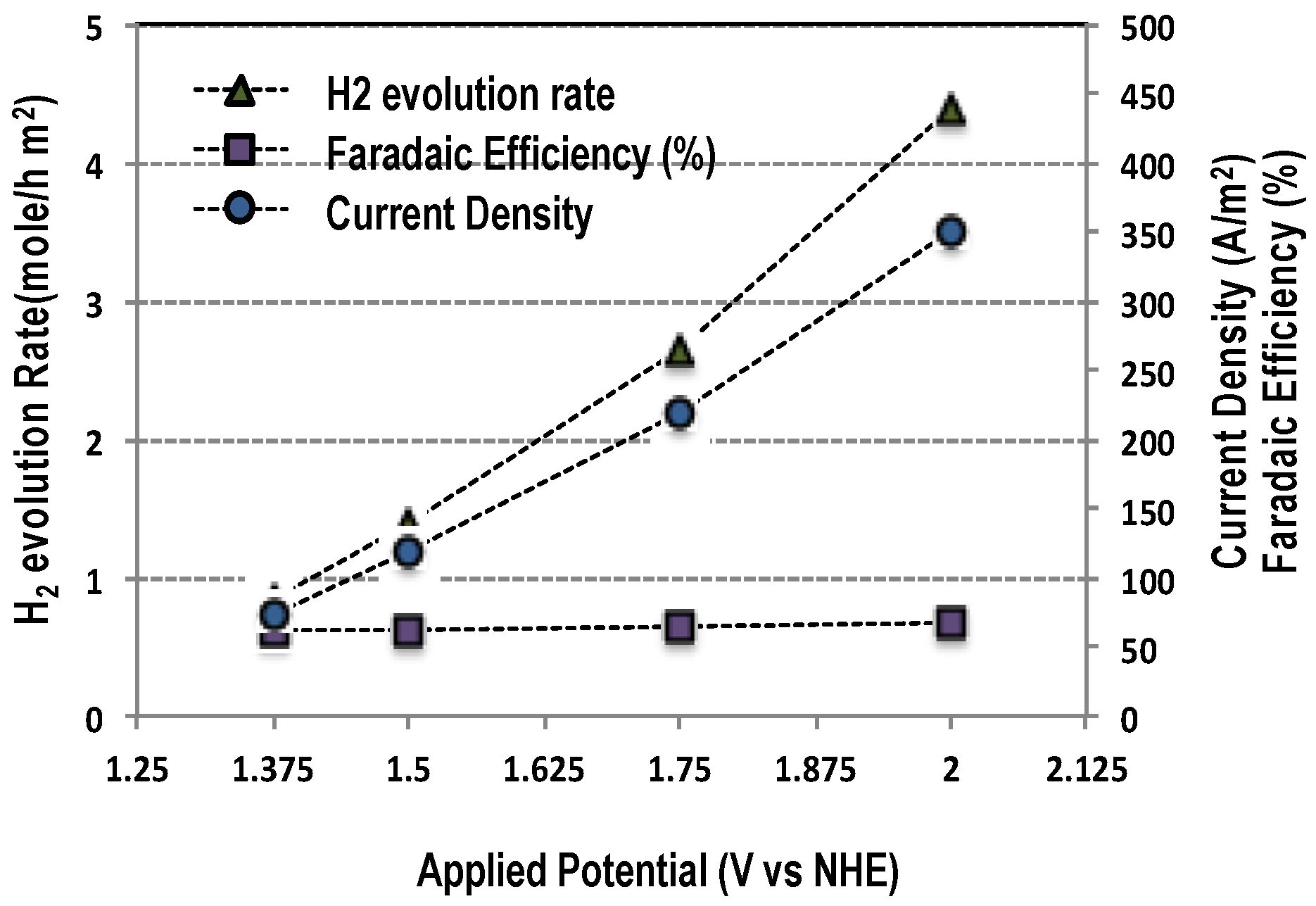
2.3. Electrocatalysis
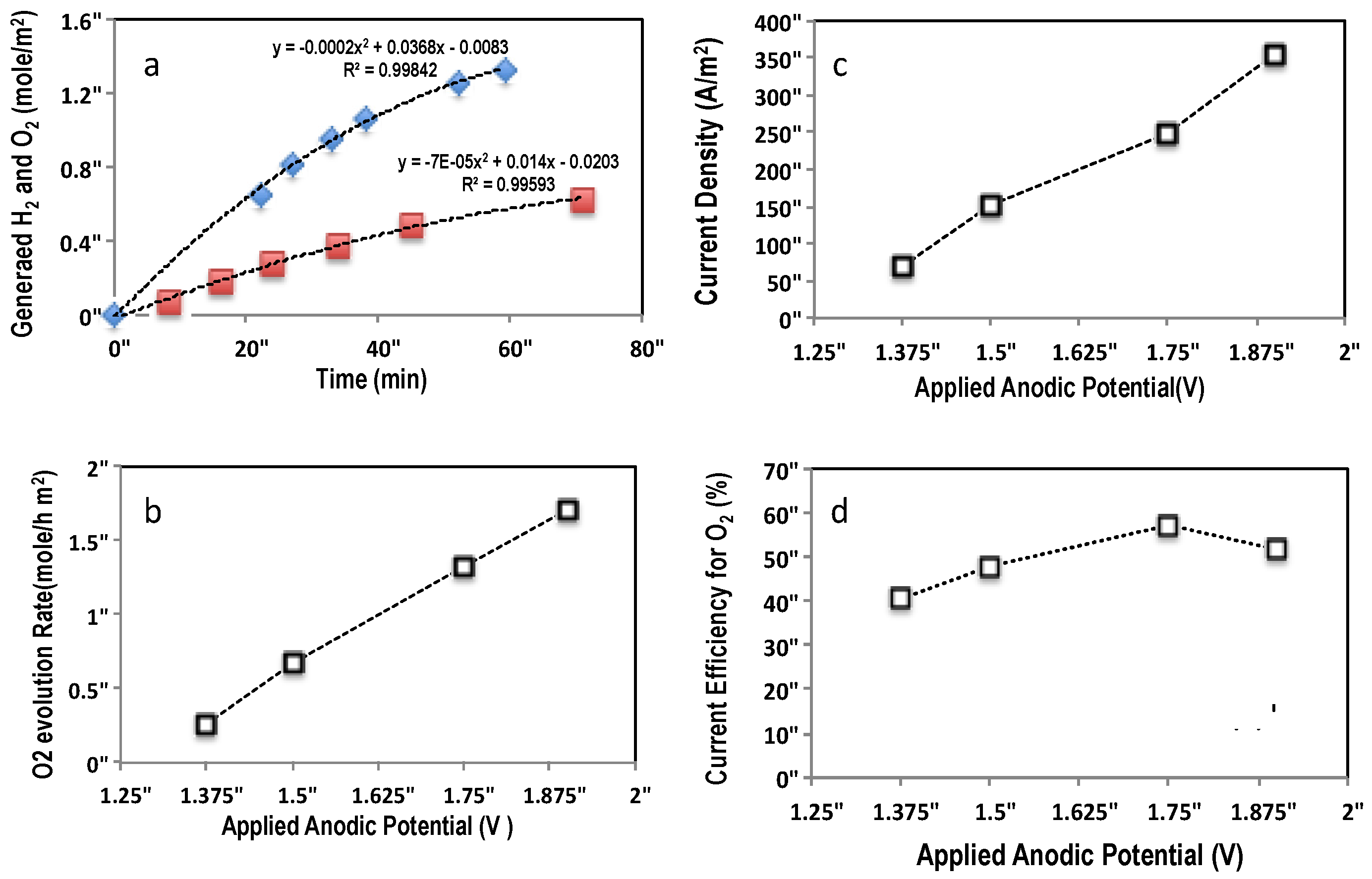
2.3.1. Water Oxidation
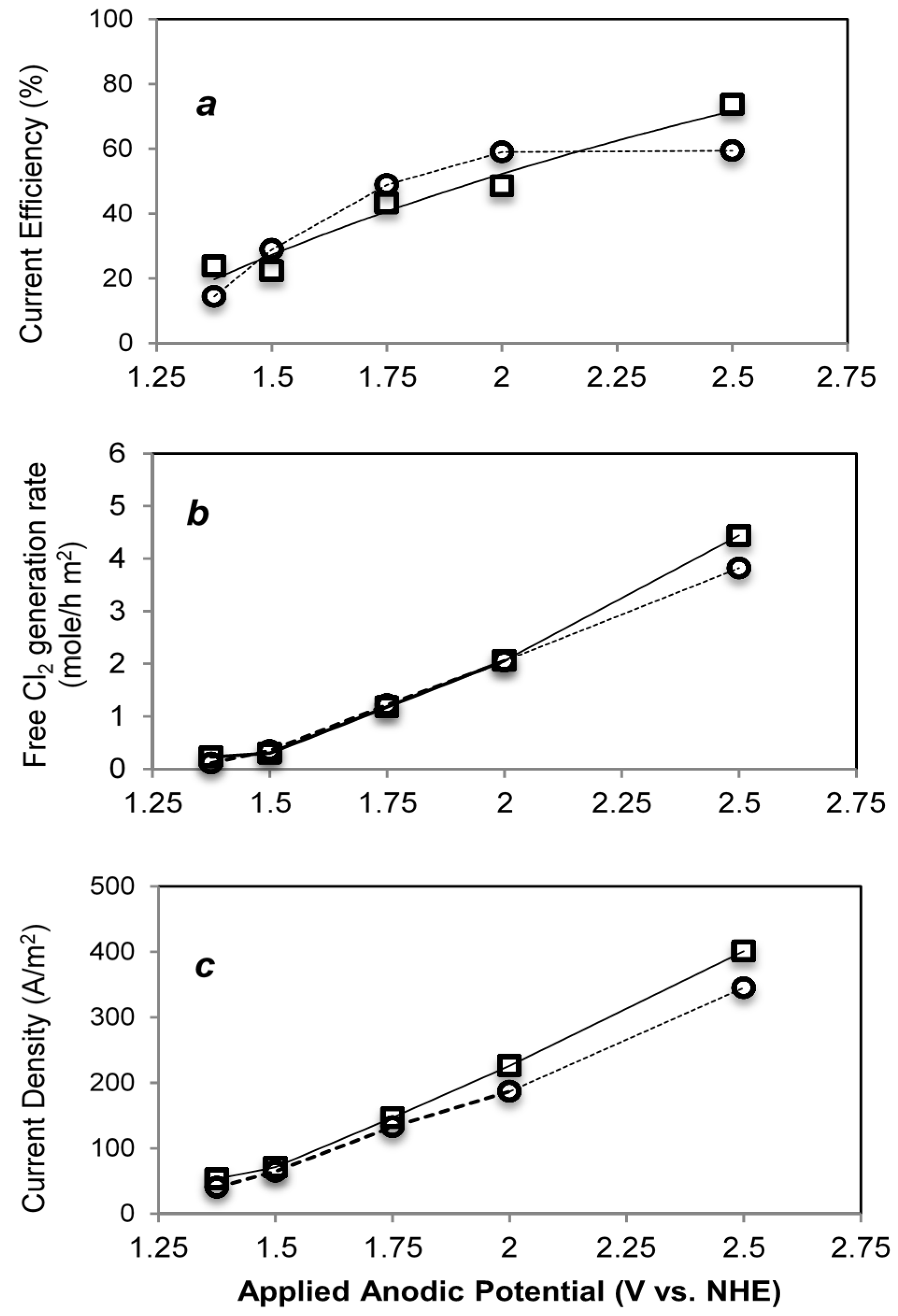
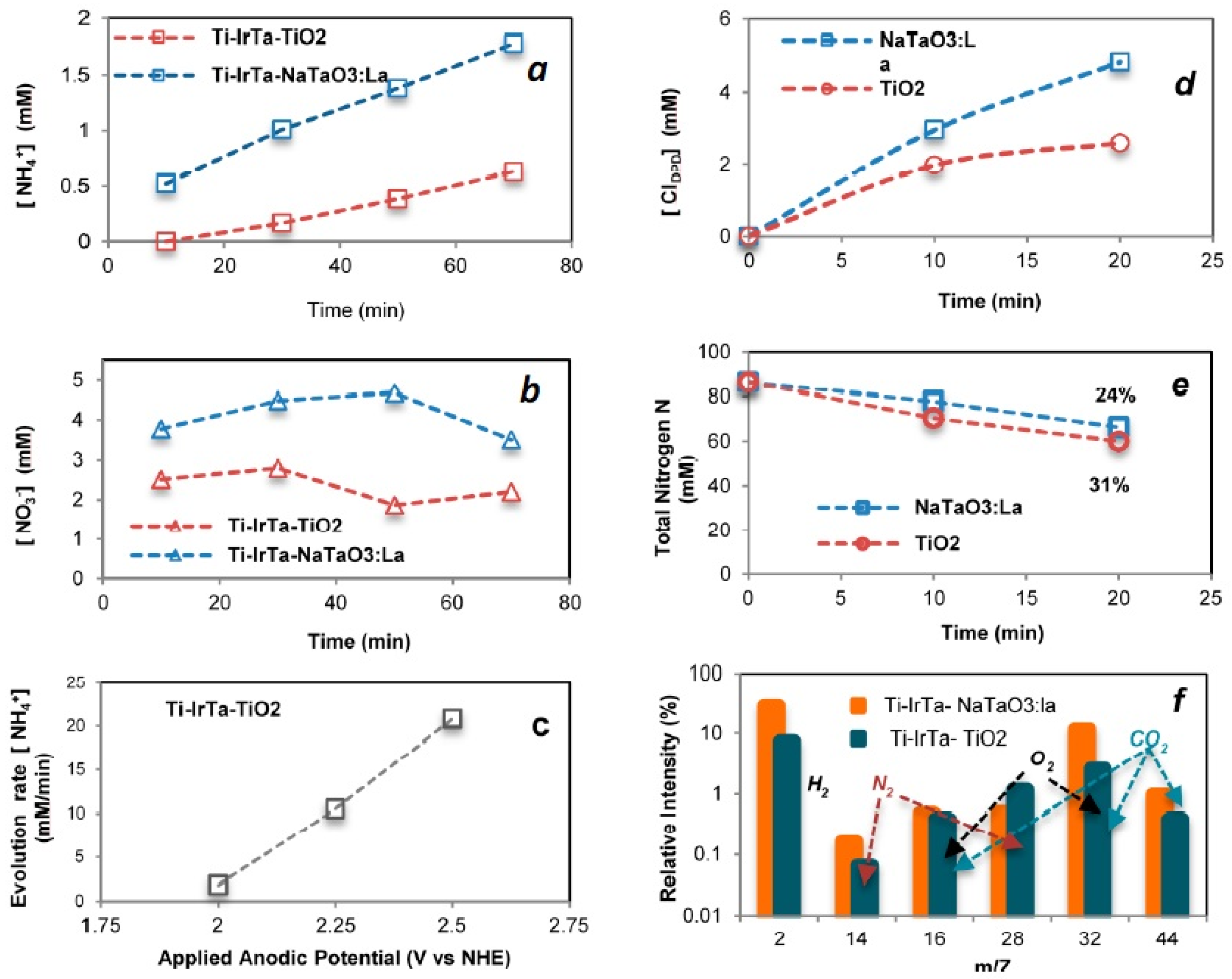
2.3.2. Wastewater Treatment
2.3.3. Urea Degradation
3. Experimental Details
3.1. Synthetic Procedures: La-Doped NaTaO3 and TiO2 Nanogel
3.2. Electrode Fabrication
3.3. Instrumentations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cho, K.; Hoffmann, M.R. Urea degradation by electrochemically generated reactive chlorine species: Products and reaction pathways. Environ. Sci. Technol. 2014, 48, 11504–11511. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Kwon, D.; Hoffmann, M.R. Electrochemical treatment of human waste coupled with molecular hydrogen production. RSC Adv. 2014, 4, 4596–4608. [Google Scholar] [CrossRef]
- Cho, K.; Qu, Y.; Kwon, D.; Zhang, H.; Cid, C.A.; Aryanfar, A.; Hoffmann, M.R. Effects of anodic potential and chloride ion on overall reactivity in electrochemical reactors designed for solar-powered wastewater treatment. Environ. Sci. Technol. 2014, 48, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Hoffmann, M.R. BixTi1−xOz functionalized heterojunction anode with an enhanced reactive chlorine generation efficiency in dilute aqueous solutions. Chem. Mater. 2015, 27, 2224–2233. [Google Scholar] [CrossRef]
- Zeng, K.; Zhang, D.K. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 2010, 36, 307–326. [Google Scholar] [CrossRef]
- Tributsch, H. Photovoltaic hydrogen generation. Int. J. Hydrog. Energy 2008, 33, 5911–5930. [Google Scholar] [CrossRef]
- Hall, D.E. Ni(OH)2 impregnated anodes for alkaline water electrolysis. J. Electrochem. Soc. 1983, 130, 317–321. [Google Scholar] [CrossRef]
- Doyle, R.L.; Godwin, I.J.; Brandon, M.P.; Lyons, M.E. Redox and electrochemical water splitting catalytic properties of hydrated metal oxide modified electrodes. Phys. Chem. Chem. Phys. PCCP 2013, 15, 13737–13783. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, W.; Lee, D.K.; Kim, K.; Park, N.G.; Ko, M.J. Pure anatase TiO2 “nanoglue”: An inorganic binding agent to improve nanoparticle interconnections in the low-temperature sintering of dye-sensitized solar cells. Appl. Phys. Lett. 2011, 98, 103301. [Google Scholar] [CrossRef]
- Lyons, M.E.; Floquet, S. Mechanism of oxygen reactions at porous oxide electrodes. Part 2-oxygen evolution at RuO2, IrO2 and IrxRu1−xO2 electrodes in aqueous acid and alkaline solution. Phys. Chem. Chem. Phys. PCCP 2011, 13, 5314–5335. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Asakura, K.; Kudo, A. Highly efficient water splitting into H2 and O2 over lanthanum-doped NaTaO3 photocatalysts with high crystallinity and surface nanostructure. J. Am. Chem. Soc. 2003, 125, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Kotz, R.; Stucki, S. Stabilization of RuO2 by IrO2 for anodic oxygen evolution in acid media. Electrochem. Acta 1986, 31, 1311–1316. [Google Scholar] [CrossRef]
- Park, H.; Vecitis, C.D.; Choi, W.; Weres, O.; Hoffmann, M.R. Solar-powered production of molecular hydrogen from water. J. Phys. Chem. C 2008, 112, 885–889. [Google Scholar] [CrossRef]
- Park, H.; Vecitis, C.D.; Hoffmann, M.R. Solar-powered electrochemical oxidation of organic compounds coupled with the cathodic production of molecular hydrogen. J. Phys. Chem. A 2008, 112, 7616–7626. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Vecitis, C.D.; Hoffmann, M.R. Electrochemical water splitting coupled with organic compound oxidation: The role of active chlorine species. J. Phys. Chem. C 2009, 113, 7935–7945. [Google Scholar] [CrossRef]
- Martinez-Huitle, C.A.; Ferro, S. Electrochemical oxidation of organic pollutants for the wastewater treatment: direct and indirect processes. Chem. Soc. Rev. 2006, 35, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Huitle, C.A.; Brillas, E. Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review. Appl. Catal. B Environ. 2009, 87, 105–145. [Google Scholar] [CrossRef]
- Raut, A.S.; Cunningham, G.B.; Parker, C.B.; Klem, E.J.D.; Stoner, B.R.; Deshusses, M.A.; Glass, J.T. Disinfection of E. coli contaminated urine using boron-doped diamond electrodes. J. Electrochem. Soc. 2014, 161, G81–G81. [Google Scholar] [CrossRef]
- Antonin, V.S.; Santos, M.C.; Garcia-Segura, S.; Brillas, E. Electrochemical incineration of the antibiotic ciprofloxacin in sulfate medium and synthetic urine matrix. Water Res. 2015, 83, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Marmanis, D.; Dermentzis, K.; Christoforidis, A.; Ouzounis, K.; Moumtzakis, A. Electrochemical treatment of actual dye house effluents using electrocoagulation process directly powered by photovoltaic energy. Desalin. Water Treat. 2015, 56, 2988–2993. [Google Scholar] [CrossRef]
- Muazu, N.D.; Jarrah, N.; Bukhari, A. Kinetic modeling of electrochemical oxidation of phenol on boron-doped diamond anode in the presence of some inorganic species. Desalin. Water Treat. 2015, 56, 3005–3012. [Google Scholar] [CrossRef]
- Radjenovic, J.; Sedlak, D.L. Challenges and opportunities for electrochemical processes as next generation technologies for the treatment of contaminated water. Environ. Sci. Technol. 2015, 49, 11292–11302. [Google Scholar] [CrossRef] [PubMed]
- Zollig, H.; Fritzsche, C.; Morgenroth, E.; Udert, K.M. Direct electrochemical oxidation of ammonia on graphite as a treatment option for stored source-separated urine. Water Res. 2015, 69, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Zollig, H.; Morgenroth, E.; Udert, K.M. Inhibition of direct electrolytic ammonia oxidation due to a change in local pH. Electrochim. Acta 2015, 165, 348–355. [Google Scholar] [CrossRef]
- Zollig, H.; Remmele, A.; Fritzsche, C.; Morgenroth, E.; Udert, K.M. Formation of chlorination byproducts and their emission pathways in chlorine mediated electro-oxidation of urine on active and nonactive type anodes. Environ. Sci. Technol. 2015, 49, 11062–11069. [Google Scholar] [CrossRef] [PubMed]
- Zeradjanin, A.R.; Menzel, N.; Schuhmann, W.; Strasser, P. On the faradaic selectivity and the role of surface inhomogeneity during the chlorine evolution reaction on ternary Ti-Ru-Ir mixed metal oxide electrocatalysts. Phys. Chem. Chem. Phys. 2014, 16, 13741–13747. [Google Scholar] [CrossRef] [PubMed]
- Exner, K.S.; Anton, J.; Jacob, T.; Over, H. Chlorine evolution reaction on RuO2(110): Ab initio atomistic thermodynamics study-Pourbaix diagrams. Electrochim. Acta 2014, 120, 460–466. [Google Scholar] [CrossRef]
- Comninellis, C. Electocatalysis in the electrochemical conversion/combustion of organic pollutants for waste water treatment. Electrochem. Acta 1994, 39, 1857–1862. [Google Scholar] [CrossRef]
- Panizza, M.; Cerisola, G. Direct and mediated anodic oxidation of organic pollutants. Chem. Rev. 2009, 109, 6541–6569. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Logan, B.E. Sustainable and efficient biohydrogen production via electrohydrogenesis. PNAS 2007, 104, 18871–18873. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.A.; Man, I.C.; Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Rossmeisl, J. Electochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 2010, 12, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Trasatti, S. Progress in the understanding of the mechanism of chlorine evolution at oxide electrodes. Electrochem. Acta 1987, 32, 369–382. [Google Scholar] [CrossRef]
- Krishtalik, L.I. Kinetics and mechanism of anodic chlorine and oxygen evolution reactions on transition metal oxide electrodes. Electrochem. Acta 1981, 26, 329–337. [Google Scholar] [CrossRef]
- Simka, W.; Piotrowski, J.; Robak, A.; Nawrat, G. Electrochemical treatment of aqueous solutions containing urea. J. Appl. Electrochem. 2009, 39, 1137–1143. [Google Scholar] [CrossRef]
- Hernlem, B.J. Electrolytic destruction of urea in dilute chloride solution using DSA electrodes in a recycled batch cell. Water Res. 2005, 39, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Carlesi Jara, C.; di Giulio, S.; Fino, D.; Spinelli, P. Combined direct and indirect electroxidation of urea containing water. J. Appl. Electrochem. 2008, 38, 915–922. [Google Scholar] [CrossRef]
- Blatchley, E.R.; Cheng, M.M. Reaction mechanism for chlorination of urea. Environ. Sci. Technol. 2010, 44, 8529–8534. [Google Scholar] [CrossRef] [PubMed]
- Jafvert, C.T.; Valentine, R.L. Reaction scheme for the chlorination of ammoniacal water. Environ. Sci. Technol. 1992, 26, 577–586. [Google Scholar] [CrossRef]
- Deborde, M.; von Gunten, U. Reactions of chlorine with inorganic and organic compounds during water treatment-Kinetics and mechanisms: A critical review. Water Res. 2008, 42, 13–51. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Potential (V vs. NHE) | Current (mA) | Current Density (A/m2) | H2 Evolution (mol/h∙m2) | O2 Evolution (mol/h∙m2) | Current Efficiency (%) for H2/O2 |
|---|---|---|---|---|---|---|
| Ti-IrTa-TiO2 | 1.38 | 43 | 71.7 | 0.834 | - | 62.4/- |
| 1.5 | 72 | 120 | 1.392 | 0.680 | 62.2/60 | |
| 1.75 | 131 | 218.3 | 2.64 | - | 64.8/- | |
| 2.0 | 210 | 350 | 4.404 | - | 67.5/- | |
| Ti-IrTa-NaTaO3 | 1.38 | 41 | 68.3 | - | 0.258 | -/40.6 |
| 1.5 | 91 | 151.7 | 1.8 | 0.678 | 62/48 | |
| 1.75 | 149 | 248.3 | - | 1.326 | -/57.2 | |
| 1.9 | 213 | 355 | - | 1.715 | -/51.8 |
| Electrode (Anode) | Potential (V vs. NHE) | Current (mA) | Current Density (A/m2) | Free Cl2 Evolution (mol/h∙m2) | Current Efficiency (%) |
|---|---|---|---|---|---|
| Ti-IrTa-TiO2 | 1.38V | 24.2 | 40.3 | 0.11 | 16.4 |
| 1.5 V | 38.8 | 64.7 | 0.35 | 31.3 | |
| 1.75 V | 79.7 | 132.8 | 1.22 | 48.9 | |
| 2.0 V | 112 | 186.7 | 2.05 | 59 | |
| 2.5 V | 207 | 345 | 3.82 | 59.4 | |
| Ti-IrTa-NaTaO3 | 1.3 V | 32 | 53.3 | 0.23 | 23.8 |
| 1.5 V | 42.6 | 71 | 0.30 | 22.3 | |
| 1.75 V | 88 | 146.7 | 1.18 | 43.25 | |
| 2.0 V | 135.6 | 226 | 2.06 | 48.5 | |
| 2.5 V | 241 | 401 | 4.44 | 73.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, S.Y.; Hoffmann, M.R. Mixed-Metal Semiconductor Anodes for Electrochemical Water Splitting and Reactive Chlorine Species Generation: Implications for Electrochemical Wastewater Treatment. Catalysts 2016, 6, 59. https://doi.org/10.3390/catal6040059
Ryu SY, Hoffmann MR. Mixed-Metal Semiconductor Anodes for Electrochemical Water Splitting and Reactive Chlorine Species Generation: Implications for Electrochemical Wastewater Treatment. Catalysts. 2016; 6(4):59. https://doi.org/10.3390/catal6040059
Chicago/Turabian StyleRyu, Su Young, and Michael R. Hoffmann. 2016. "Mixed-Metal Semiconductor Anodes for Electrochemical Water Splitting and Reactive Chlorine Species Generation: Implications for Electrochemical Wastewater Treatment" Catalysts 6, no. 4: 59. https://doi.org/10.3390/catal6040059