
Insights into SnO2 Nanoparticles Supported on Fibrous Mesoporous Silica for CO Catalytic Oxidation
 ,
,
Abstract
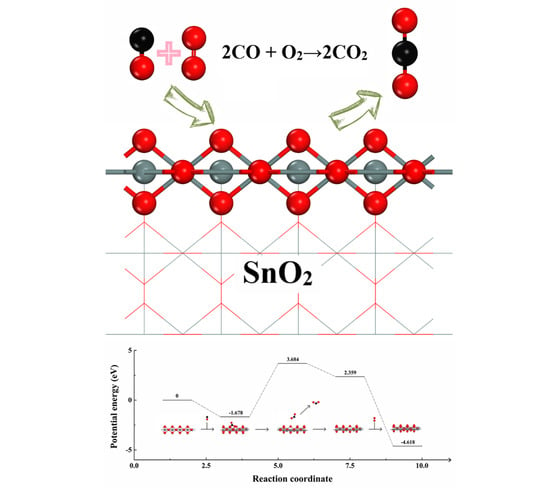
:
1. Introduction
2. Discussion
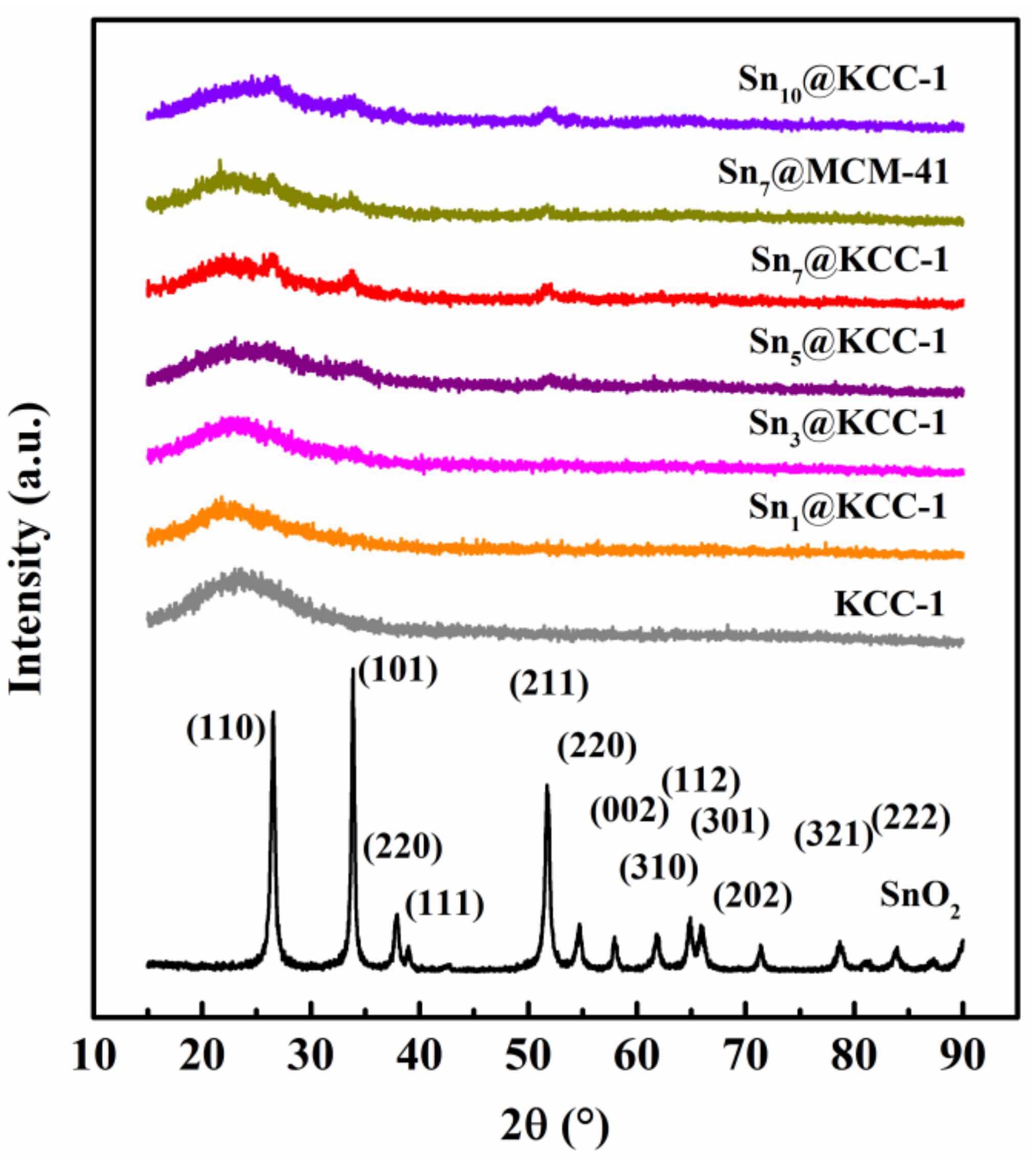
2.1. Basic Morphological Characteristics
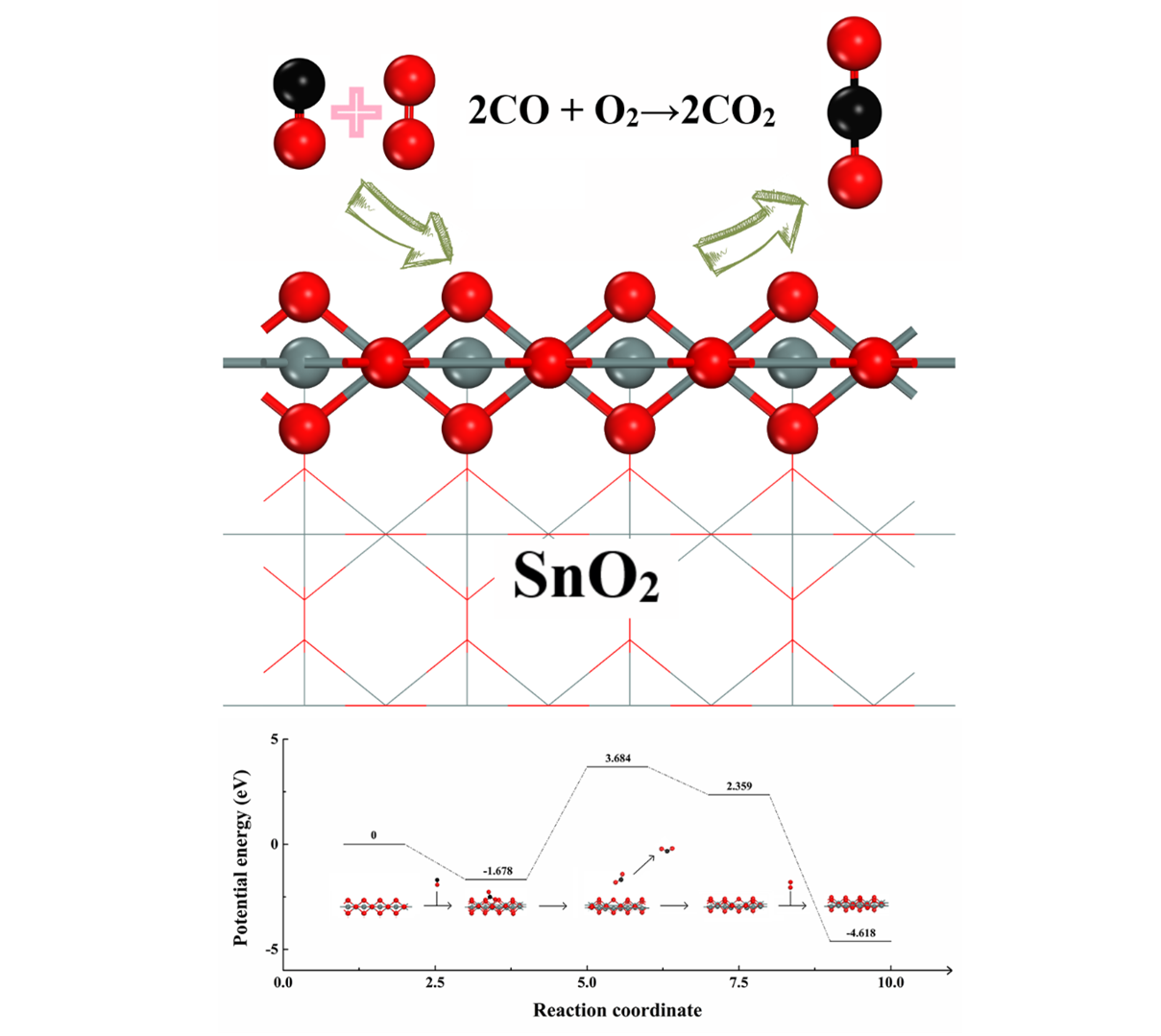
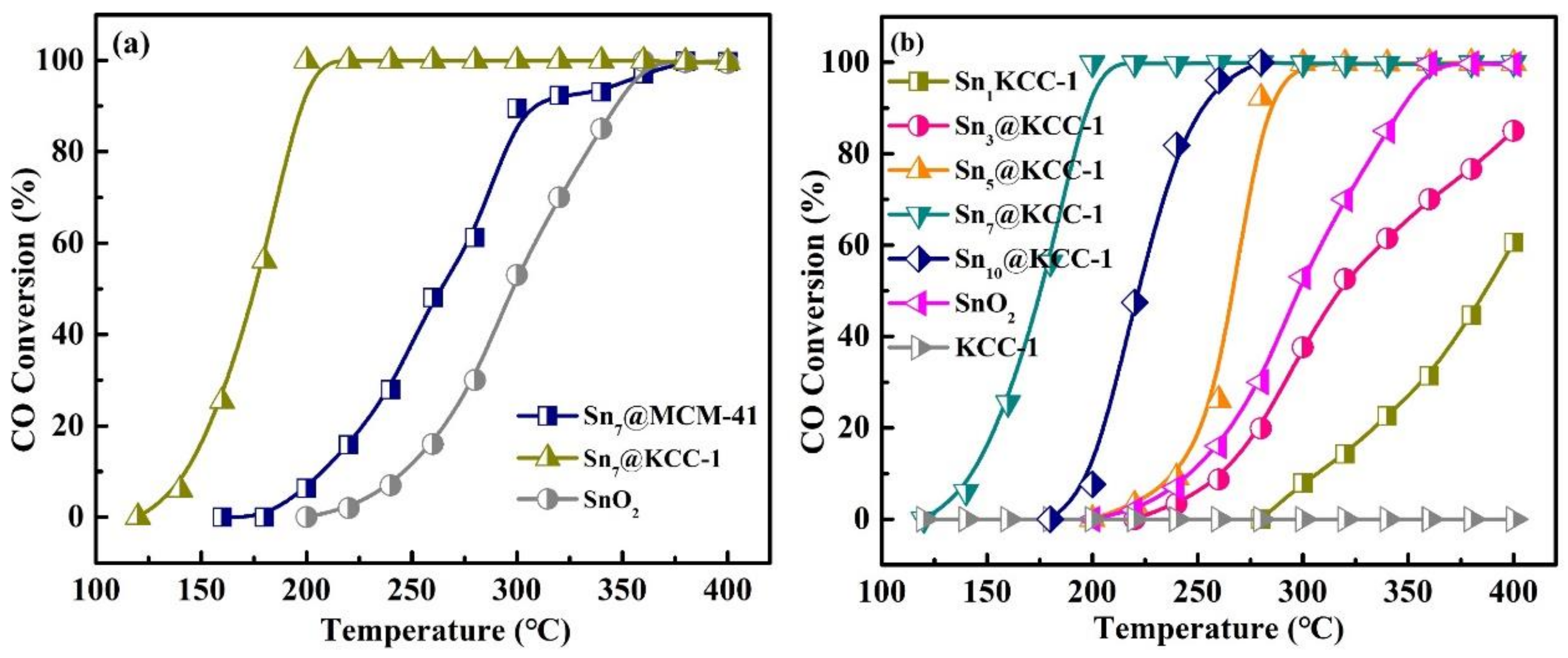
2.2. Evaluation of Catalytic Activity
2.3. Adsorption of CO and O2 on SnO2 (110) Catalyst Surface
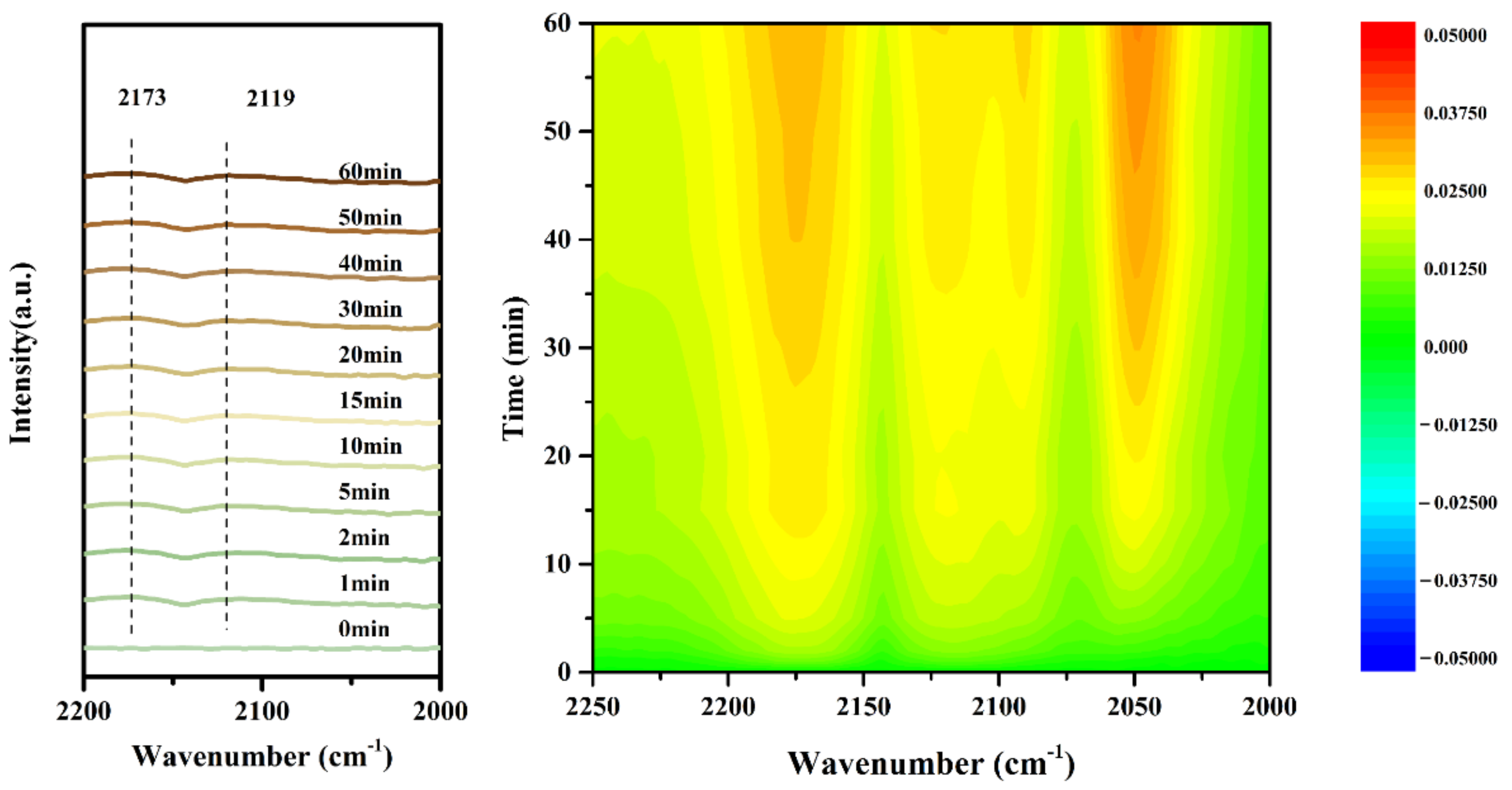
2.4. Instantaneous Intermediates on the Catalysts
3. Conclusions
4. Experimental
4.1. Catalyst Preparation
4.2. Evaluation of the Catalytic Performance
4.3. Catalyst Characterization
4.4. DFT Calculation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lu, Z.S.; Meng, S.J.; Ma, Z.Y.; Yang, M.X.; Ma, D.W.; Yang, Z.X.; Talib, S.H. Electronic and catalytic properties of Ti single atoms@SnO2 and its implications on sensing mechanism for CO. Appl. Surf. Sci. 2022, 594, 153500. [Google Scholar] [CrossRef]
- May, Y.A.; Wang, W.W.; Yan, H.; Wei, S.; Jia, C.J. Insights into facet-dependent reactivity of CuO–CeO2 nanocubes and nanorods as catalysts for CO oxidation reaction. Chin. J. Catal. 2020, 41, 1017–1027. [Google Scholar] [CrossRef]
- Qian, C.Y.; Bian, J.W.; Ju, F.; Ling, H. Effect of PdSx on sulfur resistance of Pd-Based catalysts for CO oxidation. Ind. Eng. Chem. Res. 2023, 62, 3038–3049. [Google Scholar] [CrossRef]
- Zhang, X.D.; Zhang, X.L.; Song, L.; Hou, F.L.; Yang, Y.Q.; Wang, Y.X.; Liu, N. Enhanced catalytic performance for CO oxidation and preferential CO oxidation over CuO/CeO2 catalysts synthesized from metal organic framework: Effects of preparation methods. Int. J. Hydrogen Energy 2018, 43, 18279–18288. [Google Scholar] [CrossRef]
- Twigg, M.V. Catalytic control of emissions from cars. Catal. Today 2011, 163, 33–41. [Google Scholar] [CrossRef]
- Xiang, G.H.; Zhao, S.; Wei, C.D.; Liu, C.Y.; Fei, H.L.; Liu, Z.G.; Yin, S.F. Atomically dispersed Au catalysts for preferential oxidation of CO in H2-rich stream. Appl. Catal. B Environ. 2021, 296, 120385–120396. [Google Scholar] [CrossRef]
- Xin, Y.; Zhang, N.N.; Lv, Y.N.; Wang, J.; Li, Q.; Zhang, Z.L. Room-Temperature CO oxidative coupling for oxamide production over interfacial Au/ZnO catalysts. ACS Catal. 2023, 13, 735–743. [Google Scholar]
- Liu, Q.L.; Yang, P.; Tan, W.; Yu, H.W.; Ji, J.W.; Wu, C.; Cai, Y.D.; Xie, S.H.; Liu, F.D.; Hong, S.; et al. Fabricating robust Pt clusters on Sn-Doped CeO2 for CO oxidation: A deep insight into support engineering and surface structural evolution. Chem. Eur. J. 2023, 29, 2034–2045. [Google Scholar]
- Wu, C.L.; Guo, Z.Z.; Chen, X.Y.; Liu, H. Cu/CeO2 as efficient low-temperature CO oxidation catalysts: Effects of morphological structure and Cu content. React. Kinet. Mech. Cat. 2020, 131, 691–706. [Google Scholar] [CrossRef]
- Liu, X.S.; Jin, Z.N.; Lu, J.Q.; Wang, X.X.; Luo, M.F. Highly active CuO/OMS-2 catalysts for low-temperature CO oxidation. Chem. Eng. J. 2010, 162, 151–157. [Google Scholar] [CrossRef]
- Dae, J.M.; Shin, D.; Jeong, H.; Kim, B.S.; Han, J.W.; Lee, H. Highly Water-Resistant La-doped Co3O4 Catalyst for CO oxidation. ACS Catal. 2019, 9, 10093–10100. [Google Scholar]
- Bulavchenko, O.A.; Konovalova, V.P.; Saraev, A.A.; Kremneva, A.M.; Rogov, V.A.; Gerasimov, E.Y.; Afonasenko, T.N. The catalytic performance of CO oxidation over MnOx-ZrO2 Catalysts: The role of synthetic routes. Catalysts 2022, 13, 57–72. [Google Scholar] [CrossRef]
- Liu, Y.N.; Mao, D.S.; Yu, J.; Guo, X.M.; Ma, Z. Low-temperature CO oxidation on CuO-CeO2 catalyst prepared by facile one-step solvothermal synthesis: Improved activity and moisture resistance via optimizing the activation temperature. Fuel 2023, 332, 126196. [Google Scholar] [CrossRef]
- Zhao, W.J.; Li, X.; Dang, H.; Wu, R.F.; Wang, Y.Z.; Zhao, Y.X. Effect of Sn addition on the catalytic performance of a Pd–Cu/attapulgite catalyst for room-temperature CO oxidation under moisture-rich conditions. Reac. Kinet. Mech. Cat. 2021, 134, 759–775. [Google Scholar] [CrossRef]
- Peng, H.G.; Peng, Y.; Xu, X.L.; Fang, X.Z.; Liu, Y.; Cai, J.X.; Wang, X. SnO2 nano-sheet as an efficient catalyst for CO oxidation. Chin. J. Catal. 2015, 36, 2004–2010. [Google Scholar] [CrossRef]
- Peng, H.G.; Zhang, X.H.; Han, X.; You, X.J.; Lin, S.X.; Chen, H.; Liu, W.M.; Wang, X.; Zhang, N.; Wang, Z.; et al. Catalysts in coronas: A surface spatial confinement strategy for high-performance catalysts in methane dry reforming. ACS Catal. 2019, 9, 9072–9080. [Google Scholar] [CrossRef]
- Castillejos, E.; Debouttiere, P.J.; Roiban, L.; Solhy, A.; Martinez, V.; Kihn, Y.; Ersen, O.; Philippot, K.; Chaudret, B.; Serp, P. An efficient strategy to drive nanoparticles into carbon nanotubes and the remarkable effect of confinement on their catalytic performance. Angew. Chem. Int. Ed. Eng. 2009, 48, 2529–2533. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, G.; Ma, Q.; Wu, M.; Tan, Y.; Yoneyama, Y.; Tsubaki, N. Confinement effect of carbon nanotubes: Copper nanoparticles filled carbon nanotubes for hydrogenation of methyl acetate. ACS Catal. 2012, 2, 1958–1966. [Google Scholar] [CrossRef]
- Deng, D.H.; Chen, X.Q.; Yu, L. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 2015, 1, 1500462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudouin, D.; Szeto, K.C.; Laurent, P.; Mallmann, A.D.; Fenet, B.; Veyre, L.; Rodemerck, U.; Coperet, C.; Thieuleux, C. Nickel-silicide colloid prepared under mild conditions as a versatile Ni precursor for more efficient CO2 reforming of CH4 catalysts. J. Am. Chem. Soc. 2012, 134, 20624–20627. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Suryawinata, I.Y.; Kawi, S. Highly carbon resistant multicore-shell catalyst derived from Ni-Mg phyllosilicate nanotubes@silica for dry reforming of methane. Appl. Catal. B Environ. 2016, 195, 1–8. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zhang, L.; Peng, H.G.; You, X.; Peng, C.; Xu, X.; Liu, W.M.; Fang, X.; Wang, Z.; Zhang, N.; et al. Nickel nanoparticles embedded in mesopores of AlSBA-15 with a perfect peasecod-like structure: A catalyst with superior sintering resistance and hydrothermal stability for methane dry reforming. Appl. Catal. B Environ. 2018, 224, 488–499. [Google Scholar] [CrossRef]
- Liu, D.; Quek, X.Y.; Cheo, W.N.E.; Lau, R.; Borgna, A.; Yang, Y. MCM-41 supported nickel-based bimetallic catalysts with superior stability during carbon dioxide reforming of methane: Effect of strong metal–support interaction. J. Catal. 2009, 266, 380–390. [Google Scholar] [CrossRef]
- Dadras, J.; Jimenez-Izal, E.; Alexandrova, A.N. Alloying Pt Sub-nano-clusters with boron: Sintering preventative and coke antagonist. ACS Catal. 2015, 5, 5719–5727. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Zhong, Q.; Zhao, W.; Bu, Y. Focus on the modified CexZr1−xO2 with the rigid benzene-muti-carboxylate ligands and its catalysis in oxidation of NO. Appl. Catal. B Environ. 2014, 158, 258–268. [Google Scholar] [CrossRef]
- Feng, B.; Shi, M.; Liu, J.X.; Han, X.C.; Lan, Z.J.; Gu, H.J.; Wang, X.X.; Sun, H.M.; Zhang, Q.X.; Li, H.X.; et al. An efficient defect engineering strategy to enhance catalytic performances of Co3O4 nanorods for CO oxidation. J. Hazard. Mater. 2020, 394, 122540. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Liao, X.; Liu, Y.; Hou, J.; Pham-Huu, C. Enhancing oxygen activation on high surface area Pd-SnO2 solid solution with isolated metal site catalysts for catalytic CH4 combustion. Appl. Surf. Sci. 2021, 564, 150368. [Google Scholar] [CrossRef]
- Zhang, G.; Han, W.; Zhao, H.; Zong, L.; Tang, Z. Solvothermal synthesis of well-designed ceria-tin-titanium catalysts with enhanced catalytic performance for wide temperature NH3-SCR reaction. Appl. Catal. B Environ. 2018, 226, 117–126. [Google Scholar] [CrossRef]
- Bratan, V.; Munteanu, C.; Hornoiu, C.; Vasile, A.; Papa, F.; State, R.; Preda, S.; Culita, D.; Ionescu, N.I. CO oxidation over Pd supported catalysts—In situ study of the electric and catalytic properties. Appl. Catal. B Environ. 2017, 207, 166–173. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, Z.; Brosnahan, J.T.; Zhang, S.; Guo, Y.; Guo, Y.; Wang, L.; Wang, Y.; Zhan, W. Ru/CeO2 Catalyst with Optimized CeO2 Support Morphology and Surface Facets for Propane Combustion. Environ. Sci. Technol. 2019, 53, 5349–5358. [Google Scholar] [CrossRef]
- Lu, Z.; Ma, D.; Yang, L.; Wang, X.; Xu, G.; Yang, Z. Direct CO oxidation by lattice oxygen on the SnO2 (110) surface: A DFT study. Phys. Chem. Chem. Phys. 2014, 16, 12488–12494. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ji, Q.; Imai, T.; Ariga, K.; Abe, H. Sintering-Resistant Nanoparticles in Wide-Mouthed Compartments for Sustained Catalytic Performance. Sci. Rep. 2017, 7, 41773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polshettiwar, V.; Cha, D.; Zhang, X.; Basset, J.M. High-surface-area silica nanospheres (KCC-1) with a fibrous morphology. Angew. Chem. Int. Ed. Eng. 2010, 49, 9652–9656. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Ren, X.Y.; Wang, Y.J.; Cao, J.P.; He, Z.M.; Yao, N.Y.; Bai, H.C.; Cong, H.L. Conversion of lignite-derived volatiles into aromatics over Zn@MCM-41 and ZSM-5 tandem catalysts with a high stability. Fuel 2023, 339, 127430. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Kresse, G.; FurthmãLler, J.; Hafner, J. Chemisorption of H on Pd (111): An ab initio approach with ultrasoft pseudopotentials. Phys. Rev. B Condens. Matter. 1996, 54, 2157–2166. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; NoRskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phy. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Neugebauer, J.; Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al (111). Phys. Rev. B Condens. Matter. 1992, 46, 16067–16080. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Samples | Surface Area (m2/g) a | Pore Volume (cm3/g) b | Pore Size (nm) b |
|---|---|---|---|
| KCC-1 | 552 | 1.93 | 3.5 |
| MCM-41 | 1216 | 0.96 | 3.2 |
| Sn1@KCC-1 | 529 | 1.91 | 2.7 |
| Sn3@KCC-1 | 499 | 1.74 | 2.7 |
| Sn5@KCC-1 | 422 | 1.53 | 2.7 |
| Sn7@KCC-1 | 315 | 0.96 | 2.7 |
| Sn10@KCC-1 | 269 | 0.95 | 2.7 |
| Sn7@MCM-41 | 807 | 0.61 | 2.9 |
| SnO2 | 19 | 0.16 | 10.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Zhang, Y.; Yan, J.; Luo, Y.; Wang, C.; Feng, W.; Zhang, S.; Liu, W.; Zhang, Z.; Peng, H. Insights into SnO2 Nanoparticles Supported on Fibrous Mesoporous Silica for CO Catalytic Oxidation. Catalysts 2023, 13, 1156. https://doi.org/10.3390/catal13081156
Li G, Zhang Y, Yan J, Luo Y, Wang C, Feng W, Zhang S, Liu W, Zhang Z, Peng H. Insights into SnO2 Nanoparticles Supported on Fibrous Mesoporous Silica for CO Catalytic Oxidation. Catalysts. 2023; 13(8):1156. https://doi.org/10.3390/catal13081156
Chicago/Turabian StyleLi, Guobo, Yingying Zhang, Jie Yan, Yiwei Luo, Conghui Wang, Weiwei Feng, Shule Zhang, Wenming Liu, Zehui Zhang, and Honggen Peng. 2023. "Insights into SnO2 Nanoparticles Supported on Fibrous Mesoporous Silica for CO Catalytic Oxidation" Catalysts 13, no. 8: 1156. https://doi.org/10.3390/catal13081156