
Reaction Pathways of Gamma-Valerolactone Hydroconversion over Co/SiO2 Catalyst
 , , , , and
, , , , and
Abstract
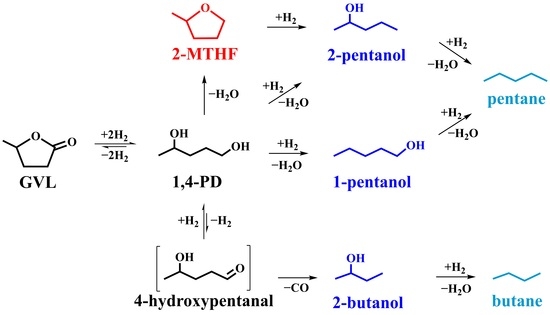
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
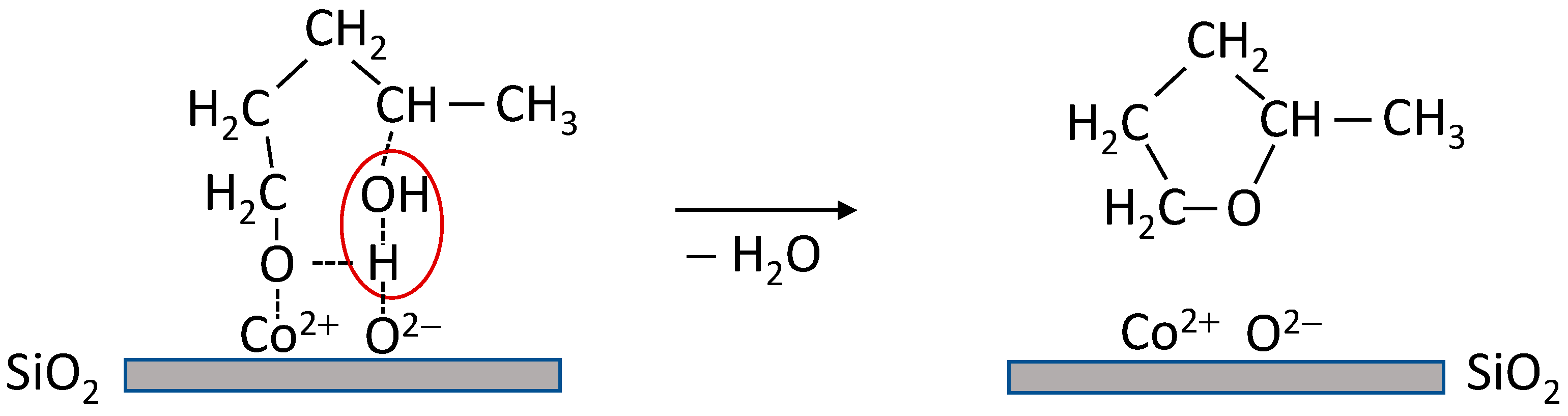{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
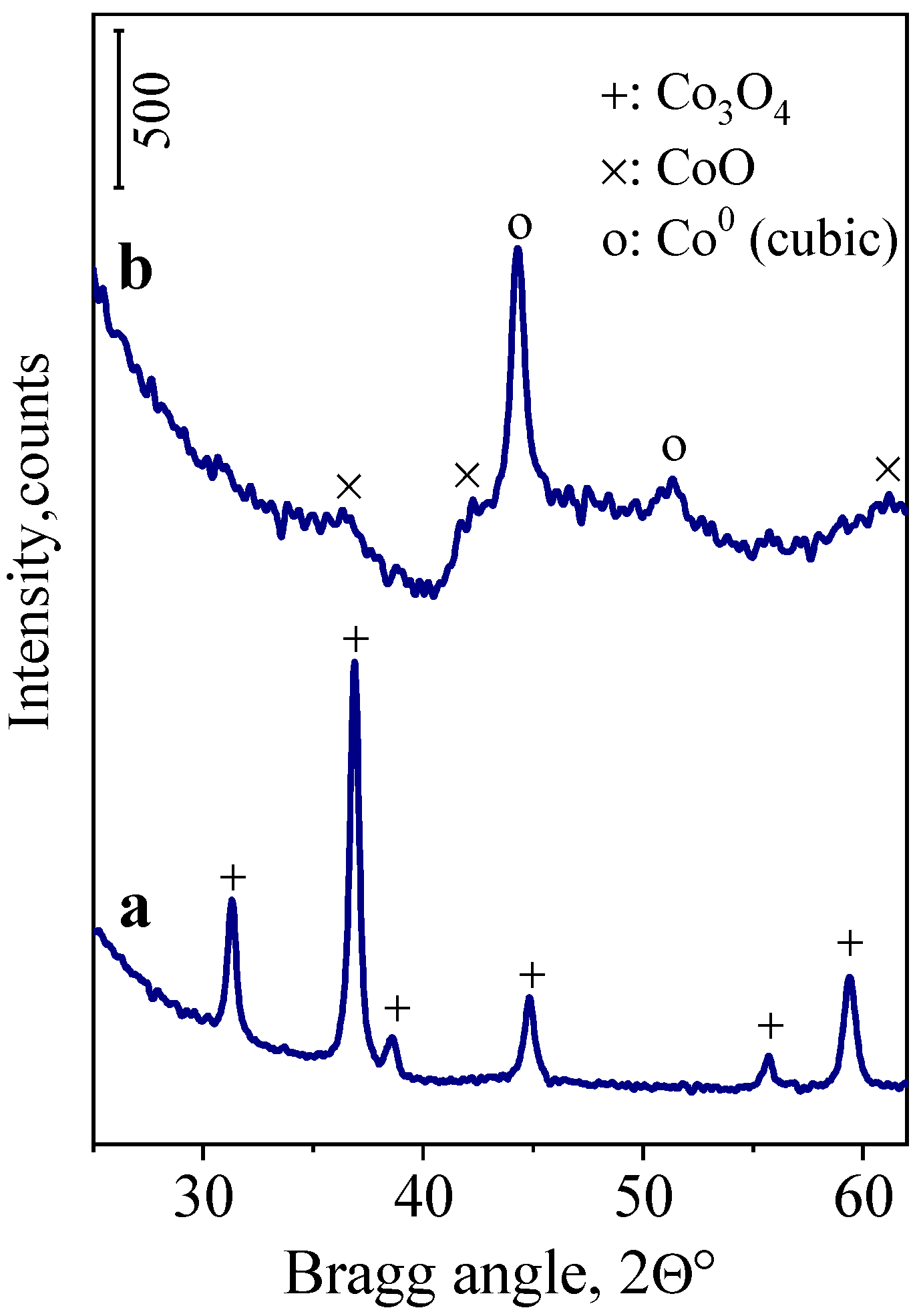
2.1. XRPD of Co/SiO2 Catalyst
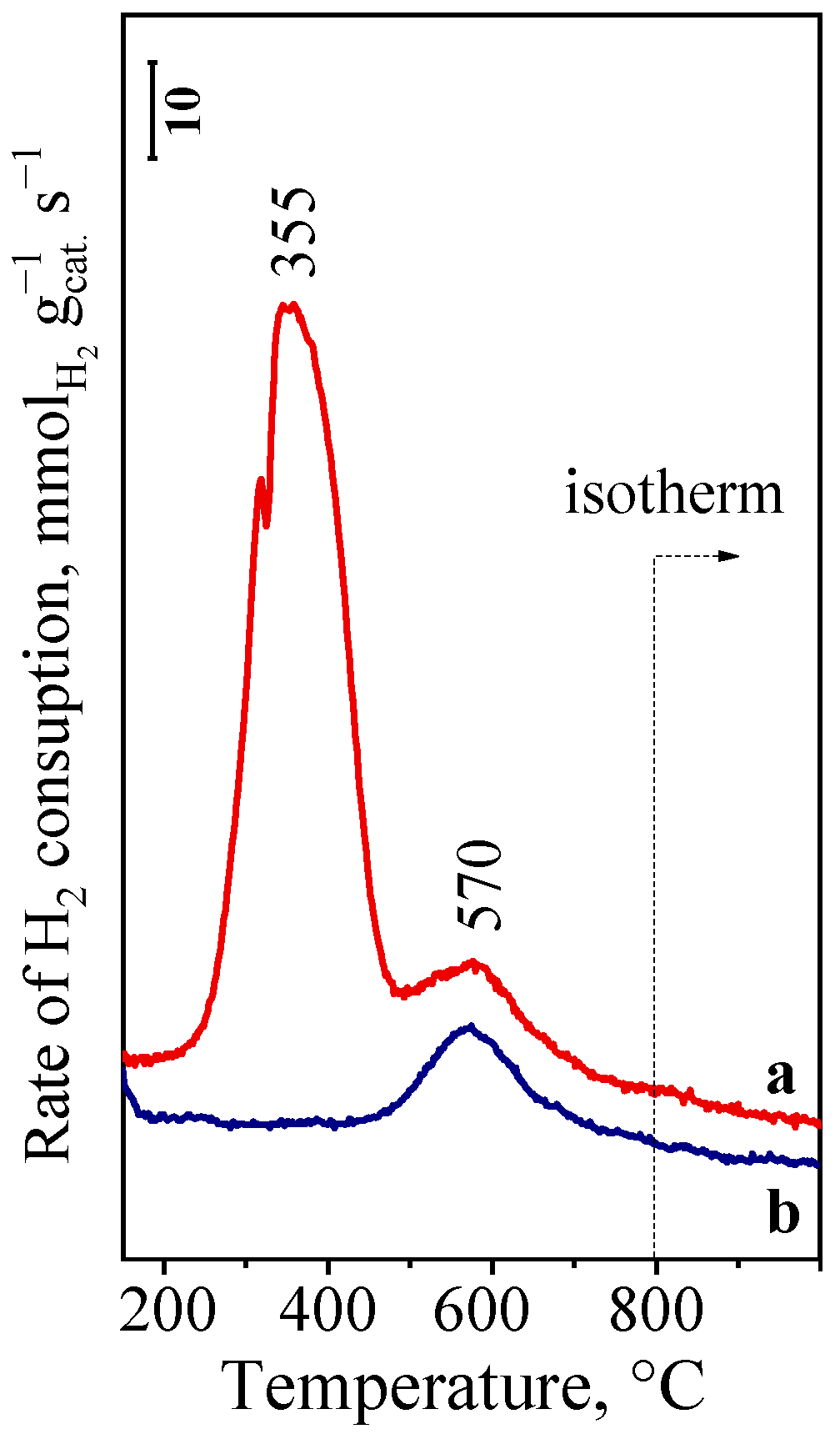
2.2. H2-TPR
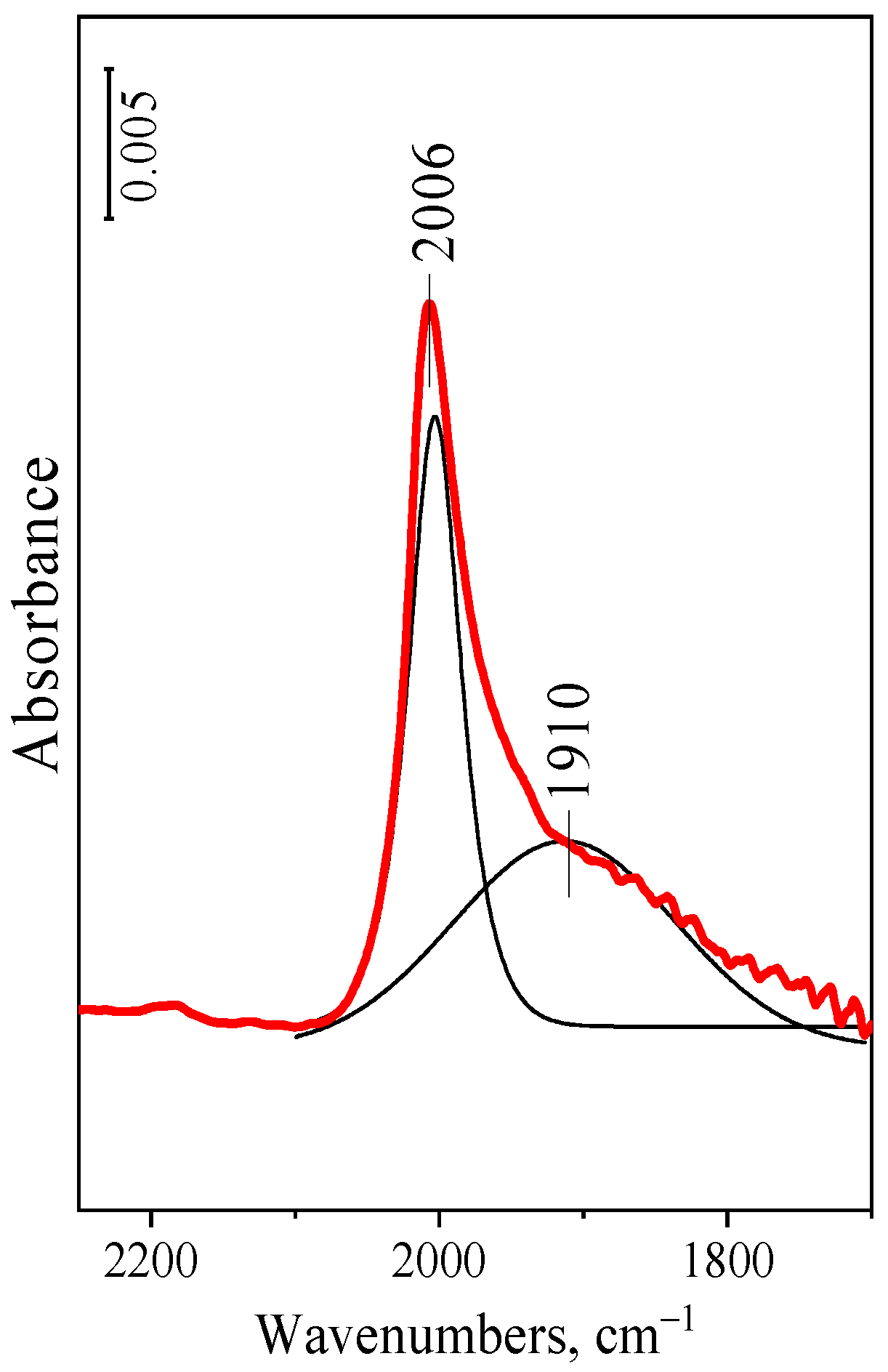
2.3. CO Chemisorption
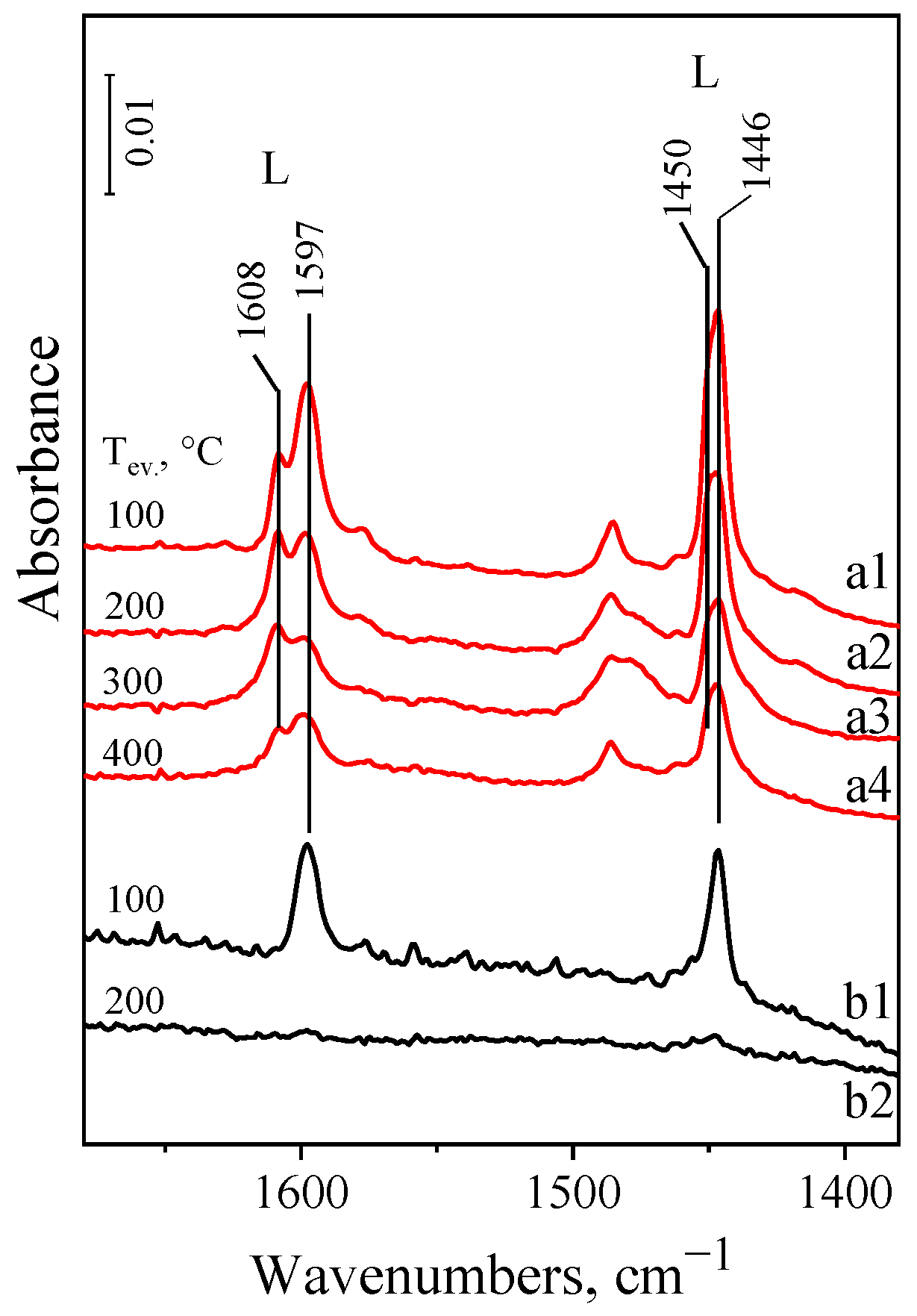
2.4. FT-IR Spectra of Adsorbed Pyridine
2.5. Catalytic Results
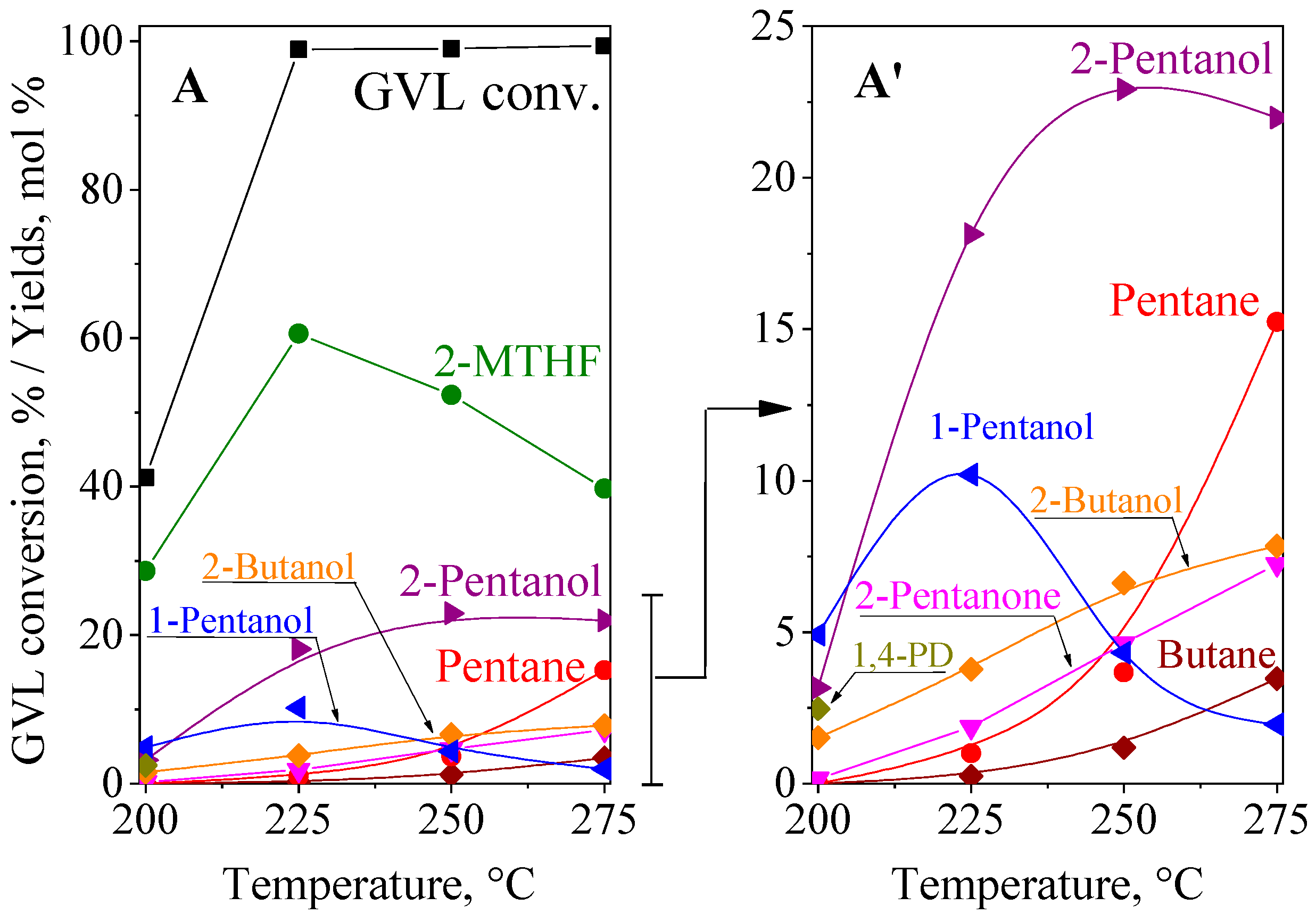
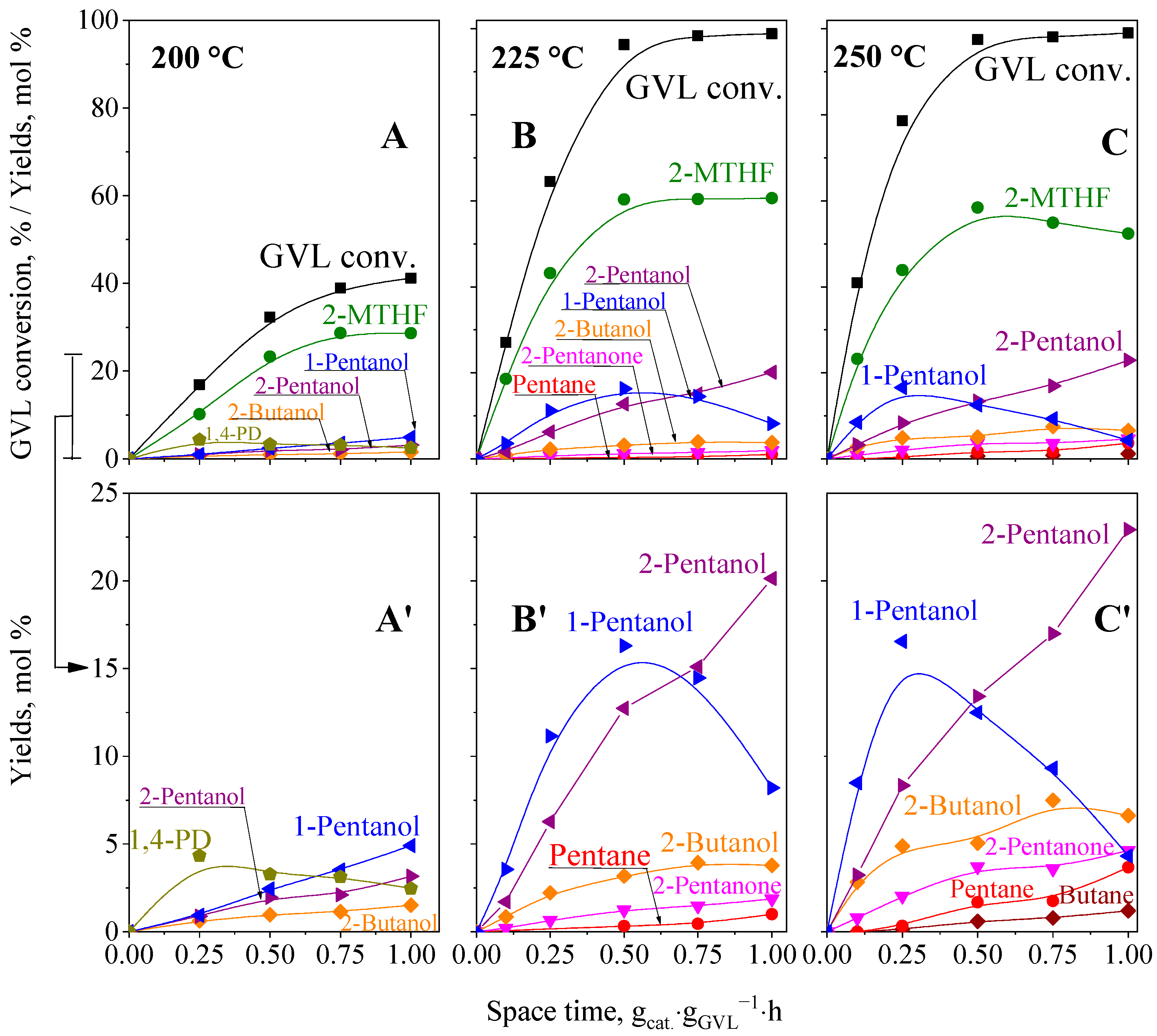
2.5.1. GVL Hydroconversion
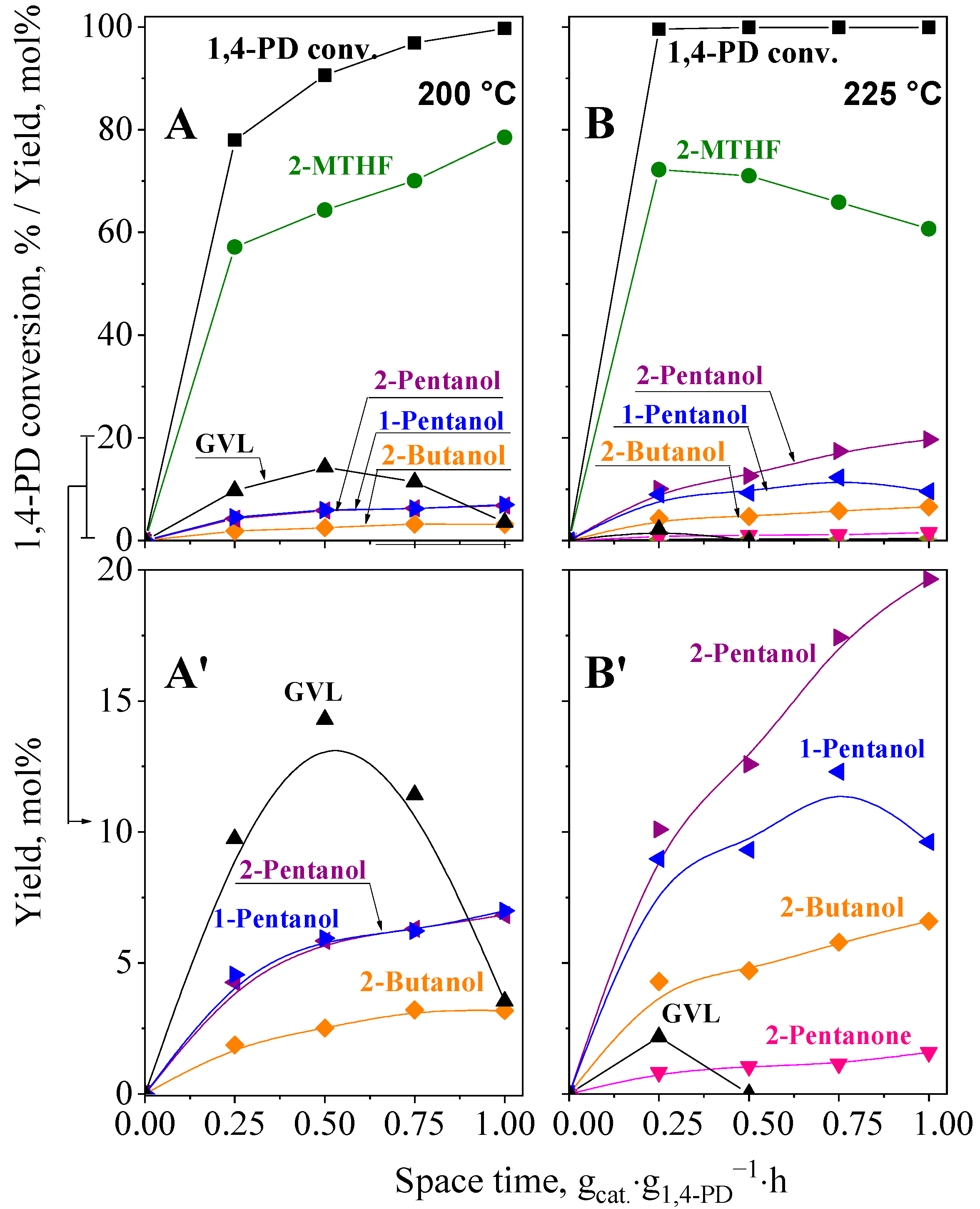
2.5.2. Hydroconversion of 1,4-PD
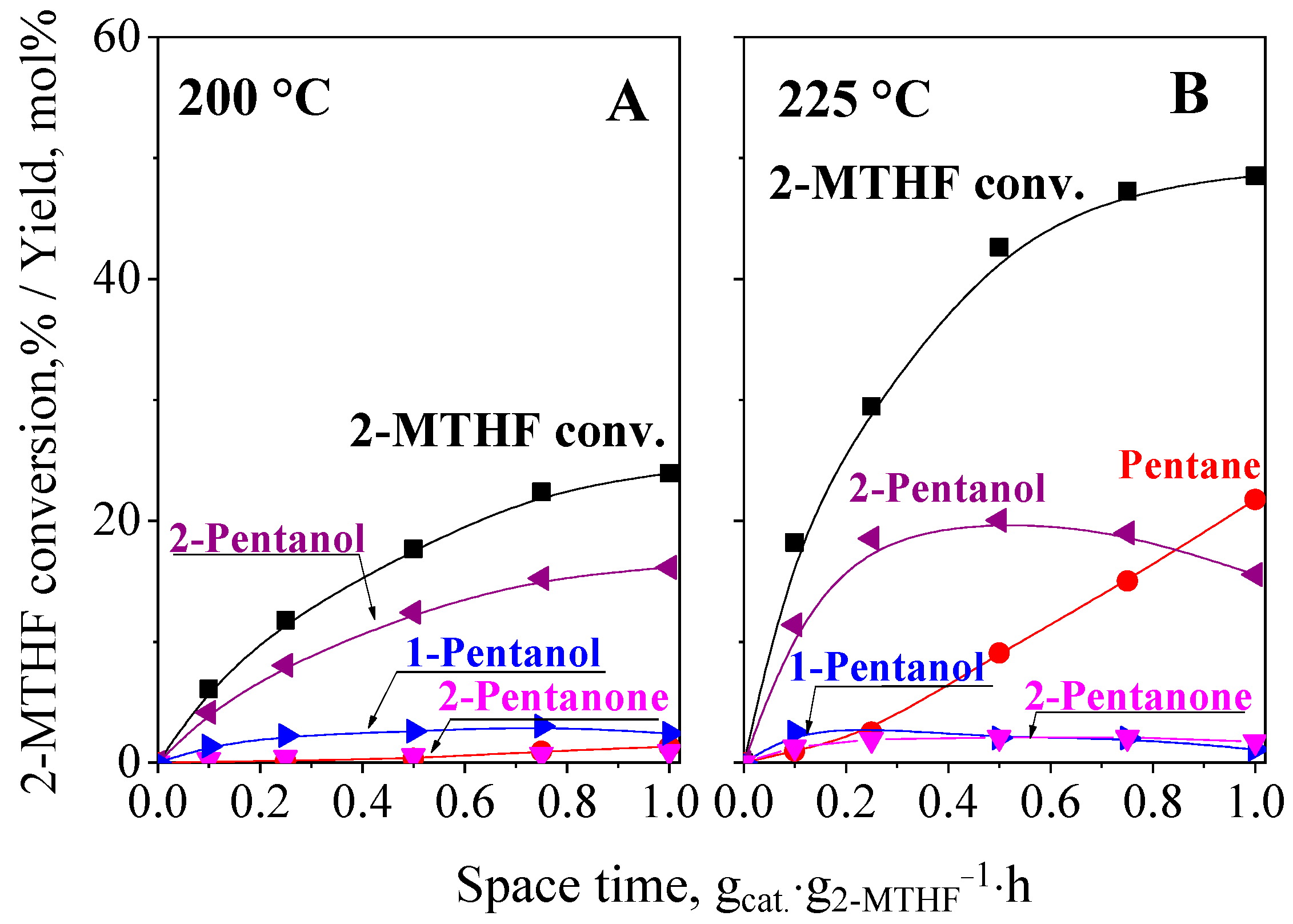
2.5.3. Hydroconversion of 2-MTHF
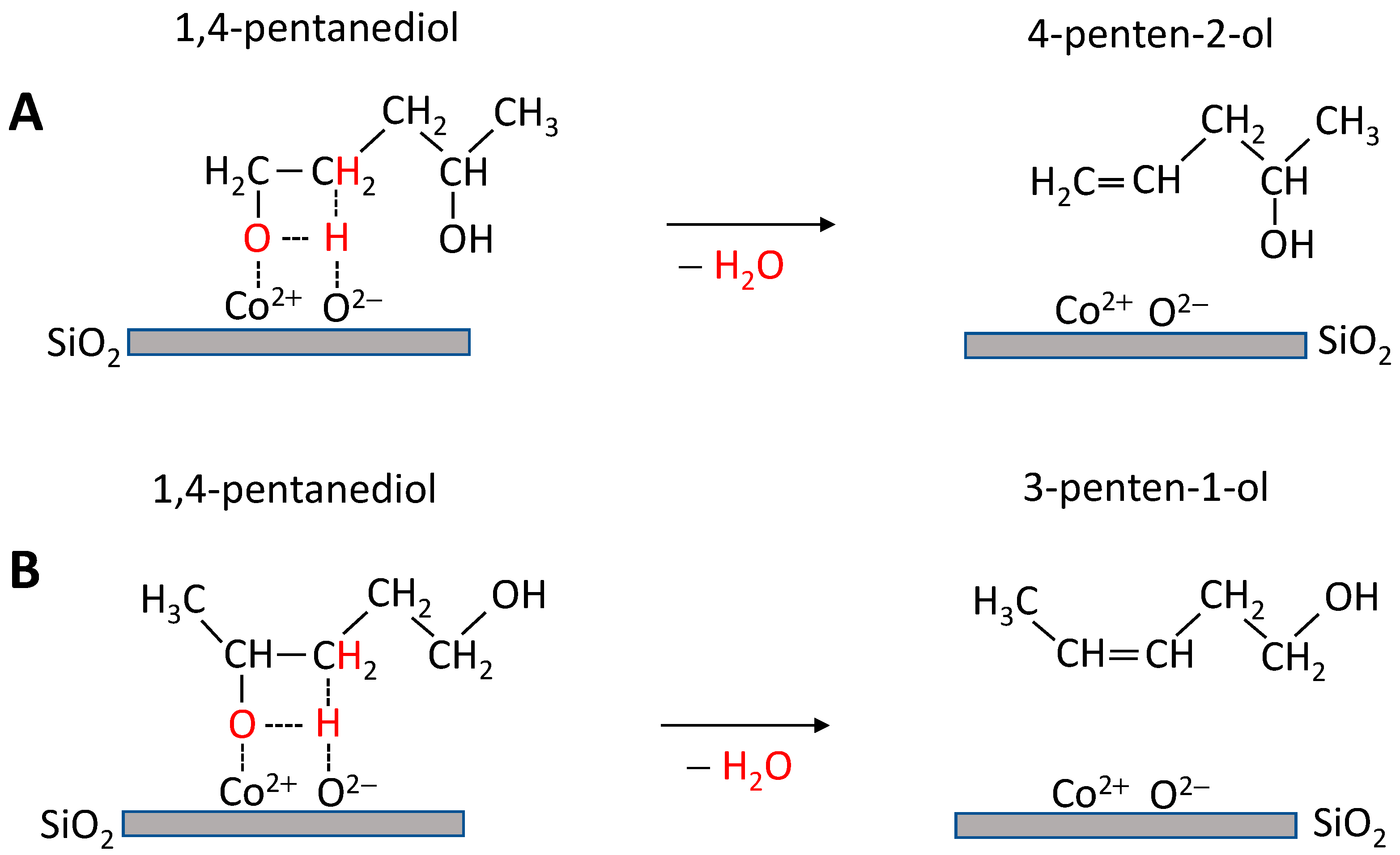
3. Discussion
4. Experimental
4.1. Catalyst Preparation
4.2. Catalyst Characterization
4.3. Catalytic Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1,4-PD | 1,4-pentanediol |
| 2-MTHF | 2-methyltetrahydrofuran |
| 2-OH-5-MTHF | 2-hydroxy-5-methyltetrahydrofuran |
| FID | Flame ionization detector |
| FT-IR | Fourier-transform infrared spectroscopy |
| FWHM | Full width at half maximum |
| GVL | γ-valerolactone |
| H2-TPR | Temperature-programmed reduction by hydrogen |
| ICP-OES | Inductively coupled plasma optical atomic emission spectroscopy |
| Py | pyridine |
| TCD | Thermal conductivity detector |
| TEM | Transmission electron microscopy |
| XRPD | X-ray powder diffraction |
References
- Hayes, D.J. An examination of biorefining processes, catalysts and challenges. Catal. Today 2009, 145, 138–151. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539. [Google Scholar] [CrossRef]
- Geilen, F.M.A.; Engendahl, B.; Harwardt, A.; Marquardt, W.; Klankermayer, J.; Leitner, W. Selective and Flexible Transformation of Biomass-Derived Platform Chemicals by a Multifunctional Catalytic System. Angew. Chem. Int. Ed. 2010, 49, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Ruiz, J.C.; Dumesic, J.A. Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels. Energy Environ. Sci. 2011, 4, 83–99. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Jarvis, C.; Gu, J.; Yan, Y. Production and catalytic transformation of levulinic acid: A platform for speciality chemicals and fuels. Renew. Sustain. Energy Rev. 2015, 51, 986–997. [Google Scholar] [CrossRef]
- Gérardy, R.; Debecker, D.P.; Estager, J.; Luis, P.; Monbaliu, J.-C.M. Continuous Flow Upgrading of Selected C2–C6 Platform Chemicals Derived from Biomass. Chem. Rev. 2020, 120, 7219–7347. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584. [Google Scholar] [CrossRef]
- Liguori, F.; Moreno-Marrodan, C.; Barbaro, P. Environmentally Friendly Synthesis of γ-Valerolactone by Direct Catalytic Conversion of Renewable Sources. ACS Catal. 2015, 5, 1882–1894. [Google Scholar] [CrossRef]
- Obregón, I.; Gandarias, I.; Al-Shaal, M.G.; Mevissen, C.; Arias, P.L.; Palkovits, R. The Role of the Hydrogen Source on the Selective Production of γ-Valerolactone and 2-Methyltetrahydrofuran from Levulinic Acid. ChemSusChem 2016, 9, 2488–2495. [Google Scholar] [CrossRef]
- Novodárszki, G.; Solt, H.E.; Valyon, J.; Lónyi, F.; Hancsók, J.; Deka, D.; Tuba, R.; Mihályi, M.R. Selective hydroconversion of levulinic acid to γ-valerolactone or 2-methyltetrahydrofuran over silica-supported cobalt catalysts. Catal. Sci. Technol. 2019, 9, 2291–2304. [Google Scholar] [CrossRef]
- Lange, J.-P.; Price, R.; Ayoub, P.M.; Louis, J.; Petrus, L.; Clarke, L.; Gosselink, H. Valeric Biofuels: A Platform of Cellulosic Transportation Fuels. Angew. Chem. Int. Ed. 2010, 49, 4479–4483. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J. Connecting Biomass and Petroleum Processing with a Chemical Bridge. Science 2010, 329, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Velisoju, V.K.; Jampaiah, D.; Gutta, N.; Bentrup, U.; Brückner, A.; Bhargava, S.K.; Akula, V. Conversion of γ-Valerolactone to Ethyl Valerate over Metal Promoted Ni/ZSM-5 Catalysts: Influence of Ni0/Ni2+ Heterojunctions on Activity and Product Selectivity. ChemCatChem 2020, 12, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Novodárszki, G.; Solt, H.E.; Lendvay, G.; Mihályi, R.M.; Vikár, A.; Lónyi, F.; Hancsók, J.; Valyon, J. Hydroconversion mechanism of biomass-derived γ-valerolactone. Catal. Today 2019, 336, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Cho, A.; Kim, H.; Iino, A.; Takagaki, A.; Ted Oyama, S. Kinetic and FTIR studies of 2-methyltetrahydrofuran hydrodeoxygenation on Ni2P/SiO2. J. Catal. 2014, 318, 151–161. [Google Scholar] [CrossRef]
- Iino, A.; Cho, A.; Takagaki, A.; Kikuchi, R.; Ted Oyama, S. Kinetic studies of hydrodeoxygenation of 2-methyltetrahydrofuran on a Ni2P/SiO2 catalyst at medium pressure. J. Catal. 2014, 311, 17–27. [Google Scholar] [CrossRef]
- Licursi, D.; Antonetti, C.; Fulignati, S.; Giannoni, M.; Raspolli Galletti, A.M. Cascade Strategy for the Tunable Catalytic Valorization of Levulinic Acid and γ-Valerolactone to 2-Methyltetrahydrofuran and Alcohols. Catalysts 2018, 8, 277. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Rong, Z.; Sun, L.; Liu, C.; Lu, A.-H.; Wang, Y.; Qu, J. Catalytic conversion of biomass-derived levulinic acid into alcohols over nanoporous Ru catalyst. Catal. Sci. Technol. 2018, 8, 975–979. [Google Scholar] [CrossRef]
- Schanke, D.; Vada, S.; Blekkan, E.A.; Hilmen, A.M.; Hoff, A.; Holmen, A. Study of Pt-Promoted Cobalt CO Hydrogenation Catalysts. J. Catal. 1995, 156, 85–95. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Bechara, R.; Griboval-Constant, A. Fischer–Tropsch synthesis over silica supported cobalt catalysts: Mesoporous structure versus cobalt surface density. Appl. Catal. A Gen. 2003, 254, 273–288. [Google Scholar] [CrossRef]
- Arnoldy, P. Temperature-programmed reduction of CoO/Al2O3 catalysts. J. Catal. 1985, 93, 38–54. [Google Scholar] [CrossRef]
- Lapidus, A.; Krylova, A.; Kazanskii, V.; Borovkov, V.; Zaitsev, A.; Rathousky, J.; Zukal, A.; Jančálková, M. Hydrocarbon synthesis from carbon monoxide and hydrogen on impregnated cobalt catalysts Part I. Physico-chemical properties of 10% cobalt/alumina and 10% cobalt/silica. Appl. Catal. 1991, 73, 65–81. [Google Scholar] [CrossRef]
- Sato, K.; Inoue, Y.; Kojima, I.; Miyazaki, E.; Yasumori, I. Infrared and X-ray photoelectron spectroscopy studies of carbon monoxide adsorbed on silica-supported cobalt catalysts. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1984, 80, 841. [Google Scholar] [CrossRef]
- Ishi, S.; Ohno, Y.; Viswanathan, B. An overview on the electronic and vibrational properties of adsorbed CO. Surf. Sci. 1985, 161, 349–372. [Google Scholar] [CrossRef]
- Sun, S.; Tsubaki, N.; Fujimoto, K. The reaction performances and characterization of Fischer–Tropsch synthesis Co/SiO2 catalysts prepared from mixed cobalt salts. Appl. Catal. A Gen. 2000, 202, 121–131. [Google Scholar] [CrossRef]
- Parry, E. An infrared study of pyridine adsorbed on acidic solids. Characterization of surface acidity. J. Catal. 1963, 2, 371–379. [Google Scholar] [CrossRef]
- Mekhemer, G.A.; Abd-Allah, H.M.; Mansour, S.A. Surface characterization of silica-supported cobalt oxide catalysts. Colloids Surfaces A Physicochem. Eng. Asp. 1999, 160, 251–259. [Google Scholar] [CrossRef]
- Kon, K.; Onodera, W.; Shimizu, K. Selective hydrogenation of levulinic acid to valeric acid and valeric biofuels by a Pt/HMFI catalyst. Catal. Sci. Technol. 2014, 4, 3227–3234. [Google Scholar] [CrossRef]
- Zaccheria, F.; Scotti, N.; Marelli, M.; Psaro, R.; Ravasio, N. Unravelling the properties of supported copper oxide: Can the particle size induce acidic behaviour? Dalt. Trans. 2013, 42, 1319–1328. [Google Scholar] [CrossRef]
- Zholobenko, V.; Freitas, C.; Jendrlin, M.; Bazin, P.; Travert, A.; Thibault-Starzyk, F. Probing the acid sites of zeolites with pyridine: Quantitative AGIR measurements of the molar absorption coefficients. J. Catal. 2020, 385, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Al-Shaal, M.G.; Dzierbinski, A.; Palkovits, R. Solvent-free γ-valerolactone hydrogenation to 2-methyltetrahydrofuran catalysed by Ru/C: A reaction network analysis. Green Chem. 2014, 16, 1358–1364. [Google Scholar] [CrossRef]
- Mihailović, M.L.; Gojković, S.; Čeković, Ž. Stereochemistry of cyclic ether formation. Part I. Stereoselective intramolecular cyclisation of aliphatic disecondary 1,4-diols and their sulphonate esters to tetrahydrofurans. J. Chem. Soc. Perkin Trans. 1972, 1, 2460–2464. [Google Scholar] [CrossRef]
- Kotkar, D.; Mahajan, S.W.; Mandal, A.K.; Ghosh, P.K. Cyclodehydration of non-aromatic diols on AlIII-montmorillonite clay: Reactivity and mechanism. J. Chem. Soc. Perkin Trans. 1988, 1, 1749. [Google Scholar] [CrossRef]
- Molnár, Á.; Felföldi, K.; Bartók, M. Studies on the conversions of diols and cyclic ethers—49. Tetrahedron 1981, 37, 2149–2151. [Google Scholar] [CrossRef]
- Novodárszki, G.; Valyon, J.; Illés, Á.; Dóbé, S.; Mihályi, M.R. Synthesis and characterization of Al-magadiite and its catalytic behavior in 1,4-pentanediol dehydration. React. Kinet. Mech. Catal. 2017, 121, 275–292. [Google Scholar] [CrossRef]
- Szegedi, Á.; Popova, M.; Mavrodinova, V.; Minchev, C. Cobalt-containing mesoporous silicas—Preparation, characterization and catalytic activity in toluene hydrogenation. Appl. Catal. A Gen. 2008, 338, 44–51. [Google Scholar] [CrossRef]
- Szegedi, Á.; Popova, M.; Minchev, C. Catalytic activity of Co/MCM-41 and Co/SBA-15 materials in toluene oxidation. J. Mater. Sci. 2009, 44, 6710–6716. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Hydrogen adsorption on supported cobalt, iron, and nickel. Catal. Lett. 1991, 7, 27–51. [Google Scholar] [CrossRef]
- Fuller, E.N.; Schettler, P.D.; Gidding, J.C. New method for prediction of binary gas-phase diffusion coefficients. Ind. Eng. Chem. 1966, 58, 18–27. [Google Scholar] [CrossRef]
- Fogler, H.S. Elements of Chemical Reaction Engineering, 3rd ed.; Prentice-Hall: Upper Saddle River, NJ, USA, 1999. [Google Scholar]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novodárszki, G.; Lónyi, F.; Mihályi, M.R.; Vikár, A.; Barthos, R.; Szabó, B.; Valyon, J.; Solt, H.E. Reaction Pathways of Gamma-Valerolactone Hydroconversion over Co/SiO2 Catalyst. Catalysts 2023, 13, 1144. https://doi.org/10.3390/catal13071144
Novodárszki G, Lónyi F, Mihályi MR, Vikár A, Barthos R, Szabó B, Valyon J, Solt HE. Reaction Pathways of Gamma-Valerolactone Hydroconversion over Co/SiO2 Catalyst. Catalysts. 2023; 13(7):1144. https://doi.org/10.3390/catal13071144
Chicago/Turabian StyleNovodárszki, Gyula, Ferenc Lónyi, Magdolna R. Mihályi, Anna Vikár, Róbert Barthos, Blanka Szabó, József Valyon, and Hanna E. Solt. 2023. "Reaction Pathways of Gamma-Valerolactone Hydroconversion over Co/SiO2 Catalyst" Catalysts 13, no. 7: 1144. https://doi.org/10.3390/catal13071144