
Structurally Rigid (8-(Arylimino)-5,6,7-trihydroquinolin-2-yl)-methyl Acetate Cobalt Complex Catalysts for Isoprene Polymerization with High Activity and cis-1,4 Selectivity

 , ,
, , 
Abstract
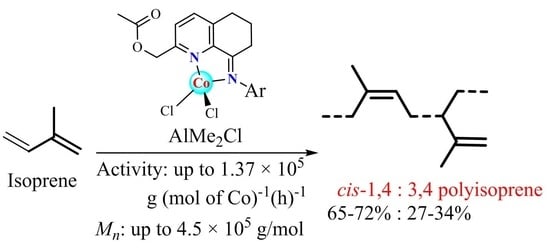
:
1. Introduction
2. Results and Discussion
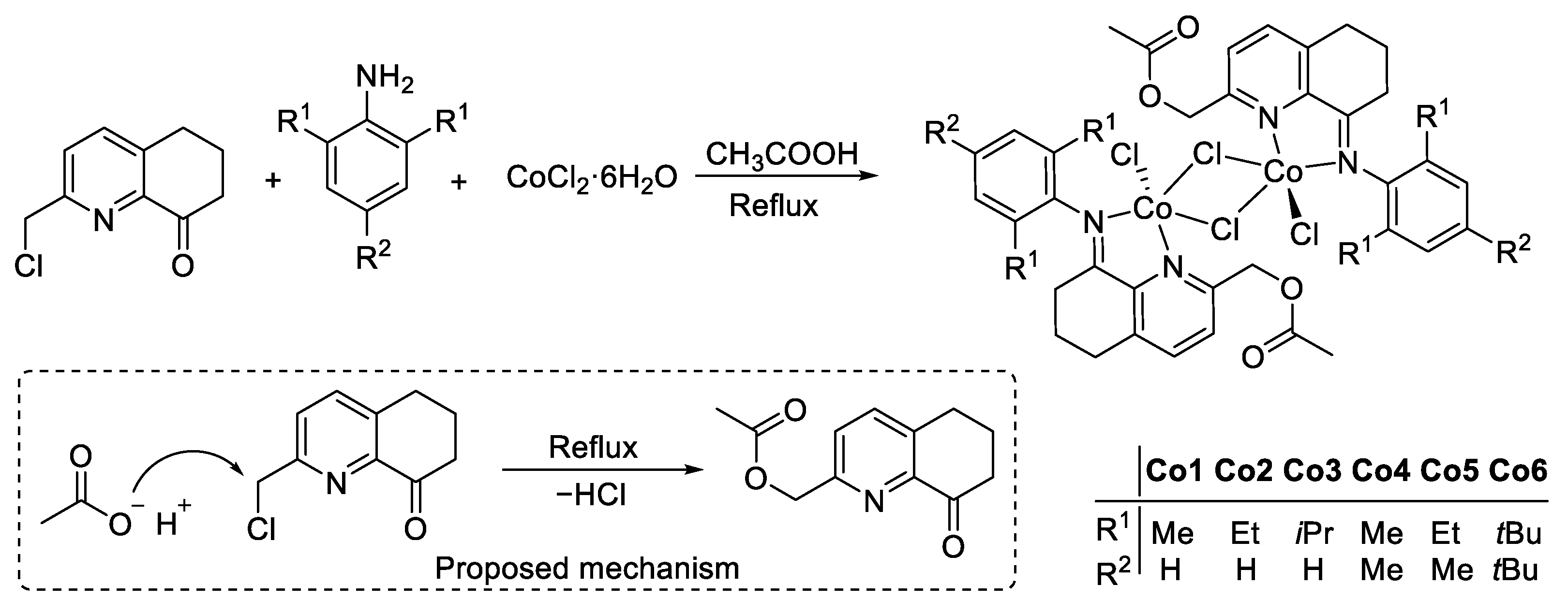
2.1. Synthesis and Characterization of Cobalt Complexes (Co1–Co6)
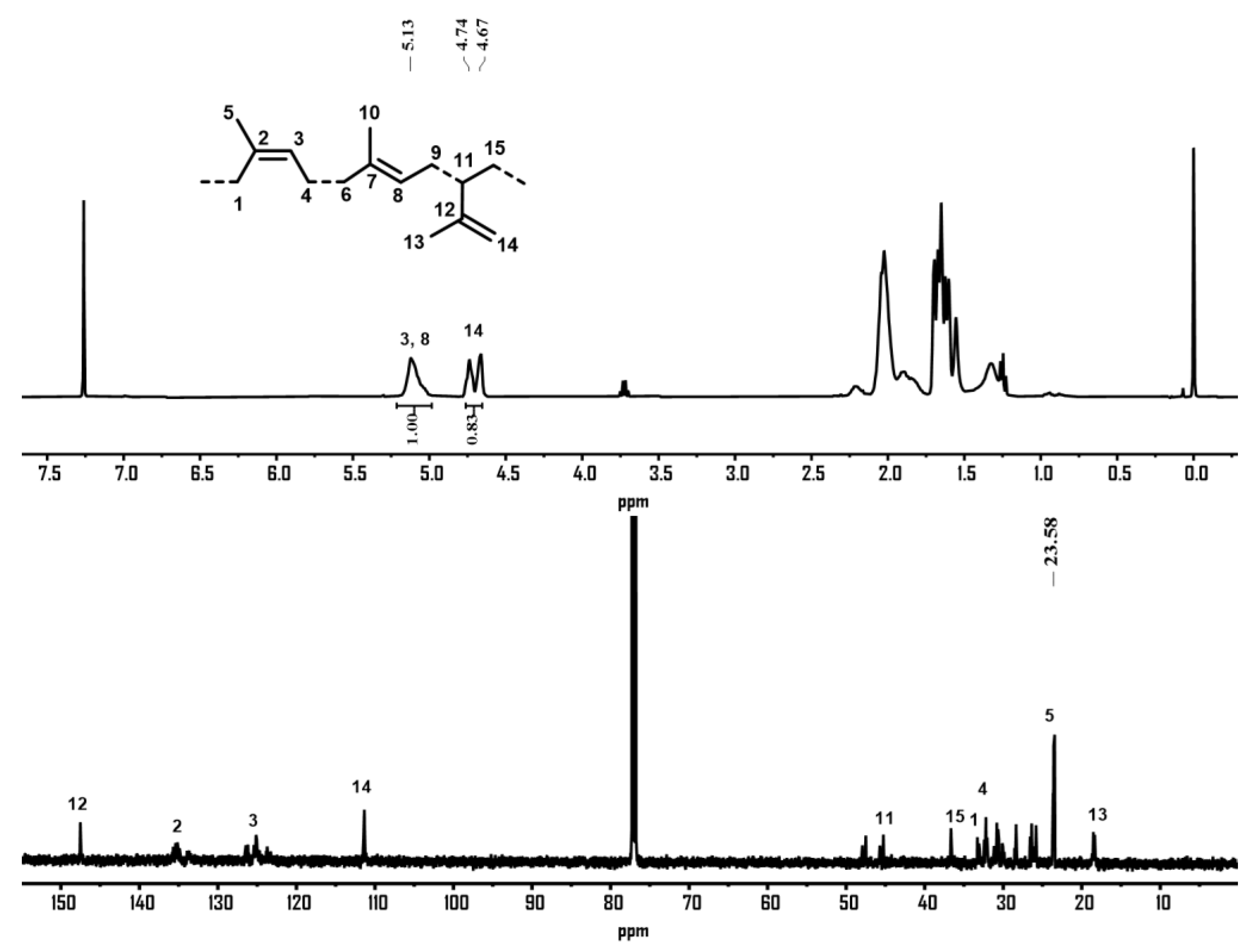
2.2. Isoprene Polymerization
2.2.1. Screening of Reaction Conditions Using Co1 as Precatalyst
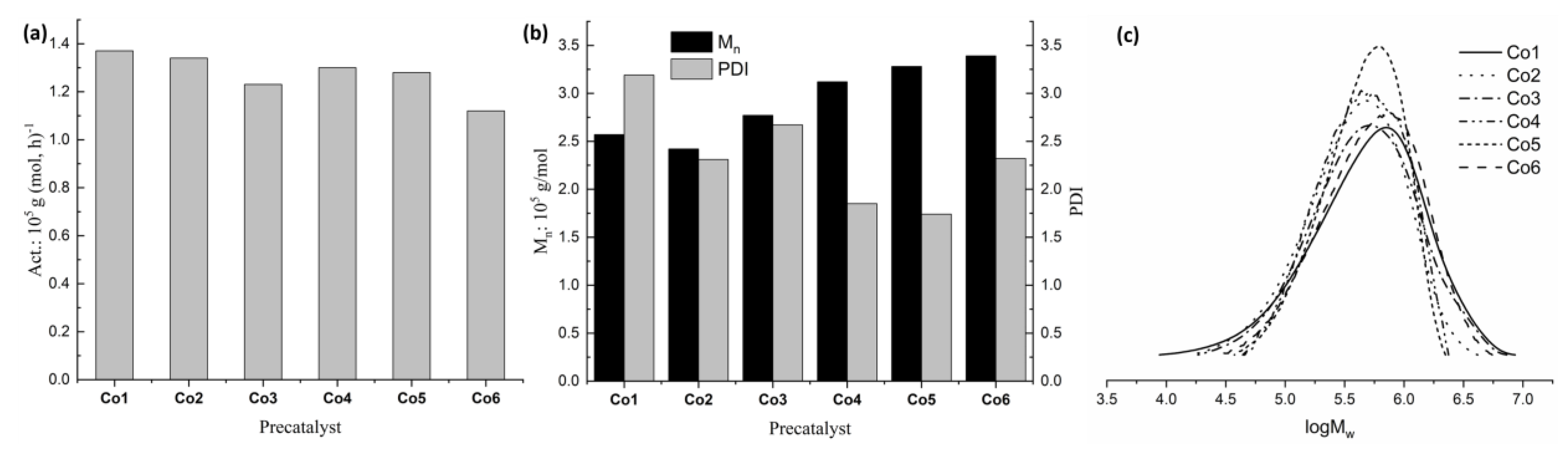
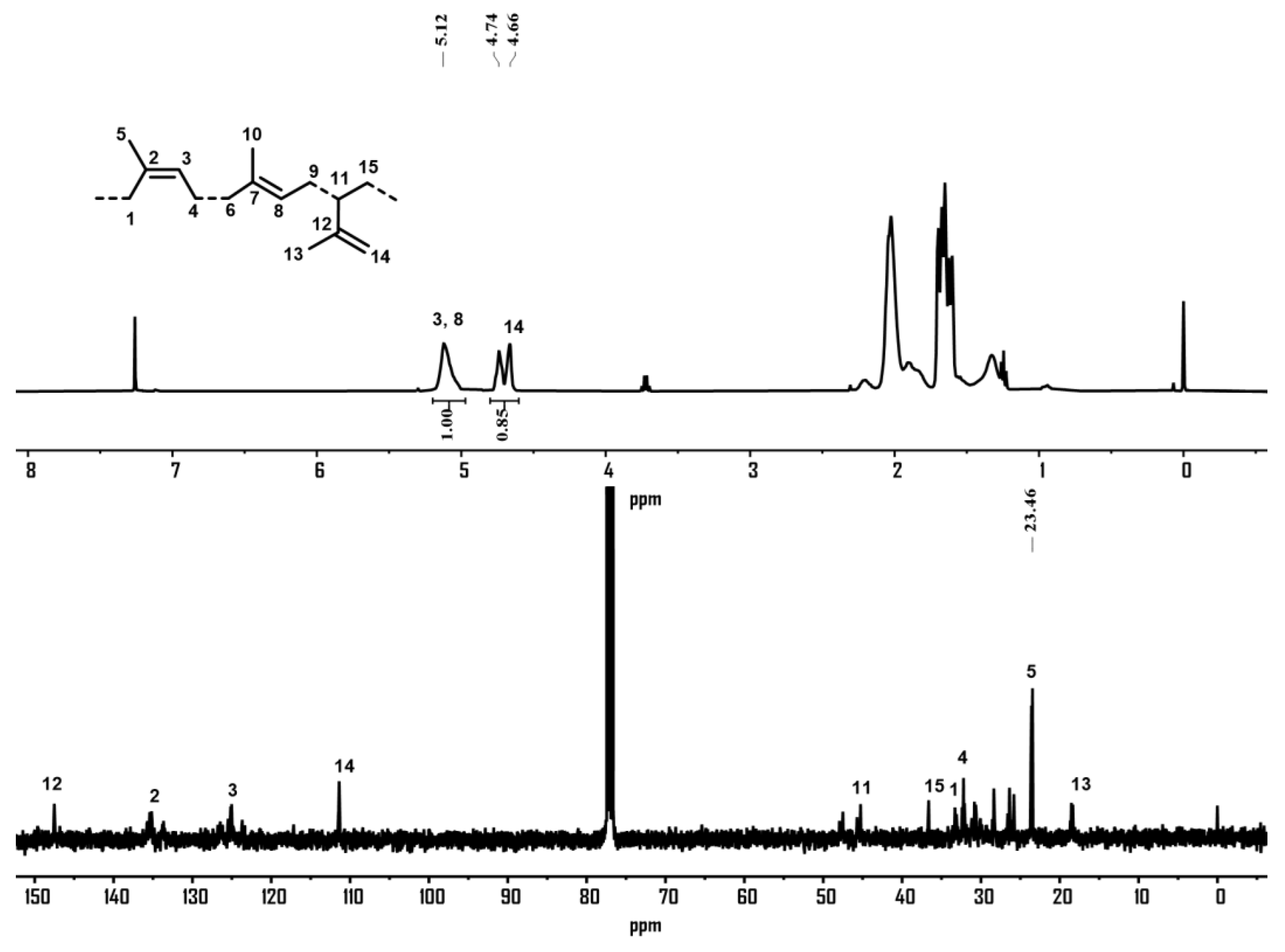
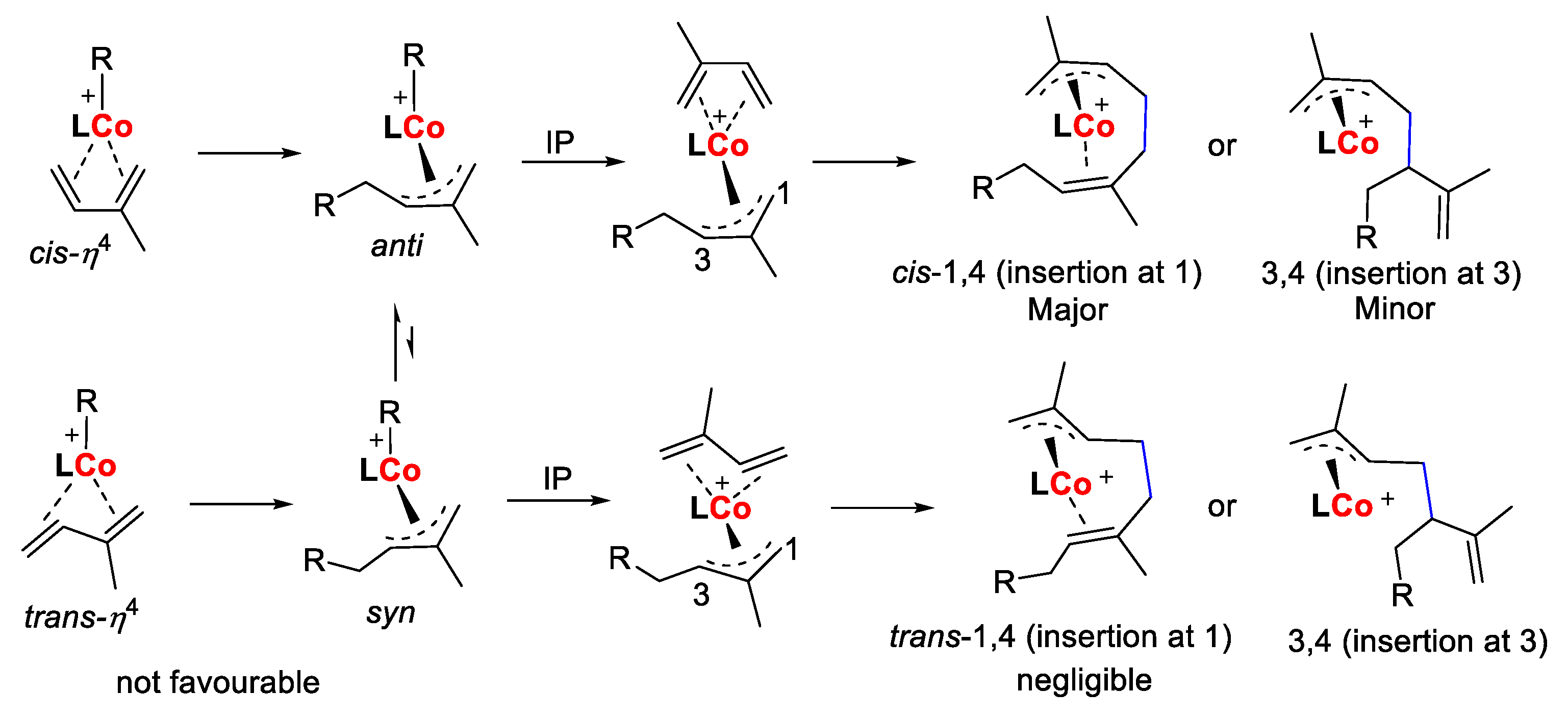
2.2.2. Isoprene Polymerization Using Complexes Co1–Co6
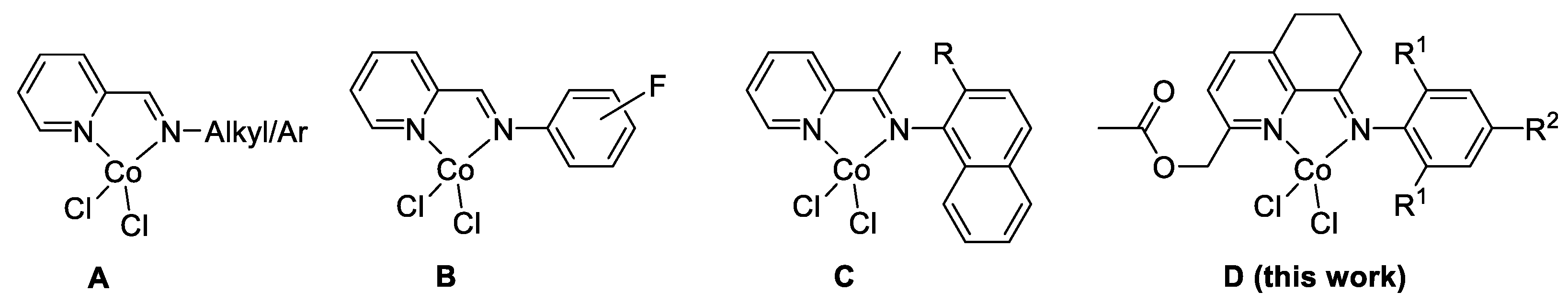
2.3. Comparison with Other Reported Iminopyridine Based Cobalt Complexes
3. Experimental Section
3.1. General Consideration and Materials
3.2. Synthesis and Characterization of (8-(Arylimino)-5,6,7-trihydroquinolin-2-yl)methyl Acetate-Cobalt Dichloride Complexes (Co1–Co6)
3.2.1. Aryl = 2,6-Me2C6H3 (Co1)
3.2.2. Aryl = 2,6-Et2C6H3 (Co2)
3.2.3. Aryl = 2,6-iPr2C6H3 (Co3)
3.2.4. Aryl = 2,4,6-Me3C6H2 (Co4)
3.2.5. Aryl = 2,6-Et2-4-MeC6H2 (Co5)
3.2.6. Aryl = 2,4,6-tBu3C6H2 (Co6)
3.3. Polymerization Procedure
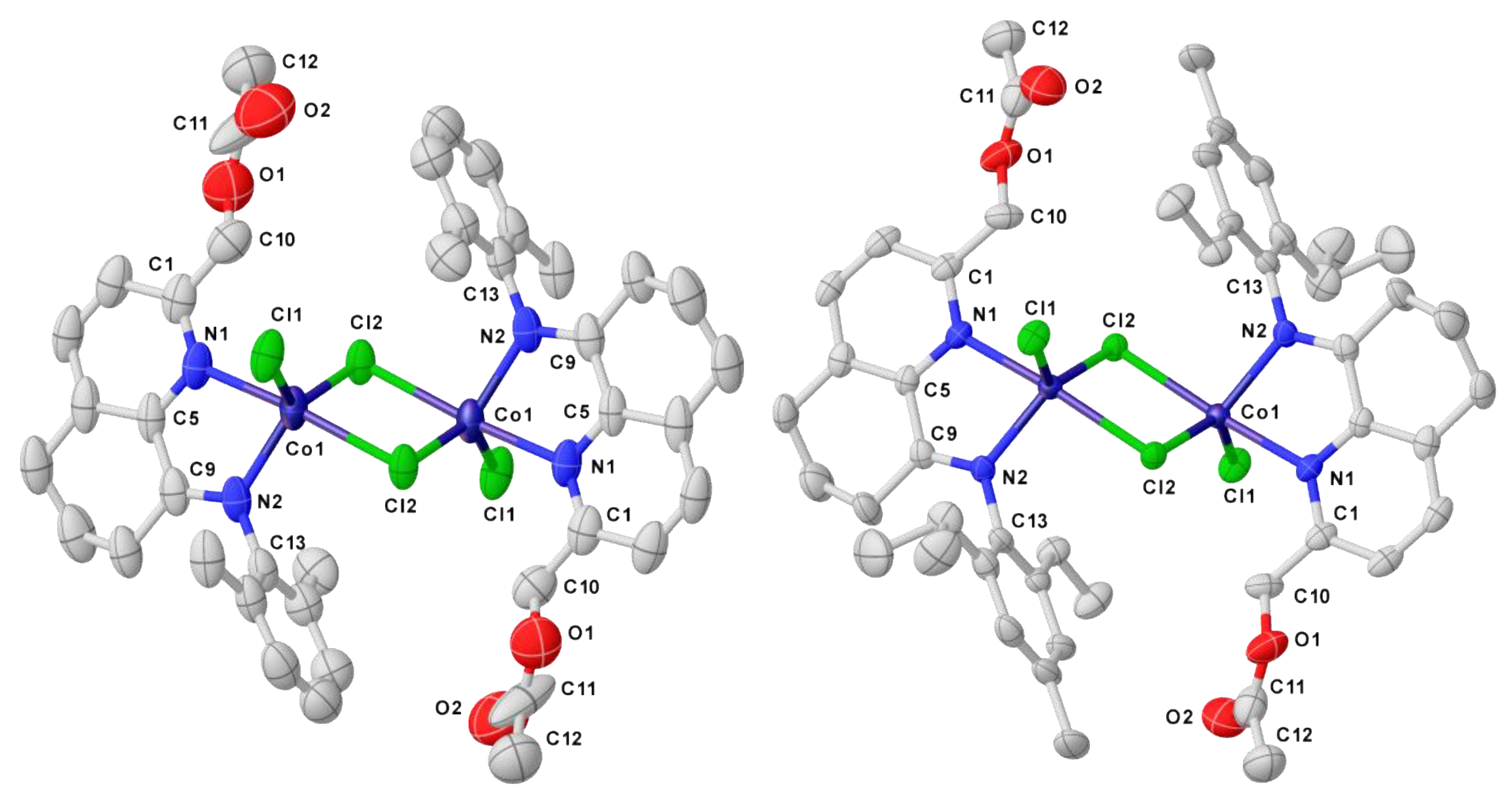
3.4. X-ray Crystallographic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, D.; Li, S.; Cui, D.; Zhang, X. β-Diketiminato rare-earth metal complexes: Structures, catalysis, and active species for highly cis-1,4-selective polymerization of isoprene. Organometallics 2010, 29, 2186–2193. [Google Scholar] [CrossRef]
- Ouardad, S.; Bakleh, M.E.; Kostjuk, S.V.; Ganachaud, F.; Puskas, J.E.; Deffieux, A.; Peruch, F. Bio-inspired cationic polymerization of isoprene and analogues: State-of-the-art. Polym. Int. 2012, 61, 149–156. [Google Scholar] [CrossRef]
- Ricci, G.; Pampaloni, G.; Sommazzi, A.; Masi, F. Dienes polymerization: Where we are and what lies ahead. Macromolecules 2021, 54, 5879–5914. [Google Scholar] [CrossRef]
- Ricci, G.; Sommazzi, A.; Masi, F.; Ricci, M.; Boglia, A.; Leone, G. Well-defined transition metal complexes with phosphorus and nitrogen ligands for 1,3-dienes polymerization. Coord. Chem. Rev. 2010, 254, 661–676. [Google Scholar] [CrossRef]
- Song, J.S.; Huang, B.C.; Yu, D.S. Progress of synthesis and application of trans-1,4-polyisoprene. J. App. Polym. Sci. 2001, 82, 81–89. [Google Scholar] [CrossRef]
- Nakajima, N. Science and Practice of Rubber Mixing; Rapra Technology Ltd.: Shawbury, Shropshire, UK, 2000. [Google Scholar]
- Friebe, L.; Nuyken, O.; Obrecht, W. Neodymium-Based ziegler/natta catalysts and their application in diene polymerization. Adv. Polym. Sci. 2006, 204, 1–154. [Google Scholar]
- Wang, B.; Cui, D.; Lv, K. Highly 3,4-selective living polymerization of isoprene with rare earth metal fluorenyl N-heterocyclic carbene precursors. Macromolecules 2008, 41, 1983–1988. [Google Scholar] [CrossRef]
- Liu, H.; He, J.; Liu, Z.; Lin, Z.; Du, G.; Zhang, S.; Li, X. Quasi-living trans-1,4-polymerization of isoprene by cationic rare earth metal alkyl species bearing a chiral (S,S)-bis(oxazolinylphenyl)amido ligand. Macromolecules 2013, 46, 3257–3265. [Google Scholar] [CrossRef]
- Jia, A.Q.; Wang, J.Q.; Hu, P.; Jin, G.X. Syntheses, reactions, and ethylene polymerization of titanium complexes with [N,N,S] ligands. Dalton Trans. 2011, 40, 7730–7736. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-based catalysts for polymerization of ethylene and α-olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Killian, C.M.; Tempel, D.J.; Johnson, L.K.; Brookhart, M. Living polymerization of α-olefins using Ni(II)-α-diimine catalysts. synthesis of new block polymers based on α-olefins. J. Am. Chem. Soc. 1996, 118, 11664–11665. [Google Scholar] [CrossRef]
- Small, B.L.; Brookhart, M.; Bennett, A.M.A. Highly active iron and cobalt catalysts for the polymerization of ethylene. J. Am. Chem. Soc. 1998, 120, 4049–4050. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Gibson, V.C.; Kimberley, B.S.; Maddox, P.J.; McTavish, S.J.; Solan, G.A.; White, A.J.P.; Williams, D.J. Novel olefin polymerization catalysts based on iron and cobalt. Chem. Commun. 1998, 7, 849–850. [Google Scholar] [CrossRef]
- Champouret, Y.; Hashmi, O.H.; Visseaux, M. Discrete iron-based complexes: Applications in homogeneous coordination-insertion polymerization catalysis. Coord. Chem. Rev. 2019, 390, 127–170. [Google Scholar] [CrossRef]
- Alnajrani, M.N.; Mair, F.S. Bidentate forms of β-triketimines: Syntheses, characterization and outstanding performance of enamine-diimine cobalt complexes in isoprene polymerization. Dalton Trans. 2016, 45, 10435–10446. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, H.; Li, W.; Jia, X.; Zhang, X.; Gong, D. Polymerization of isoprene promoted by aminophosphine(ory)-fused bipyridine cobalt complexes: Precise control of molecular weight and cis-1,4-alt-3,4 Sequence. Inorg. Chem. 2018, 57, 4088–4097. [Google Scholar] [CrossRef]
- Raynaud, J.; Wu, J.Y.; Ritter, T. Iron-catalyzed polymerization of isoprene and other 1,3-Dienes. Angew. Chem. Int. Ed. 2012, 51, 11805–11808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, X.; Hou, H.; Zhu, G.; Han, Z.; Yang, W.; Chen, X.; Wang, Q. An unsymmetrical binuclear iminopyridine-iron complex and its catalytic isoprene polymerization. Chem. Commun. 2020, 56, 8846–8849. [Google Scholar] [CrossRef]
- Fang, L.; Zhao, W.; Han, C.; Liu, H.; Hu, Y.; Zhang, X. Isoprene polymerization with pyrazolylimine cobalt (II) complexes: Manipulation 3,4-selectivities by ligand design and triphenyl phosphine. Eur. J. Inorg. Chem. 2019, 2019, 609–616. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, G.; Mahmood, Q.; Zhao, M.; Wang, L.; Jing, C.; Wang, X.; Wang, Q. Iminoimidazole-based Co(II) and Fe(II) complexes: Syntheses, characterization, and catalytic behaviors for isoprene polymerization. J. Polym. Sci. PART A Polym. Chem. 2019, 57, 767–775. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, Y.; Zhang, X.; Wang, L.; Zhu, G.; Wang, Q. Synthesis, characterization and catalytic property studies for isoprene polymerization of iron complexes bearing unionized pyridine-oxime ligands. Polymers 2022, 14, 3612. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, X.; Zhao, W.; Sun, W.-H.; Yao, W.; He, A. Catalytic behavior of Co(II) complexes with 2-(Benzimidazolyl)-6-(1-(arylimino)ethyl)pyridine ligands on isoprene stereospecific polymerization. J. Appl. Polym. Sci. 2013, 131, 1. [Google Scholar] [CrossRef]
- Jiang, X.; Wen, X.; Sun, W.-H.; He, A. Polymerization of isoprene catalyzed by 2-(Methyl-2 benzimidazolyl)-6-(1-(arylimino) ethyl) Pyridine Iron(III) trichloride with an additional donor. J. Polym. Sci. PART A Polym. Chem. 2014, 52, 2395–2398. [Google Scholar] [CrossRef]
- He, A.; Wang, G.; Zhao, W.; Jiang, X.; Yao, W.; Sun, W.-H. High cis-1,4 polyisoprene or cis-1,4/3,4 binary polyisoprene synthesized using 2-(benzimidazolyl)-6-(1-(arylimino)ethyl)pyridine cobalt(II) dichlorides. Polym. Int. 2013, 62, 1758–1766. [Google Scholar] [CrossRef]
- Alnajrani, M.N.; Mair, F.S. The behaviour of β-triketimine cobalt complexes in the polymerization of isoprene. RSC Adv. 2015, 5, 46372–46385. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.; Mahmood, Q.; Jing, C.; Zhu, G.; Zhang, X.; Wang, X.; Wang, Q. Controlled isoprene polymerization mediated by iminopyridine-iron (II) acetylacetonate pre-catalysts. Appl. Organometal. Chem. 2019, 33, e4836. [Google Scholar] [CrossRef]
- Hashmi, O.H.; Champouret, Y.; Visseaux, M. Highly active iminopyridyl iron-based catalysts for the polymerization of isoprene. Molecules 2019, 24, 3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Wang, L.; Mahmood, Q.; Zhou, L.; Wang, Q. Ligand-regulated polymerization of conjugated dienes catalyzed by confined iminopyridine iron complexes with high activity and thermal stability. Polym. Test. 2021, 102, 107317. [Google Scholar] [CrossRef]
- Ricci, G.; Leone, G.; Zanchin, G.; Palucci, B.; Boccia, A.C.; Sommazzi, A.; Masi, F.; Zacchini, S.; Guelfi, M.; Pampaloni, G. Highly stereoregular 1,3-butadiene and isoprene polymers through monoalkyl-N-aryl substituted iminopyridine iron complex-based catalysts: Synthesis and characterization. Macromolecules 2021, 54, 9947–9959. [Google Scholar] [CrossRef]
- Guo, L.; Jing, X.; Xiong, S.; Liu, W.; Liu, Y.; Liu, Z.; Chen, C. Influences of alkyl and aryl substituents on iminopyridine Fe(II)- and Co(II)-catalyzed isoprene polymerization. Polymers 2016, 8, 389. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Zhang, X.; Zhao, M.; Wang, L.; Jing, C.; Wang, P.; Wang, X.; Wang, Q. Influences of fluorine substituents on iminopyridine Fe(II)- and Co(II)-catalyzed isoprene polymerization. Polymers 2018, 10, 934. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Zhang, L.; Suo, H.; Vignesh, A.; Yousuf, N.; Hao, X.; Sun, W.-H. Synthesis of characteristic polyisoprenes using rationally designed iminopyridyl metal (Fe and Co) precatalysts: Investigation of co-catalysts and steric influence on their catalytic activity. New J. Chem. 2020, 44, 8076–8084. [Google Scholar] [CrossRef]
- Wang, X.; Fan, L.; Huang, C.; Liang, T.; Guo, C.Y.; Sun, W.-H. Highly cis-1,4 selective polymerization of isoprene promoted by α-diimine cobalt(II) chlorides. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3609–3615. [Google Scholar] [CrossRef]
- Jing, C.; Wang, L.; Zhu, G.; Hou, H.; Zhou, L.; Wang, Q. Enhancing thermal stability in aminopyridine iron(II)-catalyzed polymerization of conjugated dienes. Organometallics 2020, 39, 4019–4026. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, X.; Wang, L.; Wang, L.; Zhu, G.; Wang, Q. Pyridine-oxazoline ligated iron complexes: Synthesis, characterization, and catalytic activity for isoprene polymerization. Appl. Organomet. Chem. 2022, 36, e6848. [Google Scholar] [CrossRef]
- Yu, J.; Zeng, Y.; Huang, W.; Hao, X.; Sun, W.-H. N-(5,6,7-trihydroquinolin-8-ylidene)arylaminonickel dichlorides as highly active single-site pro-catalysts in ethylene polymerization. Dalton Trans. 2011, 40, 8436–8443. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yue, E.; Qu, M.; Oleynik, I.V.; Oleynik, I.I.; Li, K.; Liang, T.; Zhang, W.; Sun, W.-H. 8-(2-cycloalkylphenylimino)-5,6,7-trihydroquinolylnickel halides: Polymerizing ethylene to highly branched and lower molecular weight polyethylenes. Inorg. Chem. Front. 2015, 2, 223–227. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, W.; Mahmood, Q.; Zhang, R.; Sun, Y.; Sun, W.-H. Vinyl/vinylene functionalized highly Branched polyethylene waxes obtained using electronically controlled cyclohexyl-fused pyridinylimine-nickel precatalysts. J. Polym. Sci. PART A Polym. Chem. 2018, 56, 1269–1281. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q.; Hu, X.; Ma, Y.; Solan, G.A.; Sun, Y.; Sun, W.-H. 2-Acetyloxymethyl-substituted 5,6,7-trihydroquinolinyl-8-ylideneamine-Ni(II) chlorides and their application in ethylene dimerization/trimerization. Appl. Organomet. Chem. 2019, 34, e5254. [Google Scholar] [CrossRef]
- Jiang, S.; Zheng, Y.; Liu, M.; Yu, Z.; Ma, Y.; Solan, G.A.; Zhang, W.; Liang, T.; Sun, W.-H. Polyethylene waxes with short chain branching via steric and electronic tuning of an 8-(arylimino)-5,6,7-trihydroquinoline-nickel catalyst. Organometallics 2022, 41, 3197–3211. [Google Scholar] [CrossRef]
- Lin, W.; Liu, M.; Xu, L.; Ma, Y.; Zhang, L.; Flisak, Z.; Hu, X.; Liang, T.; Sun, W.-H. Nickel(II) complexes with sterically hindered 5,6,7-trihydroquinoline derivatives selectively dimerizing ethylene to 1-butene. Appl. Organomet. Chem. 2022, 36, e6596. [Google Scholar] [CrossRef]
- Xua, L.; Li, J.; Lin, W.; Ma, Y.; Hua, X.; Flisak, Z.; Sun, W.-H. Ethylene oligomerization with 2-hydroxymethyl-5,6,7-trihydroquinolinyl-8-ylideneamine-Ni(II) chlorides. J. Organom. Chem. 2021, 937, 121720. [Google Scholar] [CrossRef]
- Hou, X.; Cai, Z.; Chen, X.; Wang, L.; Redshaw, C.; Sun, W.-H. N-(5,6,7-trihydroquinolin-8-ylidene)-2-benzhydrylbenzenaminonickel halide complexes: Synthesis, characterization and catalytic behavior towards ethylene polymerization. Dalton Trans. 2012, 41, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.; Wang, L.; Mahmood, Q.; Zhao, M.; Zhu, G.; Zhang, X.; Wang, X.; Wang, Q. Synthesis and characterization of aminopyridine iron(II) chloride catalysts for isoprene polymerization: Sterically controlled monomer enchainment. Dalton Trans. 2019, 48, 7862–7874. [Google Scholar] [CrossRef]
- Zhang, L.; Hao, X.; Sun, W.-H.; Redshaw, C. Synthesis, characterization, and ethylene polymerization behavior of 8-(nitroarylamino)-5,6,7-trihydroquinolylnickel dichlorides: Influence of the nitro group and impurities on catalytic activity liping. ACS Catal. 2011, 1, 1213–1220. [Google Scholar] [CrossRef]
- Bonnet, F.; Dyer, H.E.; Kinani, Y.E.; Dietz, C.; Roussel, P.; Bria, M.; Visseaux, M.; Zinck, P.; Mountford, P. Bis(phenolate)amine-supported lanthanide borohydride complexes for styrene and trans-1,4-isoprene (co-)polymerisations. Dalton Trans. 2015, 44, 12312–12325. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL-97, Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co1 | Co5 | |
|---|---|---|
| Bond lengths (Å) | ||
| Co1–N1 | 2.172 (4) | 2.157 (3) |
| Co1–N2 | 2.056 (4) | 2.058 (3) |
| Co1–Cl1 | 2.2812 (14) | 2.2664 (11) |
| Co1–Cl2 | 2.3422 (12) | 2.3420 (10) |
| N1–C1 | 1.321 (7) | 1.334 (5) |
| N1–C5 | 1.345 (7) | 1.357 (5) |
| N2–C9 | 1.292 (6) | 1.295 (5) |
| N2–C13 | 1.440 (7) | 1.441 (5) |
| Bond Angles (o) | ||
| N1–Co1–Cl2 | 89.42 (10) | 89.57 (9) |
| N1–Co1–N2 | 77.53 (16) | 77.90 (13) |
| N2–Co1–Cl2 | 97.01 (11) | 97.52 (10) |
| Cl2–Co1–Cl2 | 85.45 (3) | 85.48 (3) |
| N2–Co1–Cl1 | 115.08 (11) | 111.70 (10) |
| Cl1–Co1–Cl2 | 128.79 (6) | 130.19 (5) |
| N1–Co1–Cl1 | 92.02 (11) | 91.29 (9) |
| Entry | Co-Cat. | Al/Co | Conv. b (%) | Act. c | Mn d (105, g/mol) | PDI d | Microstructure e | ||
|---|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 3,4 | |||||||
| 1 | MAO | 100 | trace | - | - | - | - | - | - |
| 2 | AlMe3 | 50 | trace | - | - | - | - | - | - |
| 3 | AlMe2Cl | 50 | 16 | 0.22 | 0.1 | 1.2 | 74 | 1 | 25 |
| 4 | AlMe2Cl | 40 | 22 | 0.30 | 2.9 | 1.7 | 71 | 4 | 25 |
| 5 | AlMe2Cl | 20 | 29 | 0.40 | 2.8 | 1.6 | 71 | 5 | 24 |
| 6 | AlMe2Cl | 10 | 18 | 0.25 | 2.6 | 2.3 | 70 | 2 | 28 |
| Entry | Temp. (°C) | Time (min) | Conv. (%) b | Act. c | Mnd (105 g/mol) | PDI d | Microstructure e | ||
|---|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 3,4 | |||||||
| 1 | 25 | 60 | 29 | 0.40 | 2.8 | 1.6 | 71 | 5 | 24 |
| 2 | 40 | 60 | 35 | 0.48 | 2.2 | 2.0 | 73 | 2 | 25 |
| 3 | 50 | 60 | 59 | 0.80 | 1.4 | 2.3 | 73 | 1 | 26 |
| 4 | 60 | 60 | >99 | 1.37 | 2.6 | 3.2 | 72 | 1 | 27 |
| 5 | 70 | 60 | 88 | 1.20 | 4.5 | 2.5 | 67 | 1 | 32 |
| 6 | 80 | 60 | 82 | 1.12 | 3.4 | 2.2 | 66 | 1 | 33 |
| 7 | 60 | 5 | 57 | 0.78 | 3.2 | 2.1 | 65 | 1 | 34 |
| 8 | 60 | 15 | 66 | 0.90 | 3.8 | 2.1 | 65 | 2 | 34 |
| 9 | 60 | 30 | 70 | 0.95 | 3.3 | 2.3 | 65 | 1 | 34 |
| 10 | 60 | 45 | 79 | 1.08 | 2.1 | 2.2 | 71 | 1 | 28 |
| 11 f | 60 | 60 | 53 | 1.44 | 1.5 | 3.8 | 65 | 2 | 33 |
| 12 g | 60 | 60 | 47 | 2.56 | 2.1 | 2.8 | 65 | 3 | 32 |
| Entry | Cat. | Conv. (%) b | Act. c | Mn d (105) | PDI d | Microstructure e | ||
|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 3,4 | ||||||
| 1 | Co1 | >99 | 1.37 | 2.6 | 3.2 | 72 | 1 | 27 |
| 2 | Co2 | 98 | 1.34 | 2.4 | 2.3 | 65 | 1 | 34 |
| 3 | Co3 | 90 | 1.23 | 2.8 | 2.7 | 68 | 1 | 31 |
| 4 | Co4 | 96 | 1.30 | 3.1 | 1.9 | 69 | 1 | 30 |
| 5 | Co5 | 94 | 1.28 | 3.3 | 1.7 | 67 | 1 | 32 |
| 6 | Co6 | 82 | 1.12 | 3.4 | 2.3 | 69 | 1 | 30 |
| Co1 | Co5 | |
|---|---|---|
| Identification code | 2270716 | 2270717 |
| Empirical formula | C20H22Cl2CoN2O2 | C23H28Cl2CoN2O2 |
| Formula weight | 452.24 | 494.32 |
| Temperature/K | 169.98 (10) | 169.99 (10) |
| Crystal system | monoclinic | triclinic |
| Space group | P21/n | P-1 |
| a/Å | 9.1464 (4) | 9.1668 (3) |
| b/Å | 22.0899 (15) | 10.4116 (4) |
| c/Å | 10.6237 (4) | 13.8557 (5) |
| α/° | 90 | 71.220 (3) |
| β/° | 106.870 (5) | 87.520 (3) |
| γ/° | 90 | 69.207 (3) |
| Volume/Å3 | 2054.08 (19) | 1166.68 (8) |
| Z | 4 | 2 |
| ρcalcg/cm3 | 1.462 | 1.419 |
| μ/mm−1 | 9.048 | 8.024 |
| F(000) | 928.6 | 518.0 |
| Crystal size/mm3 | 0.15 × 0.10 × 0.05 | 0.15 × 0.10 × 0.05 |
| Radiation | Cu Kα (λ = 1.54184) | Cu Kα (λ = 1.54184) |
| 2Θ range for data collection/ | 8.0 to 151.5 | 6.8 to 151.5 |
| Index ranges | −11 ≤ h ≤ 11 −25 ≤ k ≤ 27 −12 ≤ l ≤ 10 | −11 ≤ h ≤ 11 −13 ≤ k ≤ 12 −17 ≤ l ≤ 17 |
| Reflections collected | 17,017 | 13,334 |
| Independent reflections | 4097 [Rint = 0.0727, Rsigma = 0.0598] | 4645 [Rint = 0.0441, Rsigma = 0.0430] |
| Data/restraints/parameters | 4097/0/247 | 4645/1/282 |
| Goodness-of-fit on F2 | 1.032 | 1.040 |
| Final R indexes [I> = 2σ (I)] | R1 = 0.0595, wR2 = 0.1508 | R1 = 0.0636, wR2 = 0.1841 |
| Final R indexes [all data] | R1 = 0.0914, wR2 = 0.1734 | R1 = 0.0687, wR2 = 0.1884 |
| Largest diff. peak/hole/e Å−3 | 0.70/−0.72 | 1.07/−0.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousuf, N.; Ma, Y.; Mahmood, Q.; Zhang, W.; Liu, M.; Yuan, R.; Sun, W.-H. Structurally Rigid (8-(Arylimino)-5,6,7-trihydroquinolin-2-yl)-methyl Acetate Cobalt Complex Catalysts for Isoprene Polymerization with High Activity and cis-1,4 Selectivity. Catalysts 2023, 13, 1120. https://doi.org/10.3390/catal13071120
Yousuf N, Ma Y, Mahmood Q, Zhang W, Liu M, Yuan R, Sun W-H. Structurally Rigid (8-(Arylimino)-5,6,7-trihydroquinolin-2-yl)-methyl Acetate Cobalt Complex Catalysts for Isoprene Polymerization with High Activity and cis-1,4 Selectivity. Catalysts. 2023; 13(7):1120. https://doi.org/10.3390/catal13071120
Chicago/Turabian StyleYousuf, Nighat, Yanping Ma, Qaiser Mahmood, Wenjuan Zhang, Ming Liu, Rongyan Yuan, and Wen-Hua Sun. 2023. "Structurally Rigid (8-(Arylimino)-5,6,7-trihydroquinolin-2-yl)-methyl Acetate Cobalt Complex Catalysts for Isoprene Polymerization with High Activity and cis-1,4 Selectivity" Catalysts 13, no. 7: 1120. https://doi.org/10.3390/catal13071120